
Fecal Microbiota Transplantation Reduces Campylobacter jejuni Colonization in Young Broiler Chickens Challenged by Oral Gavage but Not by Seeder Birds

Abstract
:1. Introduction
2. Results
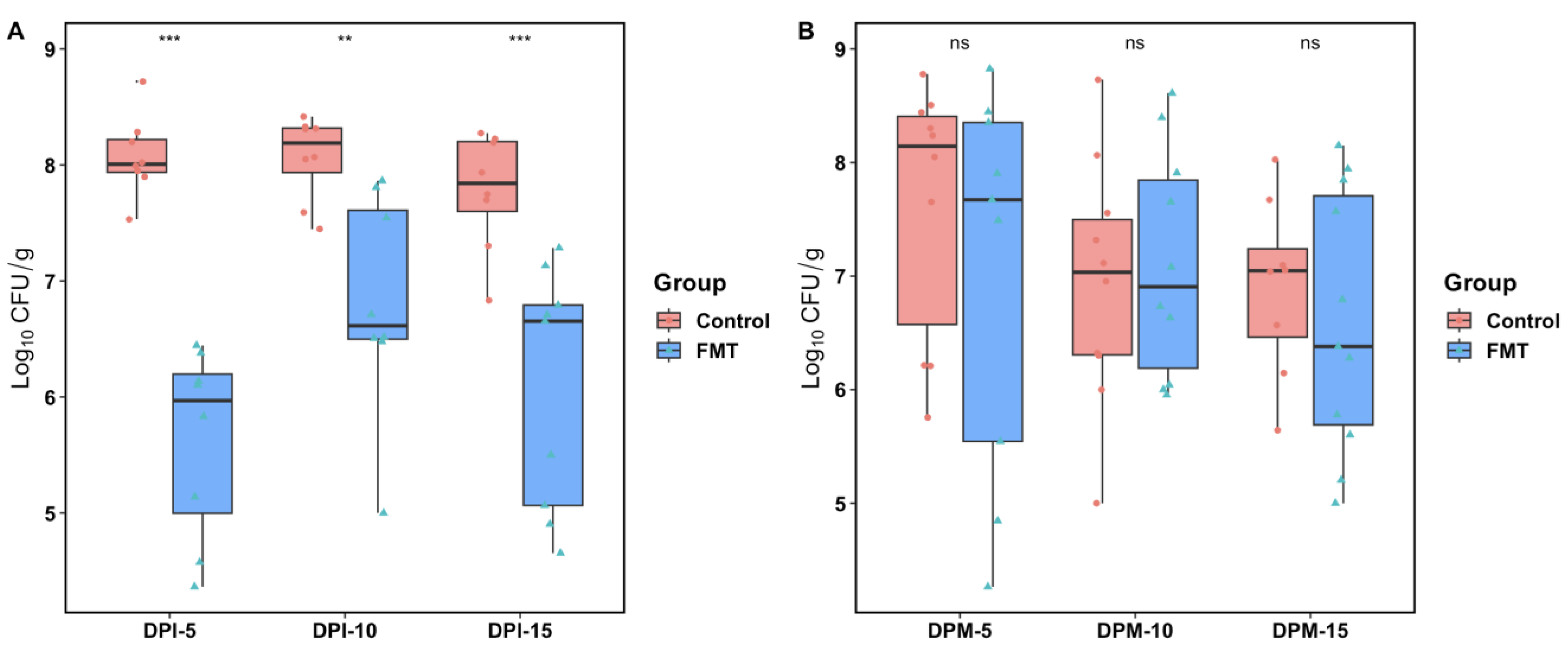
2.1. FMT Significantly Lowered C. jejuni Colonization in Broilers Challenged by Oral Gavage but Not via the Seeder Bird Infection Model
2.2. Microbiota Analysis
2.2.1. 16S rRNA Gene Sequencing Outputs Overview
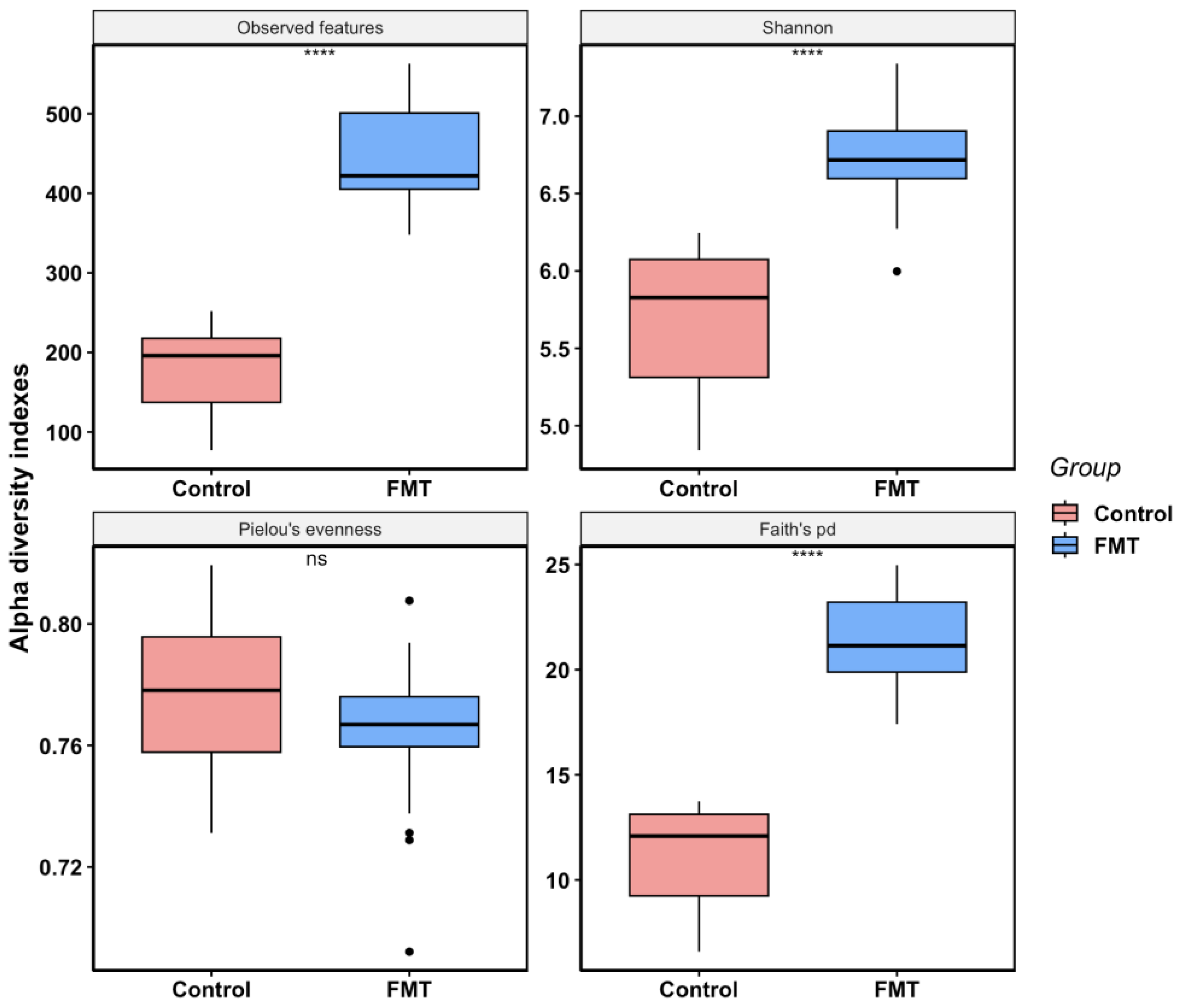
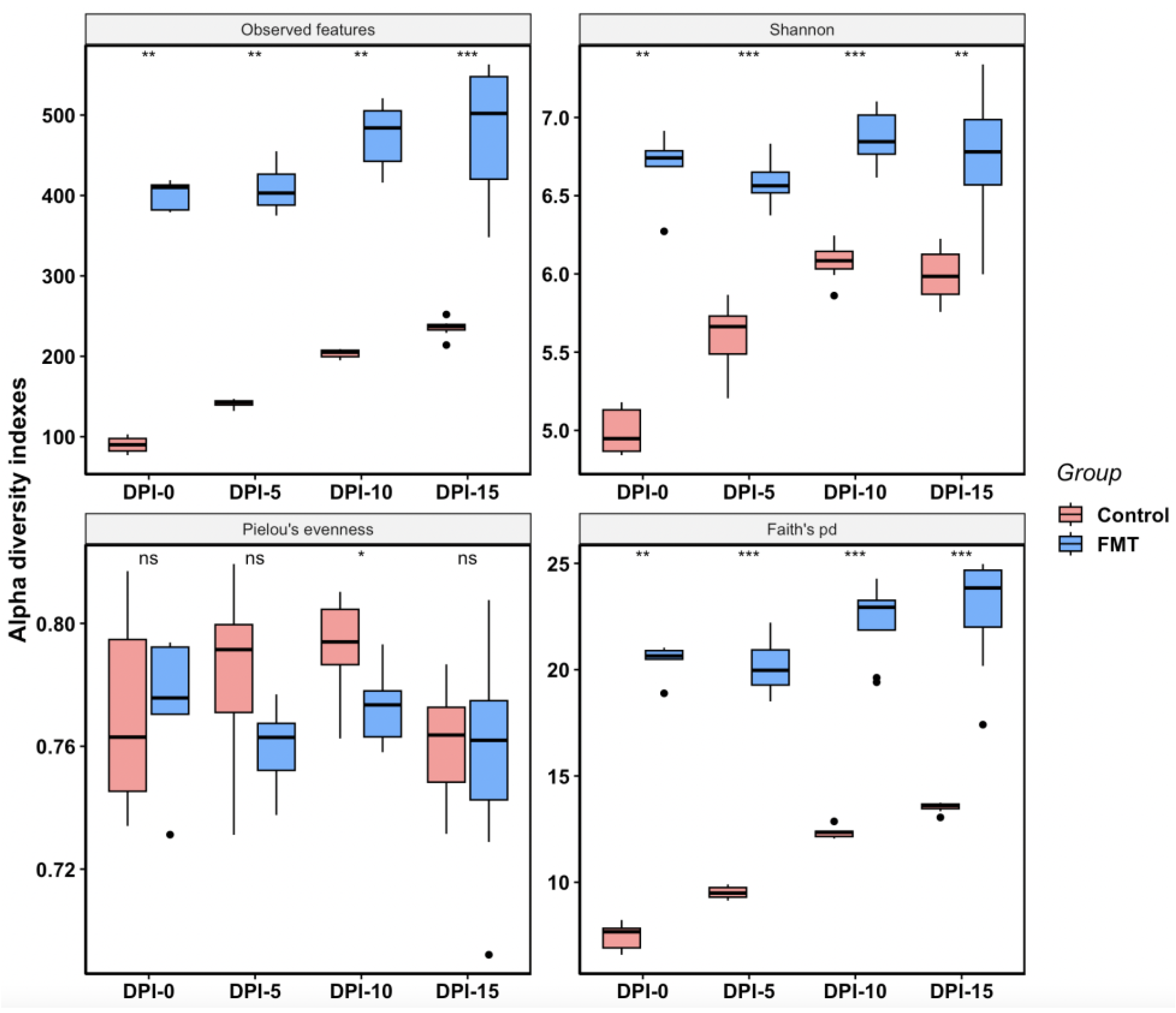
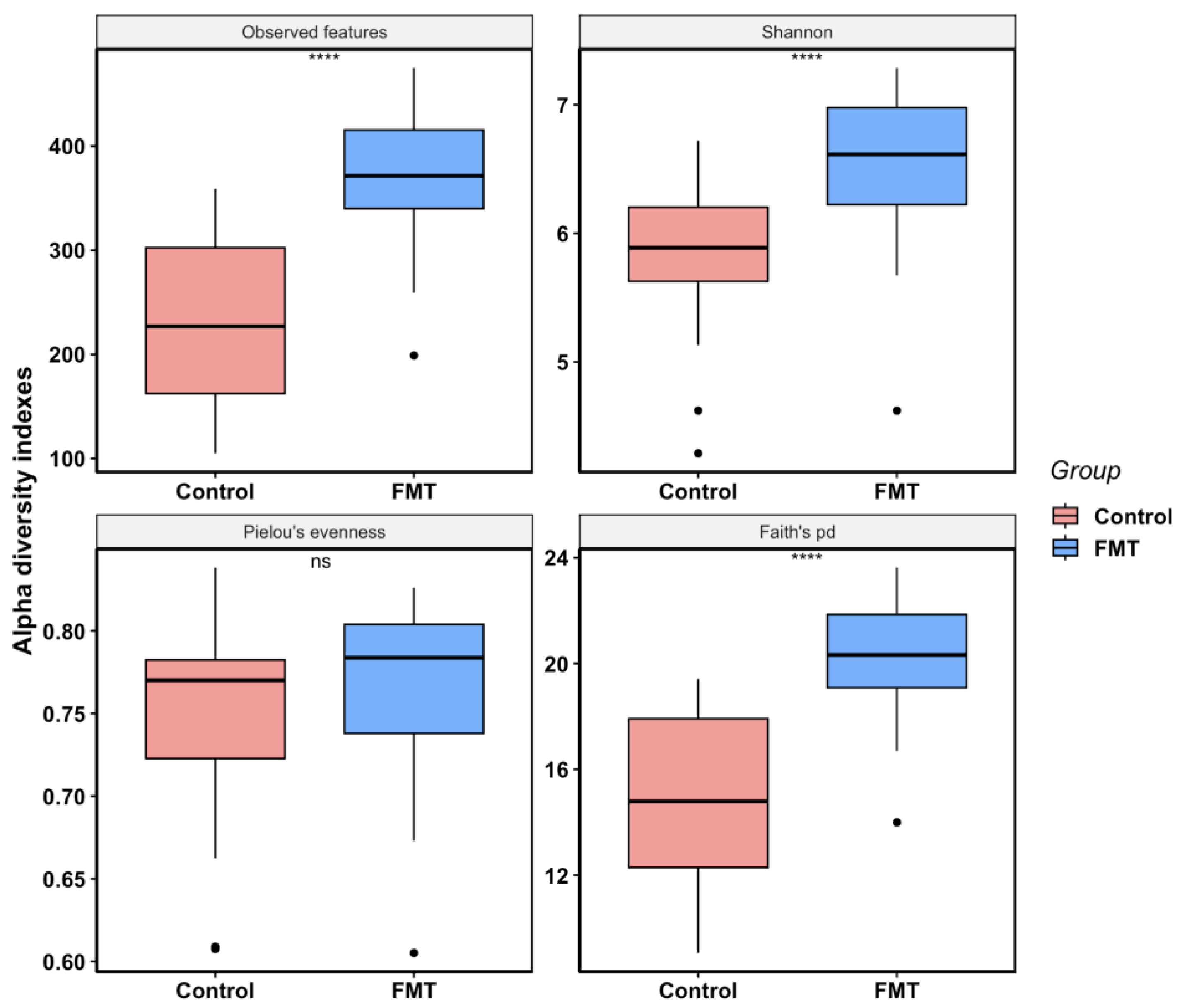
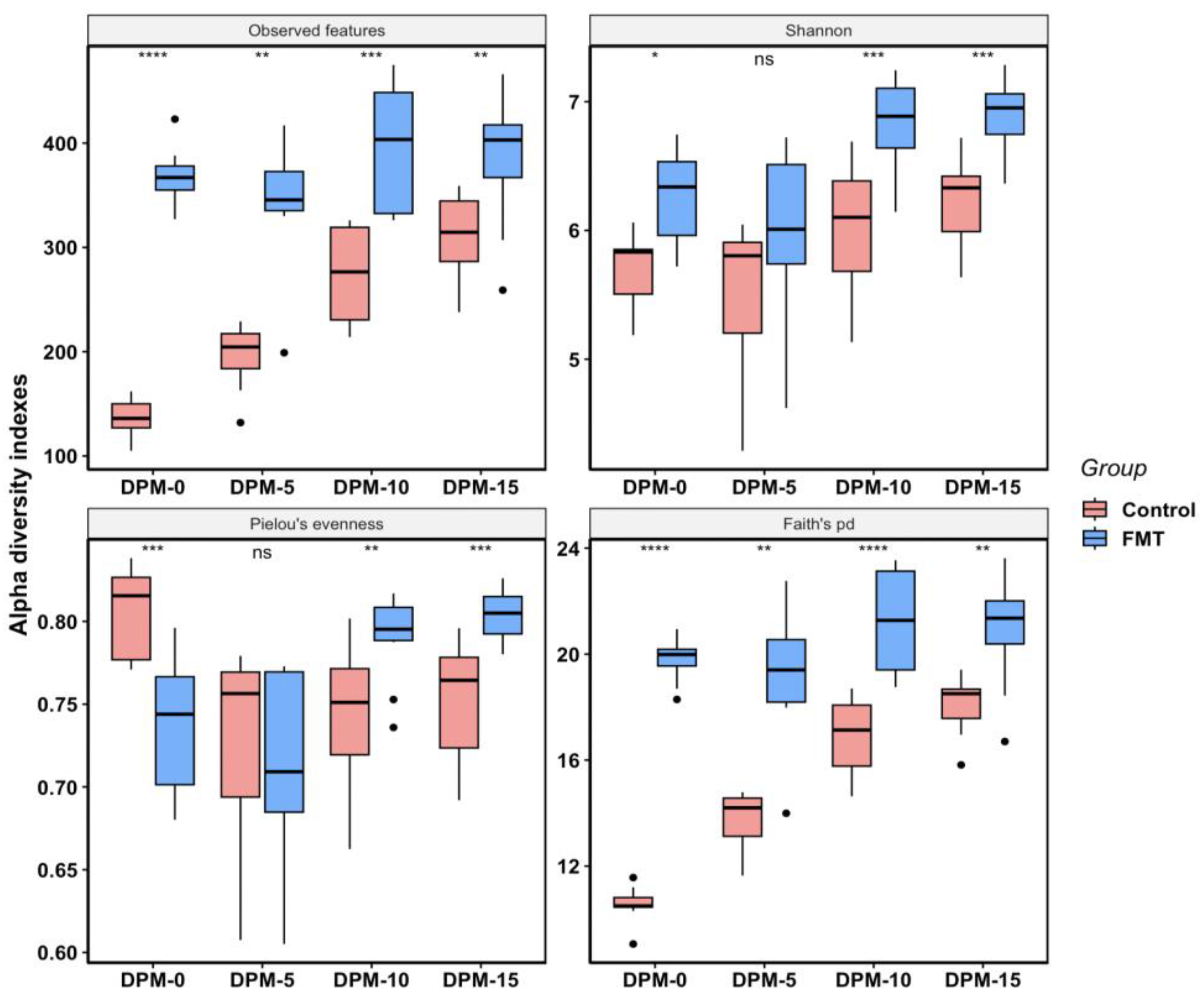
2.2.2. FMT Increased Cecal Microbial Species Richness and Phylogenetic Diversity in Young Broilers
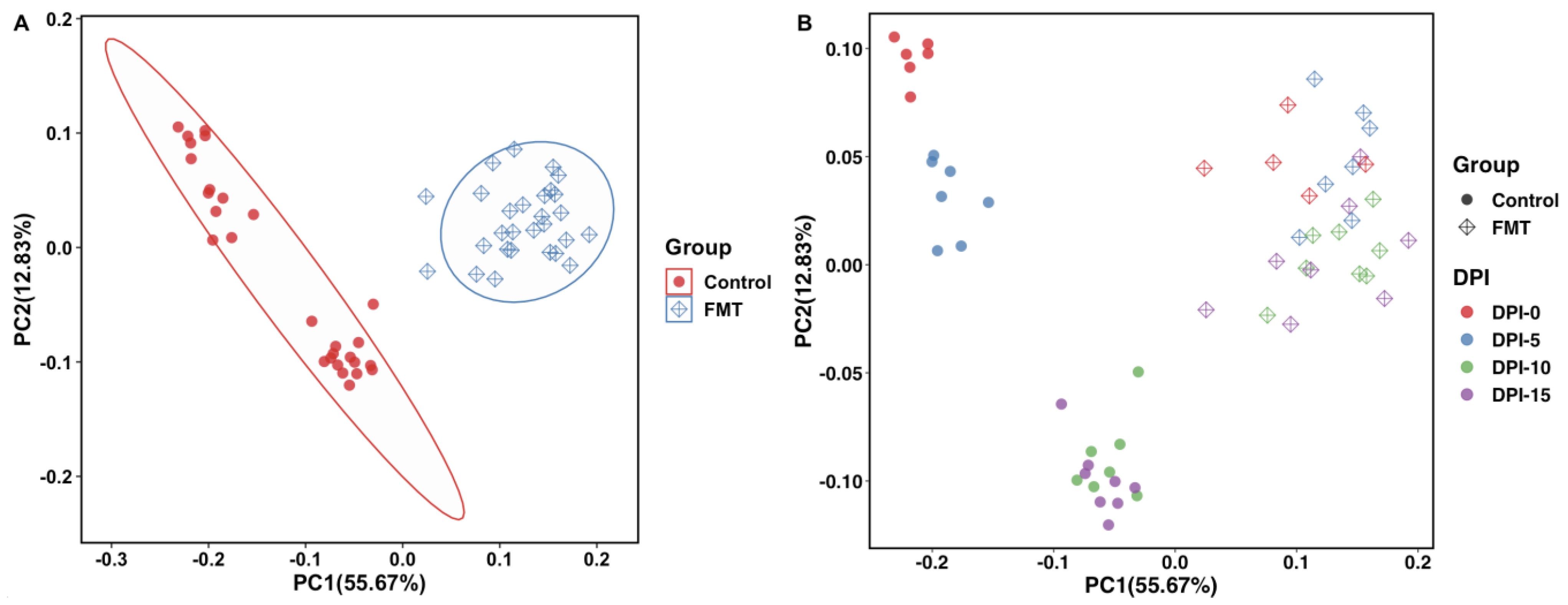
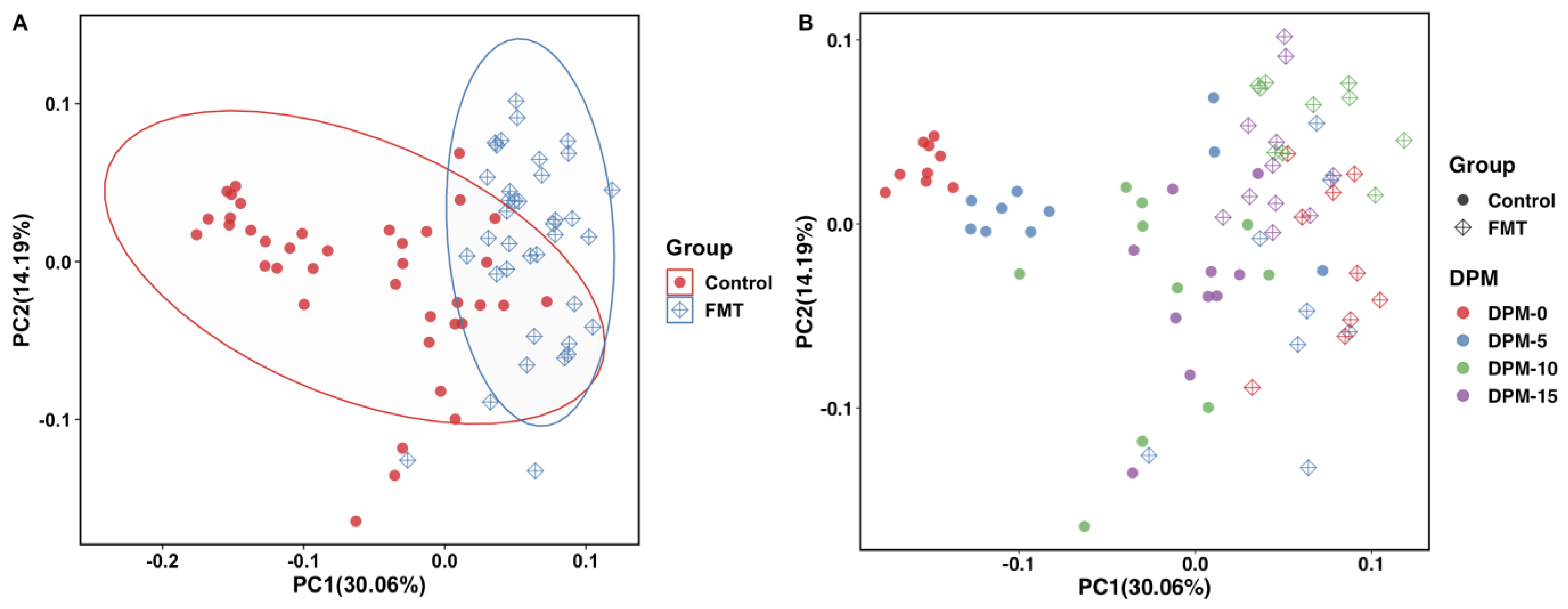
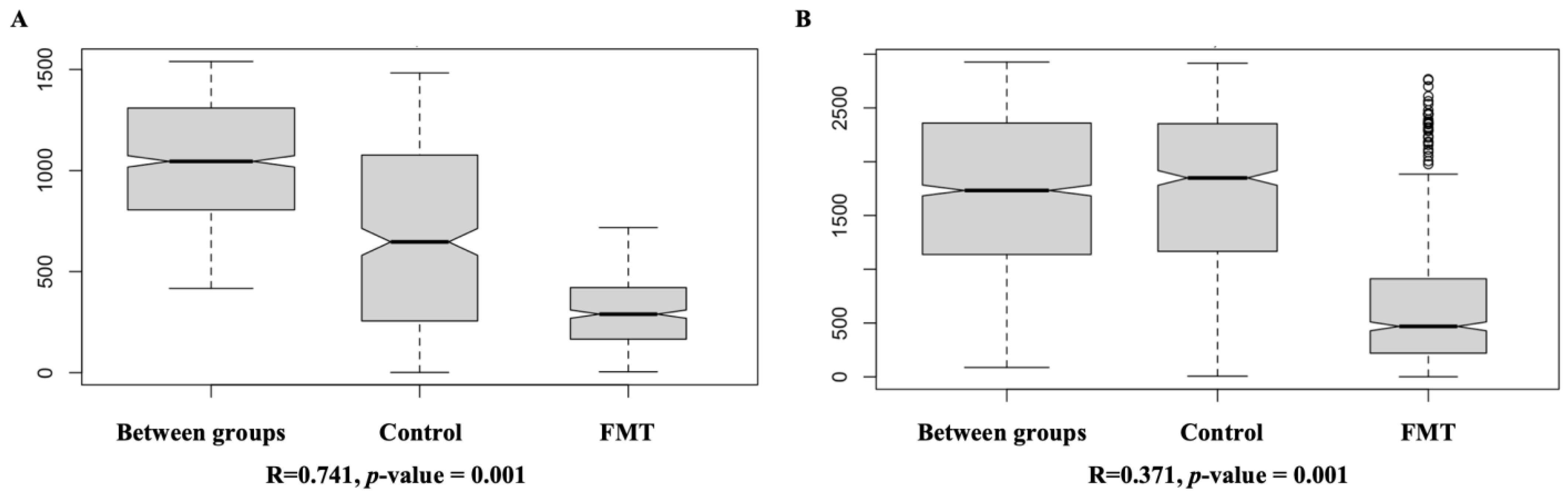
2.2.3. FMT Resulted in Significant Dissimilarity in Cecal Microbiome Composition as Revealed by Beta Diversity Analysis
2.3. Composition of Microbiotas
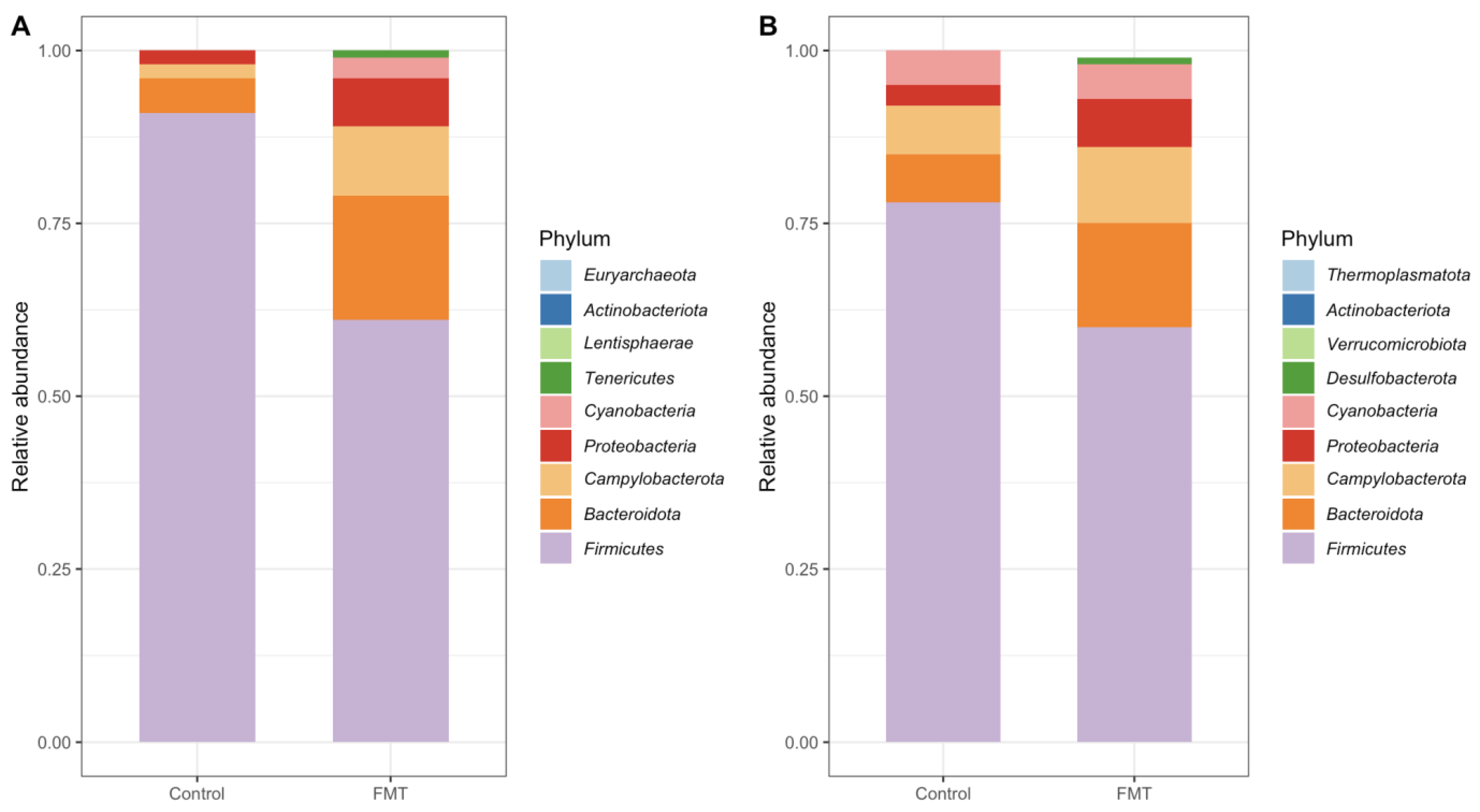
2.3.1. Composition at the Phylum Level
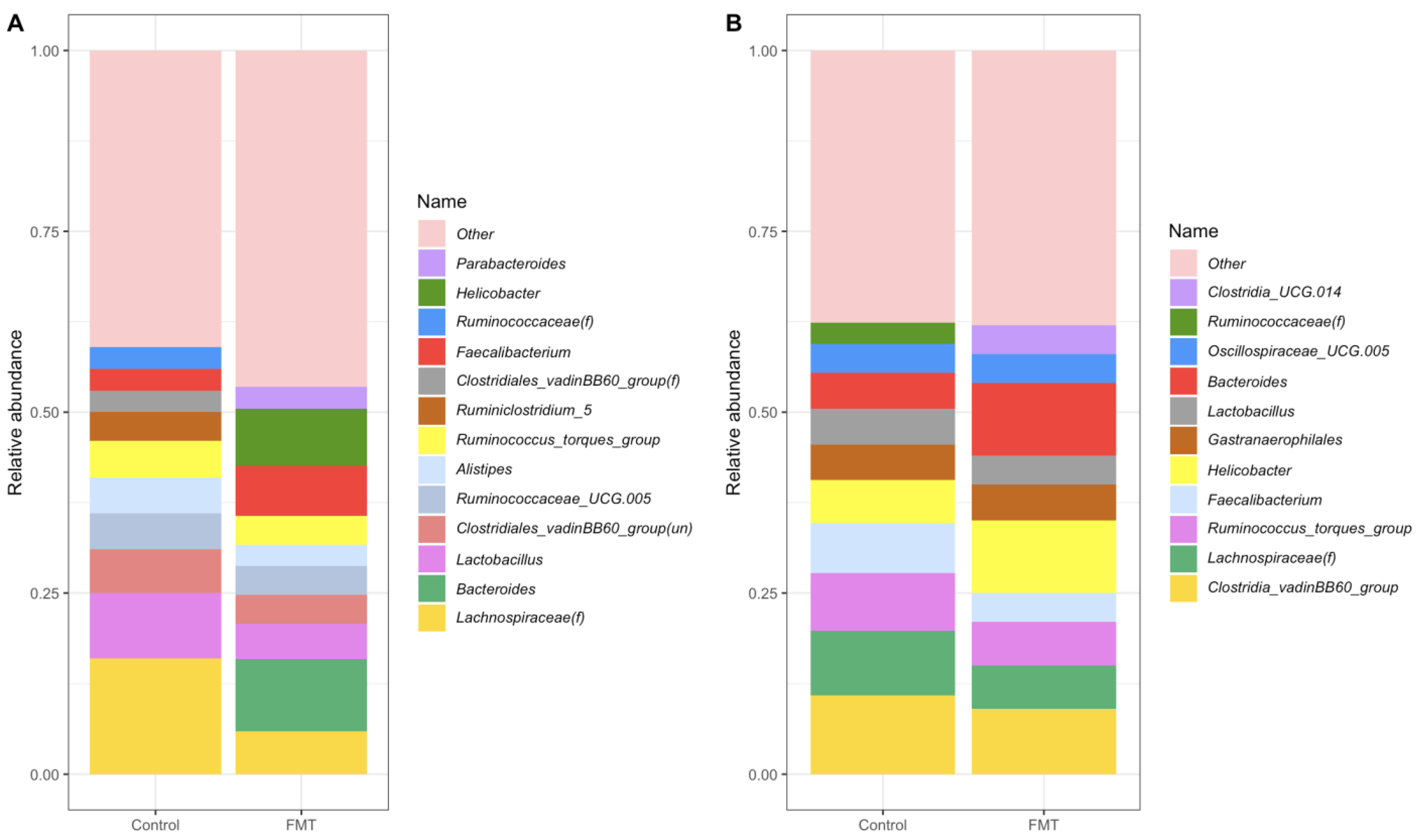
2.3.2. Composition at the Genus Level
2.3.3. Composition of the FMT Inoculum
2.4. FMT Resulted in Significant Changes in the Cecal Microbiota Composition as Revealed by ANCOM Analysis
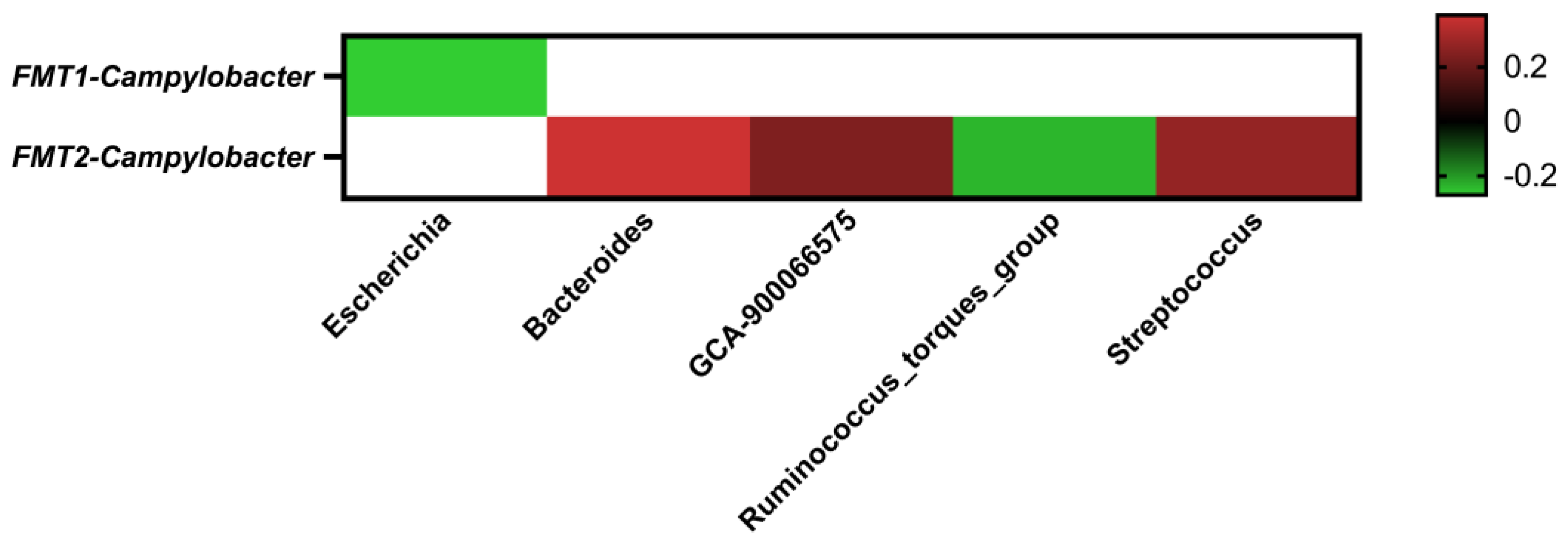
2.5. Correlations between the Core Cecal Microbial Genera
3. Discussion
4. Materials and Methods
4.1. Bacterial Strains and Culture Conditions
4.2. Preparation of Fecal Transplantation Material
4.3. Fecal Microbiota Transplantation (FMT) Trials
4.4. DNA Extraction and 16S rRNA Gene Sequencing
4.5. Bioinformatics and Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kaakoush, N.O.; Castaño-Rodríguez, N.; Mitchell, H.M.; Man, S.M. Global Epidemiology of Campylobacter Infection. Clin. Microbiol. Rev. 2015, 28, 687–720. [Google Scholar] [CrossRef]
- Delahoy, M.J. Preliminary Incidence and Trends of Infections Caused by Pathogens Transmitted Commonly through Food—Foodborne Diseases Active Surveillance Network, 10 U.S. Sites, 2022. MMWR Morb. Mortal. Wkly. Rep. 2023, 72, 701–706. [Google Scholar] [CrossRef] [PubMed]
- Igwaran, A.; Okoh, A.I. Human Campylobacteriosis: A Public Health Concern of Global Importance. Heliyon 2019, 5, e02814. [Google Scholar] [CrossRef] [PubMed]
- Acheson, D.; Allos, B.M. Campylobacter jejuni Infections: Update on Emerging Issues and Trends. Clin. Infect. Dis. 2001, 32, 1201–1206. [Google Scholar] [CrossRef] [PubMed]
- Fischer, G.H.; Paterek, E. Campylobacter. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Blaser, M.J. Epidemiologic and Clinical Features of Campylobacter jejuni Infections. J. Infect. Dis. 1997, 176 (Suppl. 2), S103–S105. [Google Scholar] [CrossRef]
- Gormley, F.J.; Little, C.L.; Rawal, N.; Gillespie, I.A.; Lebaigue, S.; Adak, G.K. A 17-Year Review of Foodborne Outbreaks: Describing the Continuing Decline in England and Wales (1992–2008). Epidemiol. Infect. 2011, 139, 688–699. [Google Scholar] [CrossRef]
- Moffatt, C.R.M.; Fearnley, E.; Bell, R.; Wright, R.; Gregory, J.; Sloan-Gardner, T.; Kirk, M.; Stafford, R. Characteristics of Campylobacter Gastroenteritis Outbreaks in Australia, 2001 to 2016. Foodborne Pathog. Dis. 2020, 17, 308–315. [Google Scholar] [CrossRef]
- Corry, J.E.L.; Atabay, H.I. Poultry as a Source of Campylobacter and Related Organisms. J. Appl. Microbiol. 2001, 90, 96S–114S. [Google Scholar] [CrossRef]
- Lee, M.D.; Newell, D.G. Campylobacter in Poultry: Filling an Ecological Niche. Avian Dis. 2006, 50, 1–9. [Google Scholar] [CrossRef]
- Sahin, O.; Morishita, T.Y.; Zhang, Q. Campylobacter Colonization in Poultry: Sources of Infection and Modes of Transmission. Anim. Health Res. Rev. 2002, 3, 95–105. [Google Scholar] [CrossRef]
- Shane, S.M. The Significance of Campylobacter jejuni Infection in Poultry: A Review. Avian Pathol. 1992, 21, 189–213. [Google Scholar] [CrossRef] [PubMed]
- Williams, L.K.; Fonseca, B.B.; Humphrey, T.J. Campylobacter jejuni in Poultry: A Commensal or a Pathogen? In Campylobacter spp. and Related Organisms in Poultry: Pathogen-Host Interactions, Diagnosis and Epidemiology; Fonseca, B.B., Fernandez, H., Rossi, D.A., Eds.; Springer International Publishing: Cham, Switzerland, 2016; pp. 75–87. ISBN 978-3-319-29907-5. [Google Scholar]
- A Critical Review of Interventions and Strategies (Both Biosecurity and Non-Biosecurity) to Reduce Campylobacter on the Poultry Farm|National Agricultural Library. Available online: https://www.nal.usda.gov/research-tools/food-safety-research-projects/critical-review-interventions-and-strategies-both (accessed on 12 November 2022).
- Luangtongkum, T.; Morishita, T.Y.; Ison, A.J.; Huang, S.; McDermott, P.F.; Zhang, Q. Effect of Conventional and Organic Production Practices on the Prevalence and Antimicrobial Resistance of Campylobacter spp. in Poultry. Appl. Environ. Microbiol. 2006, 72, 3600–3607. [Google Scholar] [CrossRef] [PubMed]
- Newell, D.G.; Fearnley, C. Sources of Campylobacter Colonization in Broiler Chickens. Appl. Environ. Microbiol. 2003, 69, 4343–4351. [Google Scholar] [CrossRef] [PubMed]
- Stern, N.J.; Fedorka-Cray, P.; Bailey, J.S.; Cox, N.A.; Craven, S.E.; Hiett, K.L.; Musgrove, M.T.; Ladely, S.; Cosby, D.; Mead, G.C. Distribution of Campylobacter spp. in Selected U.S. Poultry Production and Processing Operations. J. Food Prot. 2001, 64, 1705–1710. [Google Scholar] [CrossRef] [PubMed]
- Wieczorek, K.; Osek, J. Poultry Flocks as a Source of Campylobacter Contamination of Broiler Carcasses. Pol. J. Vet. Sci. 2015, 18, 101–106. [Google Scholar] [CrossRef] [PubMed]
- Hasan, M.M.; Talukder, S.; Mandal, A.K.; Tasmim, S.T.; Parvin, M.S.; Ali, M.Y.; Sikder, M.H.; Islam, M.T. Prevalence and Risk Factors of Campylobacter Infection in Broiler and Cockerel Flocks in Mymensingh and Gazipur Districts of Bangladesh. Prev. Vet. Med. 2020, 180, 105034. [Google Scholar] [CrossRef]
- Sahin, O.; Kassem, I.I.; Shen, Z.; Lin, J.; Rajashekara, G.; Zhang, Q. Campylobacter in Poultry: Ecology and Potential Interventions. Avian Dis. 2015, 59, 185–200. [Google Scholar] [CrossRef]
- Njoga, E.O.; Ezenduka, E.V.; Nwanta, J.A. Surveillance for Campylobacter Infections in Indigenous Poultry Reared in Nsukka, Nigeria. Not. Sci. Biol. 2020, 12, 242–250. [Google Scholar] [CrossRef]
- Facciolà, A.; Riso, R.; Avventuroso, E.; Visalli, G.; Delia, S.A.; Laganà, P. Campylobacter: From Microbiology to Prevention. J. Prev. Med. Hyg. 2017, 58, E79–E92. [Google Scholar]
- Sibanda, N.; McKenna, A.; Richmond, A.; Ricke, S.C.; Callaway, T.; Stratakos, A.C.; Gundogdu, O.; Corcionivoschi, N. A Review of the Effect of Management Practices on Campylobacter Prevalence in Poultry Farms. Front. Microbiol. 2018, 9, 2002. [Google Scholar] [CrossRef]
- Taha-Abdelaziz, K.; Singh, M.; Sharif, S.; Sharma, S.; Kulkarni, R.R.; Alizadeh, M.; Yitbarek, A.; Helmy, Y.A. Intervention Strategies to Control Campylobacter at Different Stages of the Food Chain. Microorganisms 2023, 11, 113. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Marmion, M.; Ferone, M.; Wall, P.; Scannell, A.G.M. On Farm Interventions to Minimise Campylobacter spp. Contamination in Chicken. Br. Poult. Sci. 2021, 62, 53–67. [Google Scholar] [CrossRef] [PubMed]
- Pessoa, J.; Rodrigues da Costa, M.; Nesbakken, T.; Meemken, D.; on behalf of the RIBMINS Cost Action. Assessment of the Effectiveness of Pre-Harvest Meat Safety Interventions to Control Foodborne Pathogens in Broilers: A Systematic Review. Curr. Clin. Microbiol. Rep. 2021, 8, 21–30. [Google Scholar] [CrossRef]
- Lin, J. Novel Approaches for Campylobacter Control in Poultry. Foodborne Pathog. Dis. 2009, 6, 755–765. [Google Scholar] [CrossRef]
- Meunier, M.; Guyard-Nicodème, M.; Dory, D.; Chemaly, M. Control Strategies against Campylobacter at the Poultry Production Level: Biosecurity Measures, Feed Additives and Vaccination. J. Appl. Microbiol. 2016, 120, 1139–1173. [Google Scholar] [CrossRef] [PubMed]
- Thursby, E.; Juge, N. Introduction to the Human Gut Microbiota. Biochem. J. 2017, 474, 1823–1836. [Google Scholar] [CrossRef] [PubMed]
- LeBlanc, J.G.; Milani, C.; de Giori, G.S.; Sesma, F.; van Sinderen, D.; Ventura, M. Bacteria as Vitamin Suppliers to Their Host: A Gut Microbiota Perspective. Curr. Opin. Biotechnol. 2013, 24, 160–168. [Google Scholar] [CrossRef]
- Khan, S.; Moore, R.J.; Stanley, D.; Chousalkar, K.K. The Gut Microbiota of Laying Hens and Its Manipulation with Prebiotics and Probiotics to Enhance Gut Health and Food Safety. Appl. Environ. Microbiol. 2020, 86, e00600-20. [Google Scholar] [CrossRef]
- Śmiałek, M.; Kowalczyk, J.; Koncicki, A. The Use of Probiotics in the Reduction of Campylobacter spp. Prevalence in Poultry. Animals 2021, 11, 1355. [Google Scholar] [CrossRef]
- Buffie, C.G.; Pamer, E.G. Microbiota-Mediated Colonization Resistance against Intestinal Pathogens. Nat. Rev. Immunol. 2013, 13, 790–801. [Google Scholar] [CrossRef]
- Khan, I.; Bai, Y.; Zha, L.; Ullah, N.; Ullah, H.; Shah, S.R.H.; Sun, H.; Zhang, C. Mechanism of the Gut Microbiota Colonization Resistance and Enteric Pathogen Infection. Front. Cell. Infect. Microbiol. 2021, 11, 716299. [Google Scholar] [CrossRef] [PubMed]
- Balta, I.; Butucel, E.; Stef, L.; Pet, I.; Gradisteanu-Pircalabioru, G.; Chifiriuc, C.; Gundogdu, O.; McCleery, D.; Corcionivoschi, N. Anti-Campylobacter Probiotics: Latest Mechanistic Insights. Foodborne Pathog. Dis. 2022, 19, 693–703. [Google Scholar] [CrossRef] [PubMed]
- Deng, W.; Dittoe, D.K.; Pavilidis, H.O.; Chaney, W.E.; Yang, Y.; Ricke, S.C. Current Perspectives and Potential of Probiotics to Limit Foodborne Campylobacter in Poultry. Front. Microbiol. 2020, 11, 583429. [Google Scholar] [CrossRef] [PubMed]
- Šimunović, K.; Sahin, O.; Erega, A.; Štefanič, P.; Zhang, Q.; Mandic Mulec, I.; Smole Možina, S.; Klančnik, A. Bacillus subtilis PS-216 Spores Supplemented in Broiler Chicken Drinking Water Reduce Campylobacter jejuni Colonization and Increases Weight Gain. Front. Microbiol. 2022, 13, 910616. [Google Scholar] [CrossRef]
- Metzler-Zebeli, B.U.; Siegerstetter, S.-C.; Magowan, E.; Lawlor, P.G.; O’Connell, N.E.; Zebeli, Q. Fecal Microbiota Transplant from Highly Feed Efficient Donors Affects Cecal Physiology and Microbiota in Low- and High-Feed Efficient Chickens. Front. Microbiol. 2019, 10, 1576. [Google Scholar] [CrossRef]
- Gilroy, R.; Chaloner, G.; Wedley, A.; Lacharme-Lora, L.; Jopson, S.; Wigley, P. Campylobacter jejuni Transmission and Colonisation in Broiler Chickens Is Inhibited by Faecal Microbiota Transplantation. bioRxiv 2018. [Google Scholar] [CrossRef]
- Nurmi, E.; Rantala, M. New Aspects of Salmonella Infection in Broiler Production. Nature 1973, 241, 210–211. [Google Scholar] [CrossRef] [PubMed]
- Pottenger, S.; Watts, A.; Wedley, A.; Jopson, S.; Darby, A.C.; Wigley, P. Timing and Delivery Route Effects of Cecal Microbiome Transplants on Salmonella Typhimurium Infections in Chickens: Potential for in-Hatchery Delivery of Microbial Interventions. Anim. Microbiome 2023, 5, 11. [Google Scholar] [CrossRef]
- Boyd, Y.; Herbert, E.G.; Marston, K.L.; Jones, M.A.; Barrow, P.A. Host Genes Affect Intestinal Colonisation of Newly Hatched Chickens by Campylobacter jejuni. Immunogenetics 2005, 57, 248–253. [Google Scholar] [CrossRef]
- Chintoan-Uta, C.; Wisedchanwet, T.; Glendinning, L.; Bremner, A.; Psifidi, A.; Vervelde, L.; Watson, K.; Watson, M.; Stevens, M.P. Role of Cecal Microbiota in the Differential Resistance of Inbred Chicken Lines to Colonization by Campylobacter jejuni. Appl. Environ. Microbiol. 2020, 86, e02607-19. [Google Scholar] [CrossRef]
- Aruwa, C.E.; Pillay, C.; Nyaga, M.M.; Sabiu, S. Poultry Gut Health—Microbiome Functions, Environmental Impacts, Microbiome Engineering and Advancements in Characterization Technologies. J. Anim. Sci. Biotechnol. 2021, 12, 119. [Google Scholar] [CrossRef] [PubMed]
- Pang, J.; Looft, T.; Zhang, Q.; Sahin, O. Deciphering the Association between Campylobacter Colonization and Microbiota Composition in the Intestine of Commercial Broilers. Microorganisms 2023, 11, 1724. [Google Scholar] [CrossRef]
- Romero-Barrios, P.; Hempen, M.; Messens, W.; Stella, P.; Hugas, M. Quantitative Microbiological Risk Assessment (QMRA) of Food-Borne Zoonoses at the European Level. Food Control 2013, 29, 343–349. [Google Scholar] [CrossRef]
- EFSA Panel on Biological Hazards (BIOHAZ); Koutsoumanis, K.; Allende, A.; Alvarez-Ordóñez, A.; Bolton, D.; Bover-Cid, S.; Davies, R.; De Cesare, A.; Herman, L.; Hilbert, F.; et al. Update and Review of Control Options for Campylobacter in Broilers at Primary Production. EFSA J. 2020, 18, e06090. [Google Scholar] [CrossRef] [PubMed]
- Hermans, D.; Van Deun, K.; Martel, A.; Van Immerseel, F.; Messens, W.; Heyndrickx, M.; Haesebrouck, F.; Pasmans, F. Colonization Factors of Campylobacter jejuni in the Chicken Gut. Vet. Res. 2011, 42, 82. [Google Scholar] [CrossRef] [PubMed]
- Oakley, B.B.; Lillehoj, H.S.; Kogut, M.H.; Kim, W.K.; Maurer, J.J.; Pedroso, A.; Lee, M.D.; Collett, S.R.; Johnson, T.J.; Cox, N.A. The Chicken Gastrointestinal Microbiome. FEMS Microbiol. Lett. 2014, 360, 100–112. [Google Scholar] [CrossRef]
- Rychlik, I. Composition and Function of Chicken Gut Microbiota. Animals 2020, 10, 103. [Google Scholar] [CrossRef]
- Glendinning, L.; Stewart, R.D.; Pallen, M.J.; Watson, K.A.; Watson, M. Assembly of Hundreds of Novel Bacterial Genomes from the Chicken Caecum. Genome Biol. 2020, 21, 34. [Google Scholar] [CrossRef]
- Medvecky, M.; Cejkova, D.; Polansky, O.; Karasova, D.; Kubasova, T.; Cizek, A.; Rychlik, I. Whole Genome Sequencing and Function Prediction of 133 Gut Anaerobes Isolated from Chicken Caecum in Pure Cultures. BMC Genom. 2018, 19, 561. [Google Scholar] [CrossRef]
- Wu, F.; Guo, X.; Zhang, J.; Zhang, M.; Ou, Z.; Peng, Y. Phascolarctobacterium Faecium Abundant Colonization in Human Gastrointestinal Tract. Exp. Ther. Med. 2017, 14, 3122–3126. [Google Scholar] [CrossRef]
- Ezeji, J.C.; Sarikonda, D.K.; Hopperton, A.; Erkkila, H.L.; Cohen, D.E.; Martinez, S.P.; Cominelli, F.; Kuwahara, T.; Dichosa, A.E.K.; Good, C.E.; et al. Parabacteroides Distasonis: Intriguing Aerotolerant Gut Anaerobe with Emerging Antimicrobial Resistance and Pathogenic and Probiotic Roles in Human Health. Gut Microbes 2021, 13, 1922241. [Google Scholar] [CrossRef] [PubMed]
- Nogal, A.; Valdes, A.M.; Menni, C. The Role of Short-Chain Fatty Acids in the Interplay between Gut Microbiota and Diet in Cardio-Metabolic Health. Gut Microbes 2021, 13, 1897212. [Google Scholar] [CrossRef] [PubMed]
- Helmy, Y.A.; Closs, G.; Jung, K.; Kathayat, D.; Vlasova, A.; Rajashekara, G. Effect of Probiotic E. coli Nissle 1917 Supplementation on the Growth Performance, Immune Responses, Intestinal Morphology, and Gut Microbes of Campylobacter jejuni Infected Chickens. Infect. Immun. 2022, 90, e00337-22. [Google Scholar] [CrossRef] [PubMed]
- Torok, V.A.; Hughes, R.J.; Mikkelsen, L.L.; Perez-Maldonado, R.; Balding, K.; MacAlpine, R.; Percy, N.J.; Ophel-Keller, K. Identification and Characterization of Potential Performance-Related Gut Microbiotas in Broiler Chickens across Various Feeding Trials. Appl. Environ. Microbiol. 2011, 77, 5868–5878. [Google Scholar] [CrossRef]
- Hertogs, K.; Haegeman, A.; Schaumont, D.; Gelaude, P.; De Zutter, L.; Dewulf, J.; Heyndrickx, M.; Rasschaert, G. Contamination Sources and Transmission Routes for Campylobacter on (Mixed) Broiler Farms in Belgium, and Comparison of the Gut Microbiota of Flocks Colonized and Uncolonized with Campylobacter. Pathogens 2021, 10, 66. [Google Scholar] [CrossRef]
- Vrancken, G.; Gregory, A.C.; Huys, G.R.B.; Faust, K.; Raes, J. Synthetic Ecology of the Human Gut Microbiota. Nat. Rev. Microbiol. 2019, 17, 754–763. [Google Scholar] [CrossRef]
- Clark, R.L.; Connors, B.M.; Stevenson, D.M.; Hromada, S.E.; Hamilton, J.J.; Amador-Noguez, D.; Venturelli, O.S. Design of Synthetic Human Gut Microbiome Assembly and Butyrate Production. Nat. Commun. 2021, 12, 3254. [Google Scholar] [CrossRef]
- Mabwi, H.A.; Kim, E.; Song, D.-G.; Yoon, H.S.; Pan, C.-H.; Komba, E.V.G.; Ko, G.; Cha, K.H. Synthetic Gut Microbiome: Advances and Challenges. Comput. Struct. Biotechnol. J. 2021, 19, 363–371. [Google Scholar] [CrossRef]
- Bolsega, S.; Bleich, A.; Basic, M. Synthetic Microbiomes on the Rise—Application in Deciphering the Role of Microbes in Host Health and Disease. Nutrients 2021, 13, 4173. [Google Scholar] [CrossRef]
- Plummer, P.; Sahin, O.; Burrough, E.; Sippy, R.; Mou, K.; Rabenold, J.; Yaeger, M.; Zhang, Q. Critical Role of LuxS in the Virulence of Campylobacter jejuni in a Guinea Pig Model of Abortion. Infect. Immun. 2012, 80, 585–593. [Google Scholar] [CrossRef]
- Zhang, Q.; Widmer, G.; Tzipori, S. A Pig Model of the Human Gastrointestinal Tract. Gut Microbes 2013, 4, 193–200. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Ackermann, G.; Apprill, A.; Bauer, M.; Berg-Lyons, D.; Betley, J.; Fierer, N.; Fraser, L.; Fuhrman, J.A.; Gilbert, J.A.; et al. EMP 16S Illumina Amplicon Protocol. 2018. Available online: https://www.protocols.io/view/emp-16s-illumina-amplicon-protocol-kqdg3dzzl25z/v1 (accessed on 12 November 2022).
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, Interactive, Scalable and Extensible Microbiome Data Science Using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-Resolution Sample Inference from Illumina Amplicon Data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [PubMed]
- Robeson, M.S.; O’Rourke, D.R.; Kaehler, B.D.; Ziemski, M.; Dillon, M.R.; Foster, J.T.; Bokulich, N.A. RESCRIPt: Reproducible Sequence Taxonomy Reference Database Management for the Masses. PLoS Comput. Biol. 2020, 17, e1009581. [Google Scholar] [CrossRef]
- Oksanen, J. Vegan : Community Ecology Package. R Package Version 1. 15-4. 2009. Available online: https://CRAN.R-project.org/package=vegan (accessed on 8 March 2023).
- Mandal, S.; Van Treuren, W.; White, R.A.; Eggesbø, M.; Knight, R.; Peddada, S.D. Analysis of Composition of Microbiomes: A Novel Method for Studying Microbial Composition. Microb. Ecol. Health Dis. 2015, 26, 27663. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genus | Abundances 50% | Abundances 50% | W * | Change |
|---|---|---|---|---|
| Control | FMT | FMT vs. Control | ||
| Staphylococcus | 1184 | 7 | 148 | Decreased |
| Weissella | 105.5 | 4.5 | 144 | Decreased |
| Romboutsia | 1401 | 67.5 | 144 | Decreased |
| Proteus | 36.5 | 1 | 144 | Decreased |
| Clostridium_innocuum_group | 25.5 | 1 | 142 | Decreased |
| Brachybacterium | 16 | 1 | 138 | Decreased |
| CHKCI002 | 79 | 11 | 133 | Decreased |
| Bacteroides | 1 | 7528.5 | 148 | Increased |
| Butyricimonas | 1 | 947 | 146 | Increased |
| Parabacteroides | 1 | 2487.5 | 146 | Increased |
| Parasutterella | 1 | 2173 | 145 | Increased |
| uncultured_Clostridia_bacterium | 1 | 622.5 | 144 | Increased |
| Bilophila | 1 | 564.5 | 144 | Increased |
| Fournierella | 1 | 207.5 | 141 | Increased |
| Odoribacter | 1 | 134 | 140 | Increased |
| Helicobacter | 9 | 5963.5 | 140 | Increased |
| Phascolarctobacterium | 1 | 134 | 139 | Increased |
| Genus | Abundances 50% | Abundances 50% | W * | Change |
|---|---|---|---|---|
| Control | FMT | FMT vs. Control | ||
| Dorea | 770 | 1 | 113 | Decreased |
| Proteus | 4 | 1 | 113 | Decreased |
| LachnospiraceaeFE2018group | 12 | 1 | 110 | Decreased |
| Butyricimonas | 1 | 600 | 130 | Increased |
| Parabacteroides | 1 | 1366.5 | 130 | Increased |
| Bilophila | 1 | 609.5 | 130 | Increased |
| Parasutterella | 1 | 1421.5 | 130 | Increased |
| Phascolarctobacterium | 1 | 46.5 | 124 | Increased |
| Pygmaiobacter | 1 | 42.5 | 110 | Increased |
| Ruminococcus | 97 | 639.5 | 104 | Increased |
| Fournierella | 1 | 41.5 | 102 | Increased |
| Helicobacter | 667 | 4503 | 98 | Increased |
| Trial | Group | FMT | Cloacal Swab | C. jejuni Inoculation | Necropsy |
|---|---|---|---|---|---|
| FMT trial 1 | FMT | Yes | Yes | Oral gavage | DPI-0, 5, 10, 15 |
| Control | No | Yes | Oral gavage | DPI-0, 5, 10, 15 | |
| FMT trial 2 | FMT | Yes | Yes | Seeder birds | DPM-0, 5, 10, 15 |
| Control | No | Yes | Seeder birds | DPM-0, 5, 10, 15 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pang, J.; Beyi, A.F.; Looft, T.; Zhang, Q.; Sahin, O. Fecal Microbiota Transplantation Reduces Campylobacter jejuni Colonization in Young Broiler Chickens Challenged by Oral Gavage but Not by Seeder Birds. Antibiotics 2023, 12, 1503. https://doi.org/10.3390/antibiotics12101503
Pang J, Beyi AF, Looft T, Zhang Q, Sahin O. Fecal Microbiota Transplantation Reduces Campylobacter jejuni Colonization in Young Broiler Chickens Challenged by Oral Gavage but Not by Seeder Birds. Antibiotics. 2023; 12(10):1503. https://doi.org/10.3390/antibiotics12101503
Chicago/Turabian StylePang, Jinji, Ashenafi Feyisa Beyi, Torey Looft, Qijing Zhang, and Orhan Sahin. 2023. "Fecal Microbiota Transplantation Reduces Campylobacter jejuni Colonization in Young Broiler Chickens Challenged by Oral Gavage but Not by Seeder Birds" Antibiotics 12, no. 10: 1503. https://doi.org/10.3390/antibiotics12101503