
Catalase Modulates the Radio-Sensitization of Pancreatic Cancer Cells by Pharmacological Ascorbate

 , , , and
, , , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Treatment of Cells with P-AscH−
2.3. Western Blot Analysis
2.4. Generation of RelB or Catalase CRISPR/Cas9 Knockout Cells
2.5. Catalase Activity Assay
2.6. Clonogenic Assay
2.7. Measurement of Cellular Metabolic Flux
2.8. Confocal Microscopy (Catalase Immunofluorescence)
2.9. Follow-Up of Phase I Trial Long Term Survivors
3. Results
3.1. Activation of Nuclear Factor-Erythroid 2-Related Factor 2 Nrf2
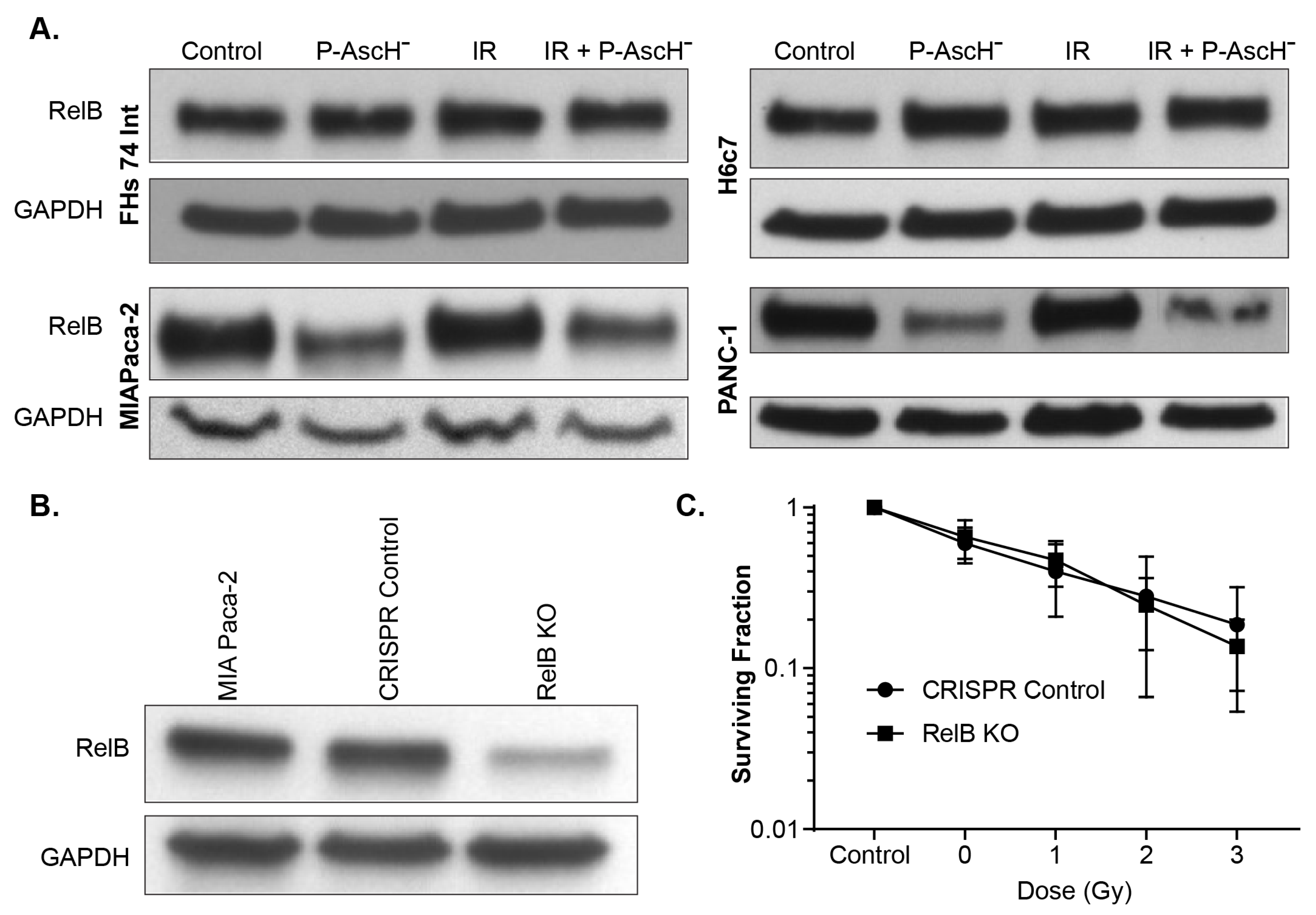
3.2. RelB in Radiation and P-AscH− Response in PDAC
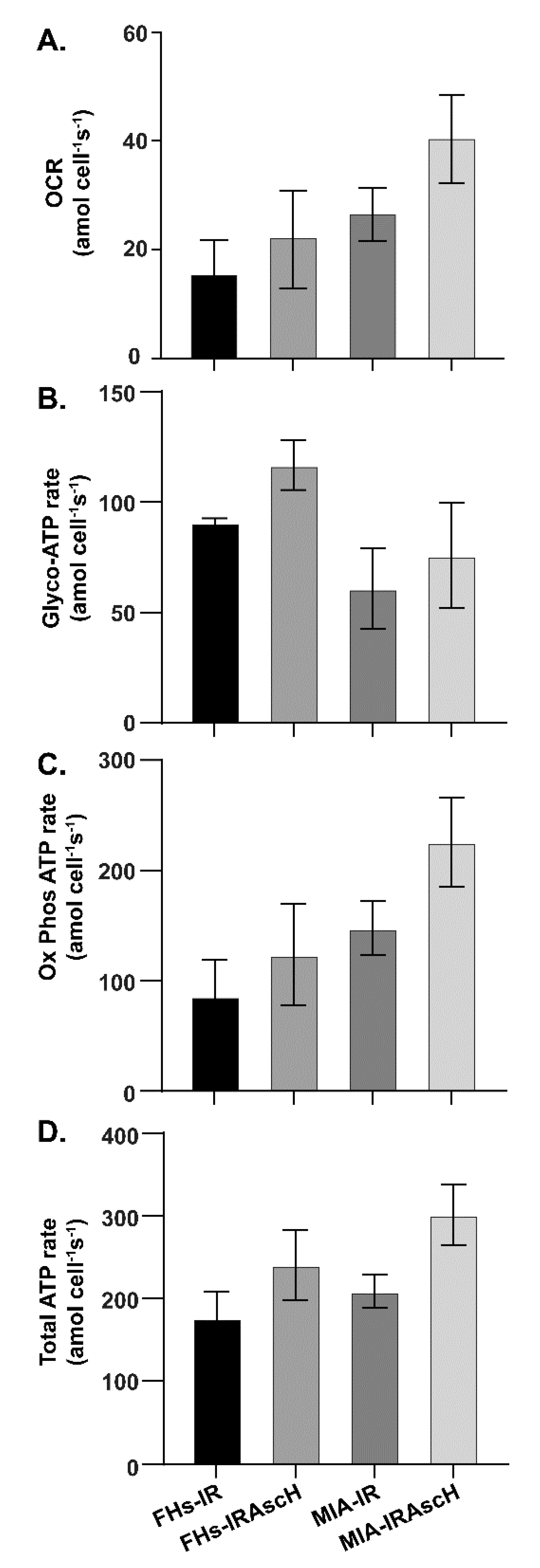
3.3. No Changes in Bioenergetics in Normal and PDAC Cells after Exposure to P-AscH− and Radiation
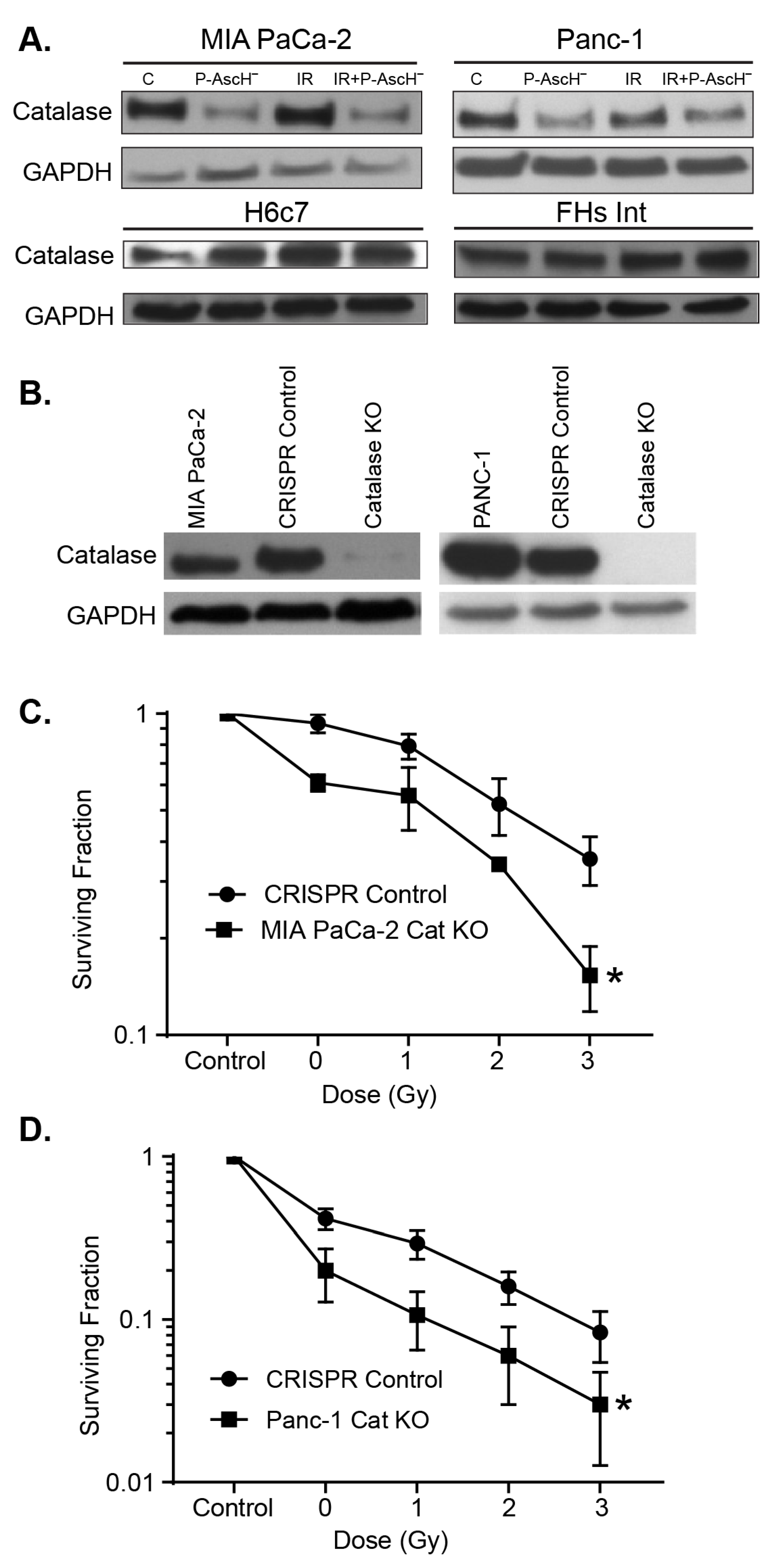
3.4. Catalase Affects Radiosensitization
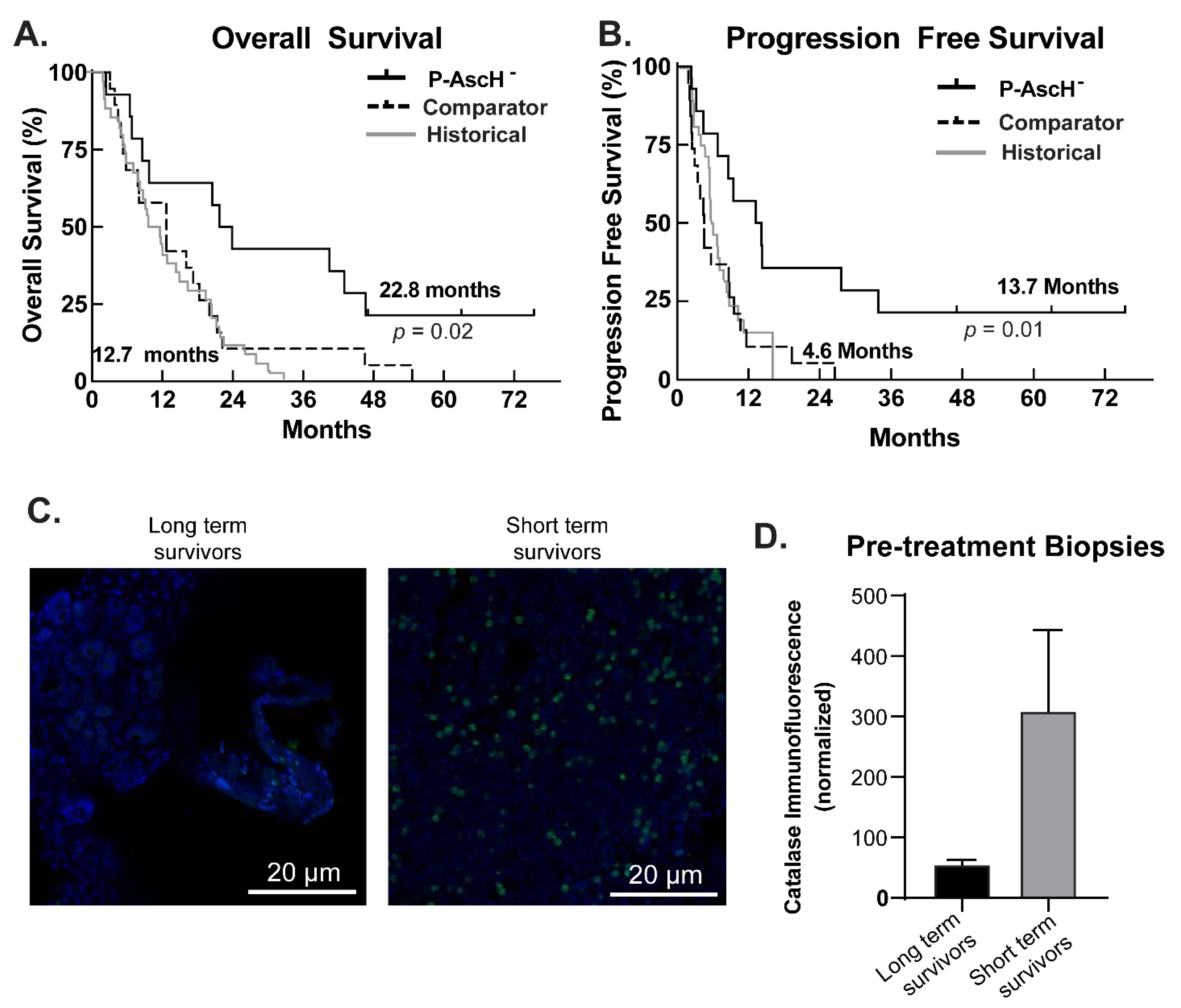
3.5. Phase I Trial, Follow-Up Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Iacobuzio-Donahue, C.A.; Fu, B.; Yachida, S.; Luo, M.; Abe, H.; Henderson, C.M.; Vilardell, F.; Wang, Z.; Keller, J.W.; Banerjee, P.; et al. DPC4 Gene Status of the Primary Carcinoma Correlates with Patterns of Failure in Patients with Pancreatic Cancer. J. Clin. Oncol. 2009, 27, 1806–1813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, J.; Nelson, E.S.; Simons, A.L.; Olney, K.E.; Moser, J.C.; Schrock, H.E.; Wagner, B.A.; Buettner, G.R.; Smith, B.J.; Teoh, M.L.; et al. Regulation of Pancreatic Cancer Growth by Superoxide. Mol. Carcinog. 2013, 52, 555–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aykin-Burns, N.; Ahmad, I.M.; Zhu, Y.; Oberley, L.W.; Spitz, D.R. Increased Levels of Superoxide and H2O2 Mediate the Differential Susceptibility of Cancer Cells vs. Normal Cells to Glucose Deprivation. Biochem. J. 2009, 418, 29–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oberley, L.W.; Buettner, G.R. Role of Superoxide Dismutase in Cancer: A Review. Cancer Res. 1979, 39, 1141–1149. [Google Scholar]
- Szatrowski, T.P.; Nathan, C.F. Production of Large Amounts of Hydrogen Peroxide by Human Tumor Cells. Cancer Res. 1991, 51, 794–798. [Google Scholar] [PubMed]
- Alexander, M.S.; Wilkes, J.G.; Schroeder, S.R.; Buettner, G.R.; Wagner, B.A.; Du, J.; Gibson-Corley, K.; O’Leary, B.R.; Spitz, D.R.; Buatti, J.M.; et al. Pharmacologic Ascorbate Reduces Radiation-Induced Normal Tissue Toxicity and Enhances Tumor Radiosensitization in Pancreatic Cancer. Cancer Res. 2018, 78, 6838–6851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, J.; Martin, S.M.; Levine, M.; Wagner, B.A.; Buettner, G.R.; Wang, S.H.; Taghiyev, A.F.; Du, C.; Knudson, C.M.; Cullen, J.J. Mechanisms of Ascorbate-induced Cytotoxicity in Pancreatic Cancer. Clin. Cancer Res. 2010, 16, 509–520. [Google Scholar] [CrossRef] [Green Version]
- Gibson, A.R.; O’Leary, B.R.; Du, J.; Sarsour, E.H.; Kalen, A.L.; Wagner, B.A.; Stolwijk, J.M.; Falls-Hubert, K.C.; Alexander, M.S.; Carroll, R.S.; et al. Dual Oxidase-Induced Sustained Generation of Hydrogen Peroxide Contributes to Pharmacologic Ascorbate-Induced Cytotoxicity. Cancer Res. 2020, 80, 1401–1413. [Google Scholar] [CrossRef]
- Welsh, J.L.; Wagner, B.A.; van’t Erve, T.J.; Zehr, P.S.; Berg, D.J.; Halfdanarson, T.R.; Yee, N.S.; Bodeker, K.L.; Du, J.; Cullen, J.J.; et al. Pharmacological Ascorbate with Gemcitabine for the Control of Metastatic and Node-positive Pancreatic Cancer (PACMAN): Results from a Phase I Clinical Trial. Cancer Chemother. Pharmacol. 2013, 71, 765–775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghanem, A.; Melzer, A.M.; Zaal, E.; Neises, L.; Baltissen, D.; Matar, O.; Glennemeier-Marke, H.; Almouhanna, F.; Theobald, J.; Abu El Maaty, M.A.; et al. Ascorbate Kills Breast Cancer Cells by Rewiring Metabolism via Redox Imbalance and Energy Crisis. Free Radic. Biol. Med. 2021, 163, 196–209. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Wagner, B.A.; Buettner, G.R.; Cullen, J.J. Role of Labile Iron in the Toxicity of Pharmacological Ascorbate. Free Radic. Biol. Med. 2015, 84, 289–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erudaitius, D.; Huang, A.; Kazmi, S.; Buettner, G.R.; Rodgers, V.G. Peroxiporin Expression Is an Important Factor for Cancer Cell Susceptibility to Therapeutic H2O2: Implications for Pharmacological Ascorbate Therapy. PLoS ONE 2017, 12, e0170442. [Google Scholar] [CrossRef] [PubMed]
- Doskey, C.M.; Buranasudja, V.; Wagner, B.A.; Wilkes, J.G.; Du, J.; Cullen, J.J.; Buettner, G.R. Tumor Cells Have Decreased Ability to Metabolize H2O2: Implications for Pharmacological Ascorbate in Cancer Therapy. Redox Biol. 2016, 10, 274–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vislisel, J.M.; Schafer, F.Q.; Buettner, G.R. A Simple and Sensitive Assay for Ascorbate Using a Plate Reader. Anal. Biochem. 2007, 365, 31–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, J.; Welsh, J.L.; Sibenaller, Z.A.; Allen, B.G.; Wagner, B.A.; Kalen, A.L.; Doskey, C.M.; Strother, R.K.; Button, A.M.; Mott, S.L.; et al. Pharmacological Ascorbate Radiosensitizes Pancreatic Cancer. Cancer Res. 2015, 75, 3314–3326. [Google Scholar] [CrossRef] [Green Version]
- Buettner, G.R.; Jurkiewicz, B.A. Catalytic Metals, Ascorbate and Free Radicals: Combinations to Avoid. Radiat. Res. 1996, 145, 532–541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weydert, C.J.; Cullen, J.J. Measurement of Superoxide Dismutase, Catalase and Glutathione Peroxidase in Cultured Cells and Tissue. Nat. Protoc. 2010, 5, 51–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doskey, C.M.; van’t Erve, T.J.; Wagner, B.A.; Buettner, G.R. Moles of a Substance per Cell Is a Highly Informative Dosing Metric in Cell Culture. PLoS ONE 2015, 10, e0132572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aebi, H. Catalase in vitro. Methods Enzymol. 1984, 105, 121–126. [Google Scholar] [PubMed]
- Beers, R.F., Jr.; Sizer, I.W. A Spectrophotometric Method for Measuring the Breakdown of Hydrogen Peroxide by Catalase. J. Biol. Chem. 1952, 195, 133–140. [Google Scholar] [CrossRef]
- Storer, B.E. Design and Analysis of Phase I Clinical Trials. Biometrics 1989, 45, 925–937. [Google Scholar] [CrossRef] [PubMed]
- Loehrer, P.J., Sr.; Feng, Y.; Cardenes, H.; Wagner, L.; Brell, J.M.; Cella, D.; Flynn, P.; Ramanathan, R.K.; Crane, C.H.; Alberts, S.R.; et al. Gemcitabine Alone vs. Gemcitabine Plus Radiotherapy in Patients with Locally Advanced Pancreatic Cancer: An Eastern Cooperative Oncology Group Trial. J. Clin. Oncol. 2011, 29, 4105–4112. [Google Scholar] [CrossRef]
- Schoenfeld, J.D.; Sibenaller, Z.A.; Mapuskar, K.A.; Wagner, B.A.; Cramer-Morales, K.L.; Furqan, M.; Sandhu, S.; Carlisle, T.L.; Smith, M.C.; Abu Hejleh, T.; et al. O2(-) and H2O2-Mediated Disruption of Fe Metabolism Causes the Differential Susceptibility of NSCLC and GBM Cancer Cells to Pharmacological Ascorbate. Cancer Cell 2017, 31, 487–500 e8. [Google Scholar] [CrossRef] [Green Version]
- Sekhar, K.R.; Freeman, M.L. Nrf2 Promotes Survival Following Exposure to Ionizing Radiation. Free Radic. Biol. Med. 2015, 88, 268–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.B.; Pandita, R.K.; Eskiocak, U.; Ly, P.; Kaisani, A.; Kumar, R.; Cornelius, C.; Wright, W.E.; Pandita, T.K.; Shay, J.W. Targeting of Nrf2 Induces DNA Damage Signaling and Protects Colonic Epithelial Cells from Ionizing Radiation. Proc. Natl. Acad. Sci. USA 2012, 109, E2949–E2955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, X.; Xu, Y.; Xu, F.F.; Chaiswing, L.; Schnell, D.; Noel, T.; Wang, C.; Chen, J.; St Clair, D.K.; St Clair, W.H. RelB Expression Determines the Differential Effects of Ascorbic Acid in Normal and Cancer Cells. Cancer Res. 2017, 77, 1345–1356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buranasudja, V.; Doskey, C.M.; Gibson, A.R.; Wagner, B.A.; Du, J.; Gordon, D.J.; Koppenhafer, S.L.; Cullen, J.J.; Buettner, G.R. Pharmacologic Ascorbate Primes Pancreatic Cancer Cells for Death by Rewiring Cellular Energetics and Inducing DNA Damage. Mol. Cancer Res. 2019, 17, 2102–2114. [Google Scholar] [CrossRef] [PubMed]
- Vidossich, P.; Alfonso-Prieto, M.; Rovira, C. Catalases vs. Peroxidases: DFT Investigation of H(2)O(2) Oxidation in Models Systems and Implications for Heme Protein Engineering. J. Inorg. Biochem. 2012, 117, 292–297. [Google Scholar] [CrossRef] [PubMed]
- Winterbourn, C.C. The Biological Chemistry of Hydrogen Peroxide. Methods Enzymol. 2013, 528, 3–25. [Google Scholar] [PubMed]
- Makino, N.; Sasaki, K.; Hashida, K.; Sakakura, Y. A Metabolic Model Describing the H2O2 Elimination by Mammalian Cells Including H2O2 Permeation through Cytoplasmic and Peroxisomal Membranes: Comparison with Experimental Data. Biochim. Biophys. Acta 2004, 1673, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Espey, M.G.; Krishna, M.C.; Mitchell, J.B.; Corpe, C.P.; Buettner, G.R.; Shacter, E.; Levine, M. Pharmacologic Ascorbic Acid Concentrations Selectively Kill Cancer Cells: Action as a Pro-drug to Deliver Hydrogen Peroxide to Tissues. Proc. Natl. Acad. Sci. USA 2005, 102, 13604–13609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rawal, M.; Schroeder, S.R.; Wagner, B.A.; Cushing, C.M.; Welsh, J.L.; Button, A.M.; Du, J.; Sibenaller, Z.A.; Buettner, G.R.; Cullen, J.J. Manganoporphyrins Increase Ascorbate-induced Cytotoxicity by Enhancing H2O2 Generation. Cancer Res. 2013, 73, 5232–5241. [Google Scholar] [CrossRef] [Green Version]
- Schafer, F.Q.; Buettner, G.R. Redox Environment of the Cell as Viewed through the Redox State of the Glutathione Disulfide/Glutathione Couple. Free Radic. Biol. Med. 2001, 30, 1191–1212. [Google Scholar] [CrossRef]
- MacMillan-Crow, L.A.; Greendorfer, J.S.; Vickers, S.M.; Thompson, J.A. Tyrosine Nitration of c-SRC Tyrosine Kinase in Human Pancreatic Ductal Adenocarcinoma. Arch. Biochem. Biophys. 2000, 377, 350–356. [Google Scholar] [CrossRef]
- Chen, Q.; Espey, M.G.; Sun, A.Y.; Lee, J.H.; Krishna, M.C.; Shacter, E.; Choyke, P.L.; Pooput, C.; Kirk, K.L.; Buettner, G.R.; et al. Ascorbate in Pharmacologic Concentrations Selectively Generates Ascorbate Radical and Hydrogen Peroxide in Extracellular Fluid in vivo. Proc. Natl. Acad. Sci. USA 2007, 104, 8749–8754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klingelhoeffer, C.; Kammerer, U.; Koospal, M.; Muhling, B.; Schneider, M.; Kapp, M.; Kubler, A.; Germer, C.T.; Otto, C. Natural resistance to ascorbic acid induced oxidative stress is mainly mediated by catalase activity in human cancer cells and catalase-silencing sensitizes to oxidative stress. BMC Complement. Altern. Med. 2012, 12, 61. [Google Scholar] [CrossRef] [PubMed] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Du, J.; Carroll, R.S.; Steers, G.J.; Wagner, B.A.; O’Leary, B.R.; Jensen, C.S.; Buettner, G.R.; Cullen, J.J. Catalase Modulates the Radio-Sensitization of Pancreatic Cancer Cells by Pharmacological Ascorbate. Antioxidants 2021, 10, 614. https://doi.org/10.3390/antiox10040614
Du J, Carroll RS, Steers GJ, Wagner BA, O’Leary BR, Jensen CS, Buettner GR, Cullen JJ. Catalase Modulates the Radio-Sensitization of Pancreatic Cancer Cells by Pharmacological Ascorbate. Antioxidants. 2021; 10(4):614. https://doi.org/10.3390/antiox10040614
Chicago/Turabian StyleDu, Juan, Rory S. Carroll, Garett J. Steers, Brett A. Wagner, Brianne R. O’Leary, Chris S. Jensen, Garry R. Buettner, and Joseph J. Cullen. 2021. "Catalase Modulates the Radio-Sensitization of Pancreatic Cancer Cells by Pharmacological Ascorbate" Antioxidants 10, no. 4: 614. https://doi.org/10.3390/antiox10040614