
Identification of Staphylococcus aureus Penicillin Binding Protein 4 (PBP4) Inhibitors

 , , , , ,
, , , , ,
Abstract
:1. Introduction
2. Results
2.1. Screening for Small Molecule S. aureus PBP4 Inhibitors
2.2. Potentiation of β-Lactams
2.3. Human Cell Cytotoxicity Measures of Putative PBP4 Inhibitors
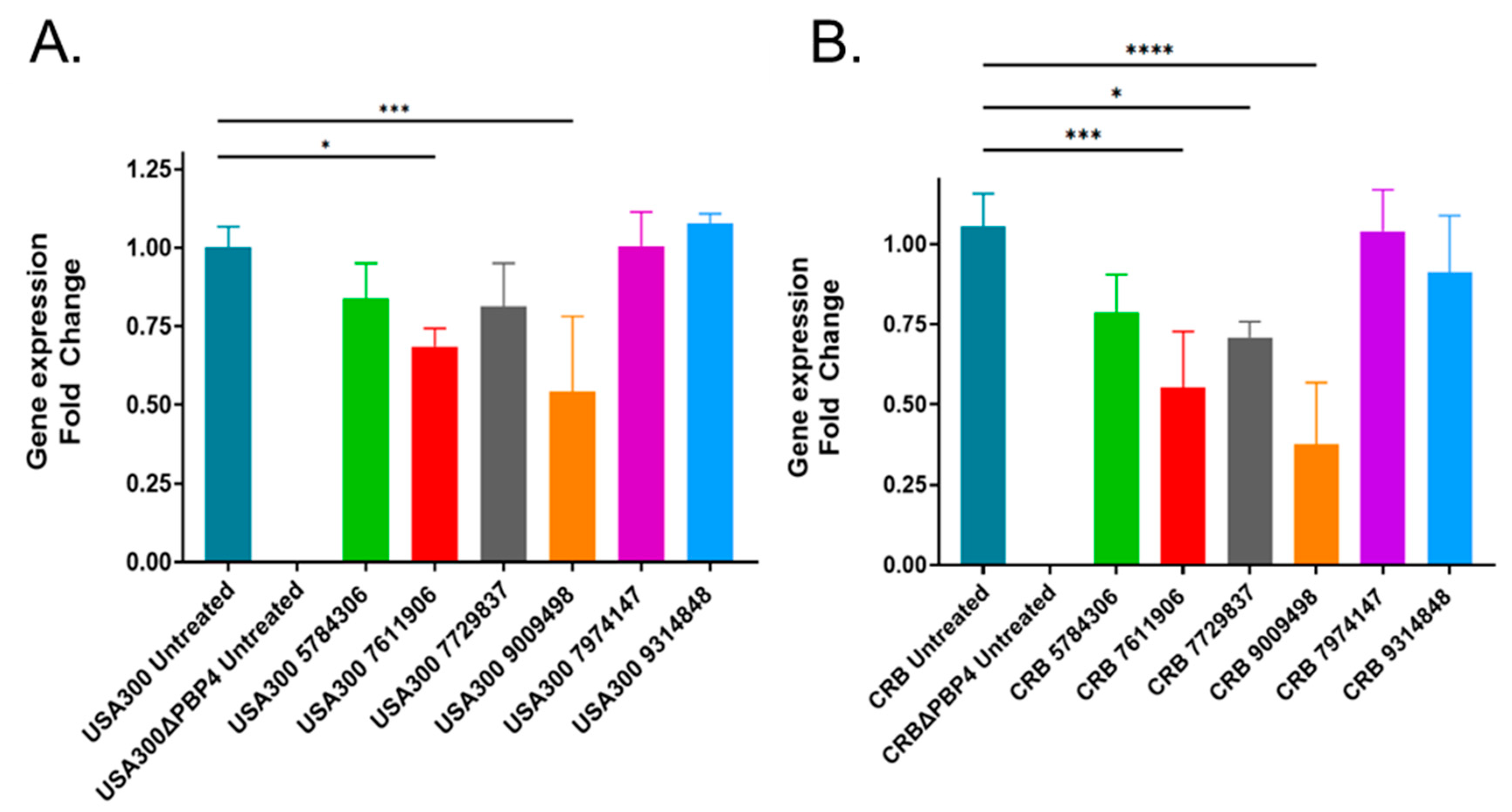
2.4. Effects of Putative PBP4 Inhibitors on pbp4 Expression
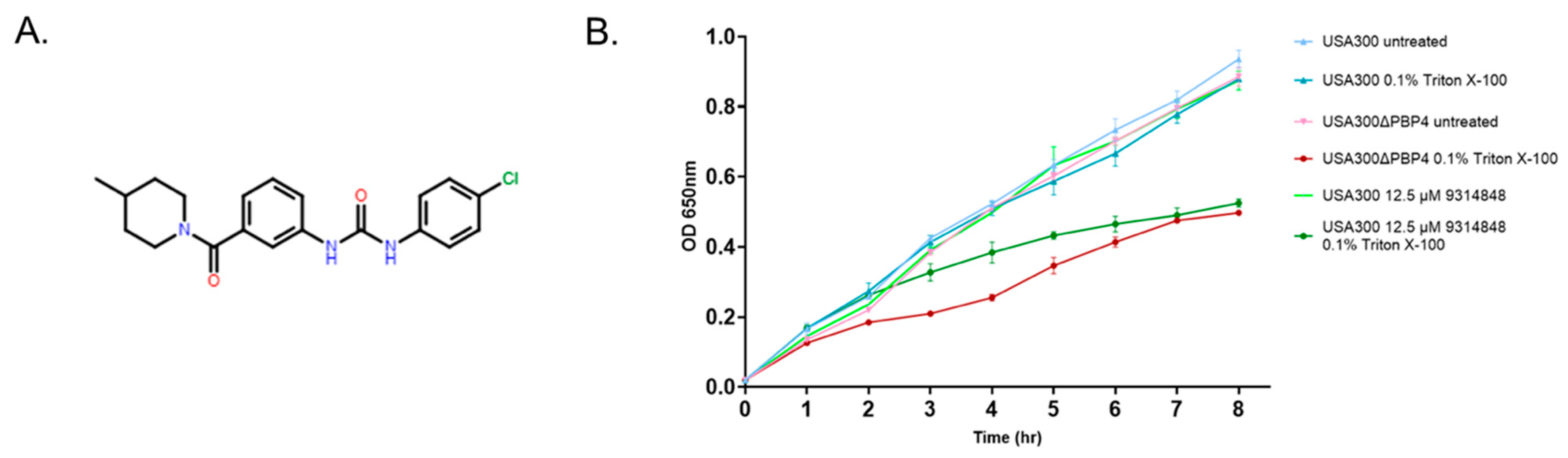
2.5. Effect of PBP4 Inhibitors on Triton X-100 Susceptibility
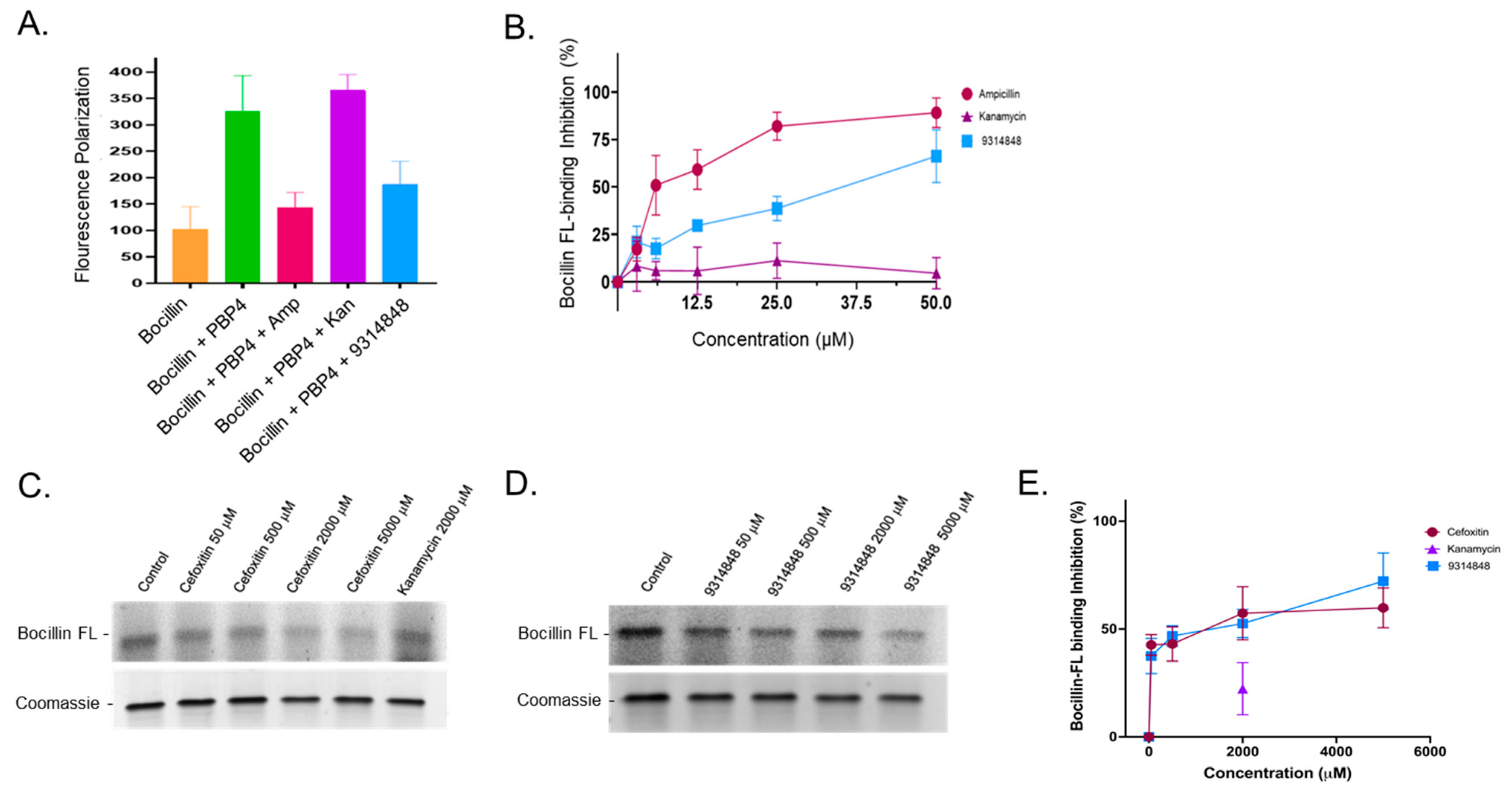
2.6. Ability of PBP4 Inhibitors to Bind to PBP4 or PBP2A
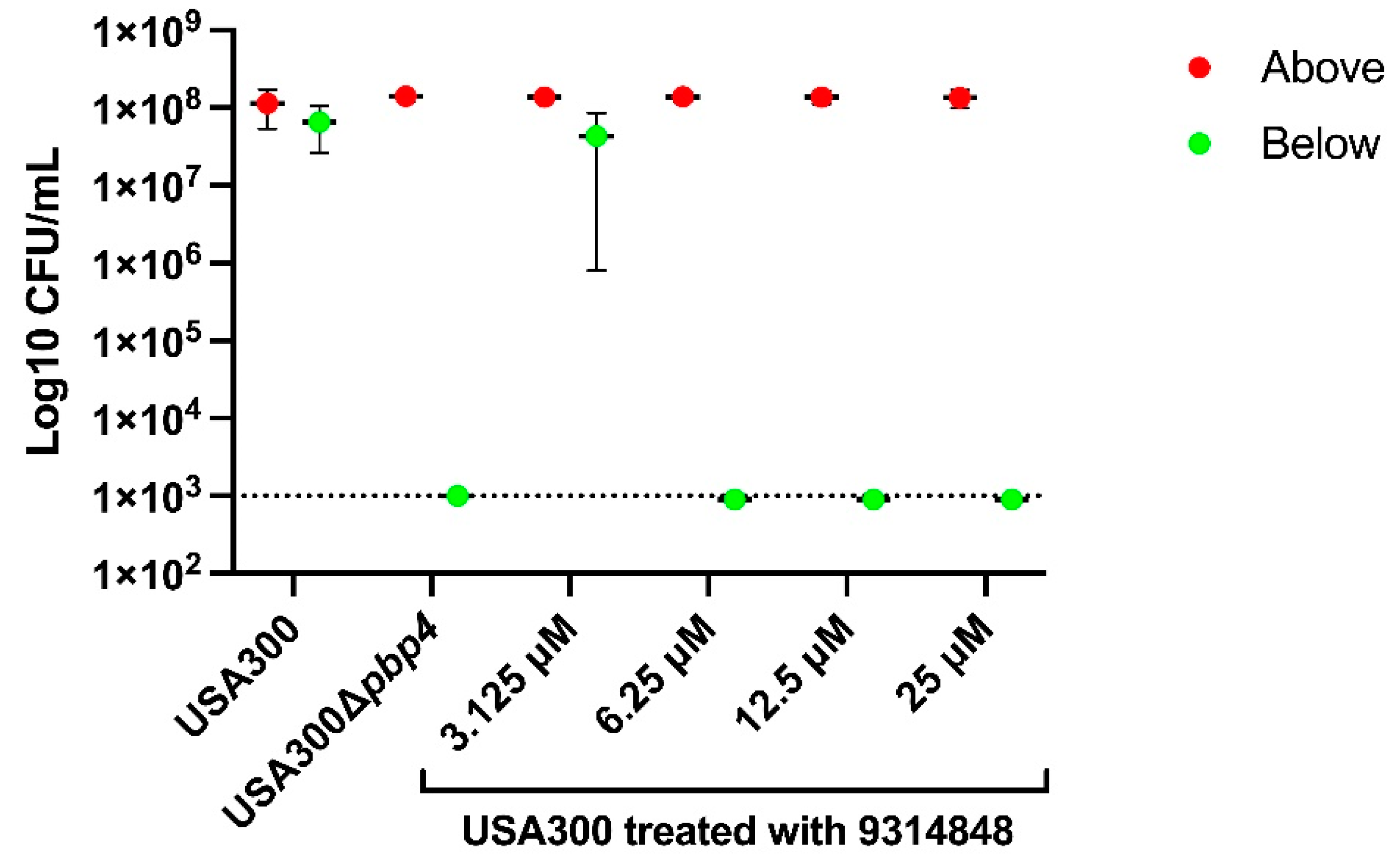
2.7. PBP4 Inhibitors Prevent Propagation in µSiM-CA Canaliculi Model
3. Discussion
4. Materials and Methods
4.1. Bacterial Strains and Chemicals
4.2. High Throughput Screen for PBP4 Inhibitors
4.3. Eukaryotic Toxicity Testing
4.4. Fractional Inhibitory Concentration Index (FICI)
4.5. S. aureus µSiM-CA Transmigration
4.6. Quantitative Reverse Transcriptase PCR
4.7. Triton X-100 Susceptibility Studies
4.8. PBP4 BOCILLIN FL Binding Competition Assays
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chambers, H.F.; DeLeo, F.R. Waves of resistance: Staphylococcus aureus in the antibiotic era. Nat. Rev. Microbiol. 2009, 7, 629–641. [Google Scholar] [CrossRef] [PubMed]
- Bai, A.D.; Lo, C.K.; Komorowski, A.S.; Suresh, M.; Guo, K.; Garg, A.; Tandon, P.; Senecal, J.; Corpo, O.D.; Stefanova, I.; et al. Staphylococcus aureus bacteremia mortality across country income groups: A secondary analysis of a systematic review. Int. J. Infect. Dis. 2022, 122, 405–411. [Google Scholar] [CrossRef] [PubMed]
- Talha, K.M.; Dayer, M.J.; Thornhill, M.H.; Tariq, W.; Arshad, V.; Tleyjeh, I.M.; Bailey, K.R.; Palraj, R.; Anavekar, N.S.; Rizwan Sohail, M.; et al. Temporal Trends of Infective Endocarditis in North America From 2000 to 2017-A Systematic Review. Open Forum Infect. Dis. 2021, 8, ofab479. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.W.; Somerville, T.; Kaye, S.B.; Romano, V. Staphylococcus aureus Keratitis: Incidence, Pathophysiology, Risk Factors and Novel Strategies for Treatment. J. Clin. Med. 2021, 10, 758. [Google Scholar] [CrossRef]
- Masters, E.A.; Ricciardi, B.F.; Bentley, K.L.M.; Moriarty, T.F.; Schwarz, E.M.; Muthukrishnan, G. Skeletal infections: Microbial pathogenesis, immunity and clinical management. Nat. Rev. Microbiol. 2022, 20, 385–400. [Google Scholar] [CrossRef]
- Kaye, K.S.; Petty, L.A.; Shorr, A.F.; Zilberberg, M.D. Current Epidemiology, Etiology, and Burden of Acute Skin Infections in the United States. Clin. Infect. Dis. 2019, 68, S193–S199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tipper, D.J.; Strominger, J.L. Mechanism of action of penicillins: A proposal based on their structural similarity to acyl-D-alanyl-D-alanine. Proc. Natl. Acad. Sci. USA 1965, 54, 1133–1141. [Google Scholar] [CrossRef] [Green Version]
- Georgopapadakou, N.; Hammarström, S.; Strominger, J.L. Isolation of the penicillin-binding peptide from D-alanine carboxypeptidase of Bacillus subtilis. Proc. Natl. Acad. Sci. USA 1977, 74, 1009–1012. [Google Scholar] [CrossRef] [Green Version]
- Georgopapadakou, N.H.; Liu, F.Y. Penicillin-binding proteins in bacteria. Antimicrob. Agents Chemother. 1980, 18, 148–157. [Google Scholar] [CrossRef] [Green Version]
- García-Álvarez, L.; Holden, M.T.; Lindsay, H.; Webb, C.R.; Brown, D.F.; Curran, M.D.; Walpole, E.; Brooks, K.; Pickard, D.J.; Teale, C.; et al. Meticillin-resistant Staphylococcus aureus with a novel mecA homologue in human and bovine populations in the UK and Denmark: A descriptive study. Lancet Infect. Dis. 2011, 11, 595–603. [Google Scholar] [CrossRef]
- Brown, D.F.; Reynolds, P.E. Intrinsic resistance to beta-lactam antibiotics in Staphylococcus aureus. FEBS Lett. 1980, 122, 275–278. [Google Scholar] [CrossRef] [Green Version]
- Hartman, B.J.; Tomasz, A. Low-affinity penicillin-binding protein associated with beta-lactam resistance in Staphylococcus aureus. J. Bacteriol. 1984, 158, 513–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lovering, A.L.; de Castro, L.H.; Lim, D.; Strynadka, N.C. Structural insight into the transglycosylation step of bacterial cell-wall biosynthesis. Science 2007, 315, 1402–1405. [Google Scholar] [CrossRef] [PubMed]
- Lim, D.; Strynadka, N.C. Structural basis for the beta lactam resistance of PBP2a from methicillin-resistant Staphylococcus aureus. Nat. Struct. Biol. 2002, 9, 870–876. [Google Scholar] [CrossRef]
- Lovering, A.L.; Gretes, M.C.; Safadi, S.S.; Danel, F.; de Castro, L.; Page, M.G.; Strynadka, N.C. Structural insights into the anti-methicillin-resistant Staphylococcus aureus (MRSA) activity of ceftobiprole. J. Biol. Chem. 2012, 287, 32096–32102. [Google Scholar] [CrossRef] [Green Version]
- Otero, L.H.; Rojas-Altuve, A.; Llarrull, L.I.; Carrasco-López, C.; Kumarasiri, M.; Lastochkin, E.; Fishovitz, J.; Dawley, M.; Hesek, D.; Lee, M.; et al. How allosteric control of Staphylococcus aureus penicillin binding protein 2a enables methicillin resistance and physiological function. Proc. Natl. Acad. Sci. USA 2013, 110, 16808–16813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelley, W.L.; Jousselin, A.; Barras, C.; Lelong, E.; Renzoni, A. Missense mutations in PBP2A Affecting ceftaroline susceptibility detected in epidemic hospital-acquired methicillin-resistant Staphylococcus aureus clonotypes ST228 and ST247 in Western Switzerland archived since 1998. Antimicrob. Agents Chemother. 2015, 59, 1922–1930. [Google Scholar] [CrossRef] [Green Version]
- Nigo, M.; Diaz, L.; Carvajal, L.P.; Tran, T.T.; Rios, R.; Panesso, D.; Garavito, J.D.; Miller, W.R.; Wanger, A.; Weinstock, G.; et al. Ceftaroline-Resistant, Daptomycin-Tolerant, and Heterogeneous Vancomycin-Intermediate Methicillin-Resistant Staphylococcus aureus Causing Infective Endocarditis. Antimicrob. Agents Chemother. 2017, 61, e01235-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banerjee, R.; Gretes, M.; Basuino, L.; Strynadka, N.; Chambers, H.F. In vitro selection and characterization of ceftobiprole-resistant methicillin-resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 2008, 52, 2089–2096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banerjee, R.; Gretes, M.; Harlem, C.; Basuino, L.; Chambers, H.F. A mecA-negative strain of methicillin-resistant Staphylococcus aureus with high-level β-lactam resistance contains mutations in three genes. Antimicrob. Agents Chemother. 2010, 54, 4900–4902. [Google Scholar] [CrossRef] [PubMed]
- Chan, L.C.; Gilbert, A.; Basuino, L.; da Costa, T.M.; Hamilton, S.M.; Dos Santos, K.R.; Chambers, H.F.; Chatterjee, S.S. PBP 4 Mediates High-Level Resistance to New-Generation Cephalosporins in Staphylococcus aureus. Antimicrob. Agents Chemother. 2016, 60, 3934–3941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chatterjee, S.S.; Chen, L.; Gilbert, A.; da Costa, T.M.; Nair, V.; Datta, S.K.; Kreiswirth, B.N.; Chambers, H.F. PBP4 Mediates β-Lactam Resistance by Altered Function. Antimicrob. Agents Chemother. 2017, 61, e00932-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greninger, A.L.; Chatterjee, S.S.; Chan, L.C.; Hamilton, S.M.; Chambers, H.F.; Chiu, C.Y. Whole-Genome Sequencing of Methicillin-Resistant Staphylococcus aureus Resistant to Fifth-Generation Cephalosporins Reveals Potential Non-mecA Mechanisms of Resistance. PLoS ONE 2016, 11, e0149541. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, S.M.; Alexander, J.A.N.; Choo, E.J.; Basuino, L.; da Costa, T.M.; Severin, A.; Chung, M.; Aedo, S.; Strynadka, N.C.J.; Tomasz, A.; et al. High-Level Resistance of Staphylococcus aureus to β-Lactam Antibiotics Mediated by Penicillin-Binding Protein 4 (PBP4). Antimicrob. Agents Chemother. 2017, 61, e02727-16. [Google Scholar] [CrossRef] [Green Version]
- Memmi, G.; Filipe, S.R.; Pinho, M.G.; Fu, Z.; Cheung, A. Staphylococcus aureus PBP4 is essential for beta-lactam resistance in community-acquired methicillin-resistant strains. Antimicrob. Agents Chemother. 2008, 52, 3955–3966. [Google Scholar] [CrossRef] [Green Version]
- Nair, D.R.; Monteiro, J.M.; Memmi, G.; Thanassi, J.; Pucci, M.; Schwartzman, J.; Pinho, M.G.; Cheung, A.L. Characterization of a novel small molecule that potentiates β-lactam activity against gram-positive and gram-negative pathogens. Antimicrob. Agents Chemother. 2015, 59, 1876–1885. [Google Scholar] [CrossRef] [Green Version]
- de Mesy Bentley, K.L.; MacDonald, A.; Schwarz, E.M.; Oh, I. Chronic Osteomyelitis with Staphylococcus aureus Deformation in Submicron Canaliculi of Osteocytes: A Case Report. JBJS Case Connect. 2018, 8, e8. [Google Scholar] [CrossRef]
- de Mesy Bentley, K.L.; Trombetta, R.; Nishitani, K.; Bello-Irizarry, S.N.; Ninomiya, M.; Zhang, L.; Chung, H.L.; McGrath, J.L.; Daiss, J.L.; Awad, H.A.; et al. Evidence of Staphylococcus aureus Deformation, Proliferation, and Migration in Canaliculi of Live Cortical Bone in Murine Models of Osteomyelitis. J. Bone Miner Res. 2017, 32, 985–990. [Google Scholar] [CrossRef] [Green Version]
- Masters, E.A.; Trombetta, R.P.; de Mesy Bentley, K.L.; Boyce, B.F.; Gill, A.L.; Gill, S.R.; Nishitani, K.; Ishikawa, M.; Morita, Y.; Ito, H.; et al. Evolving concepts in bone infection: Redefining “biofilm”, “acute vs. chronic osteomyelitis”, “the immune proteome” and “local antibiotic therapy”. Bone Res. 2019, 7, 20. [Google Scholar] [CrossRef] [Green Version]
- Schwarz, E.M.; McLaren, A.C.; Sculco, T.P.; Brause, B.; Bostrom, M.; Kates, S.L.; Parvizi, J.; Alt, V.; Arnold, W.V.; Carli, A.; et al. Adjuvant antibiotic-loaded bone cement: Concerns with current use and research to make it work. J. Orthop. Res. 2021, 39, 227–239. [Google Scholar] [CrossRef]
- Yu, B.; Pacureanu, A.; Olivier, C.; Cloetens, P.; Peyrin, F. Assessment of the human bone lacuno-canalicular network at the nanoscale and impact of spatial resolution. Sci. Rep. 2020, 10, 4567. [Google Scholar] [CrossRef] [Green Version]
- Masters, E.A.; de Mesy Bentley, K.L.; Gill, A.L.; Hao, S.P.; Galloway, C.A.; Salminen, A.T.; Guy, D.R.; McGrath, J.L.; Awad, H.A.; Gill, S.R.; et al. Identification of Penicillin Binding Protein 4 (PBP4) as a critical factor for Staphylococcus aureus bone invasion during osteomyelitis in mice. PLoS Pathog. 2020, 16, e1008988. [Google Scholar] [CrossRef]
- Katayama, Y.; Zhang, H.Z.; Hong, D.; Chambers, H.F. Jumping the barrier to beta-lactam resistance in Staphylococcus aureus. J. Bacteriol. 2003, 185, 5465–5472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- ISO 10993-5:2009; Biological Evaluation of Medical Devices-Part 5: Tests for In Vitro Cytotoxicity. International Organization for Standardization: Geneva, Switzerland, 2009; p. 34.
- Fedarovich, A.; Djordjevic, K.A.; Swanson, S.M.; Peterson, Y.K.; Nicholas, R.A.; Davies, C. High-throughput screening for novel inhibitors of Neisseria gonorrhoeae penicillin-binding protein 2. PLoS ONE 2012, 7, e44918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masters, E.A.; Muthukrishnan, G.; Ho, L.; Gill, A.L.; de Mesy Bentley, K.L.; Galloway, C.A.; McGrath, J.L.; Awad, H.A.; Gill, S.R.; Schwarz, E.M. Staphylococcus aureus Cell Wall Biosynthesis Modulates Bone Invasion and Osteomyelitis Pathogenesis. Front. Microbiol. 2021, 12, 723498. [Google Scholar] [CrossRef] [PubMed]
- Sawhney, S.S.; Ransom, E.M.; Wallace, M.A.; Reich, P.J.; Dantas, G.; Burnham, C.D. Comparative Genomics of Borderline Oxacillin-Resistant Staphylococcus aureus Detected during a Pseudo-outbreak of Methicillin-Resistant S. aureus in a Neonatal Intensive Care Unit. mBio 2022, 13, e0319621. [Google Scholar] [CrossRef] [PubMed]
- Alexander, J.A.N.; Chatterjee, S.S.; Hamilton, S.M.; Eltis, L.D.; Chambers, H.F.; Strynadka, N.C.J. Structural and kinetic analyses of penicillin-binding protein 4 (PBP4)-mediated antibiotic resistance in Staphylococcus aureus. J. Biol. Chem. 2018, 293, 19854–19865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kremers, H.M.; Nwojo, M.E.; Ransom, J.E.; Wood-Wentz, C.M.; Melton, L.J., III; Huddleston, P.M., III. Trends in the epidemiology of osteomyelitis: A population-based study, 1969 to 2009. J. Bone Joint Surg. Am. 2015, 97, 837–845. [Google Scholar] [CrossRef] [Green Version]
- Loskill, P.; Pereira, P.M.; Jung, P.; Bischoff, M.; Herrmann, M.; Pinho, M.G.; Jacobs, K. Reduction of the peptidoglycan crosslinking causes a decrease in stiffness of the Staphylococcus aureus cell envelope. Biophys. J. 2014, 107, 1082–1089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reichmann, N.T.; Tavares, A.C.; Saraiva, B.M.; Jousselin, A.; Reed, P.; Pereira, A.R.; Monteiro, J.M.; Sobral, R.G.; VanNieuwenhze, M.S.; Fernandes, F.; et al. SEDS-bPBP pairs direct lateral and septal peptidoglycan synthesis in Staphylococcus aureus. Nat. Microbiol. 2019, 4, 1368–1377. [Google Scholar] [CrossRef]
- McDougal, L.K.; Steward, C.D.; Killgore, G.E.; Chaitram, J.M.; McAllister, S.K.; Tenover, F.C. Pulsed-field gel electrophoresis typing of oxacillin-resistant Staphylococcus aureus isolates from the United States: Establishing a national database. J. Clin. Microbiol. 2003, 41, 5113–5120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dyke, K.G.; Jevons, M.P.; Parker, M.T. Penicillinase production and intrinsic resistance to penicillins in Staphylococcus aures. Lancet 1966, 1, 835–838. [Google Scholar] [CrossRef]
- Roth, K.M.; Wolf, M.K.; Rossi, M.; Butler, J.S. The nuclear exosome contributes to autogenous control of NAB2 mRNA levels. Mol. Cell. Biol. 2005, 25, 1577–1585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chojnacki, M.; Philbrick, A.; Wucher, B.; Reed, J.N.; Tomaras, A.; Dunman, P.M.; Wozniak, R.A. Development of a broad-spectrum antimicrobial combination for the treatment of Staphylococcus aureus and Pseudomonas aeruginosa corneal infections. Antimicrob. Agents Chemother. 2019, 63, e01929-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Odds, F.C. Synergy, antagonism, and what the chequerboard puts between them. J. Antimicrob. Chemother. 2003, 52, 1. [Google Scholar] [CrossRef]
- Chojnacki, M.; Cao, X.; Flaherty, D.P.; Dunman, P.M. Optimization of 2-acylaminocycloalkylthiophene derivatives for activity against Staphylococcus aureus RnpA. Antibiotics 2021, 10, 369. [Google Scholar] [CrossRef]
- Goldberg, J.A.; Nguyen, H.; Kumar, V.; Spencer, E.J.; Hoyer, D.; Marshall, E.K.; Cmolik, A.; O’Shea, M.; Marshall, S.H.; Hujer, A.M.; et al. A γ-Lactam Siderophore Antibiotic Effective against Multidrug-Resistant Gram-Negative Bacilli. J. Med. Chem. 2020, 63, 5990–6002. [Google Scholar] [CrossRef]
- Kumar, V.; Viviani, S.L.; Ismail, J.; Agarwal, S.; Bonomo, R.A.; van den Akker, F. Structural analysis of the boronic acid β-lactamase inhibitor vaborbactam binding to Pseudomonas aeruginosa penicillin-binding protein 3. PLoS ONE 2021, 16, e0258359. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain | NAF | MER | BPR | CIP | LEV | TOB | MIN | MUP | COL |
|---|---|---|---|---|---|---|---|---|---|
| USA300 | 8 | 4 | 0.5 | 4 | 1 | 1 | 0.5 | 0.125 | 128 |
| USA300∆pbp4 | 4 | 2 | 0.25 | 1 | 0.5 | 1 | 0.5 | 0.125 | 128 |
| USA300∆pbp4 pPBP4 | 8 | 4 | 0.5 | 2 | 1 | 1 | 0.5 | 0.125 | 128 |
| COLnex | 0.25 | 1 | 0.25 | 0.25 | 0.25 | 0.06 | 0.5 | 0.03 | 128 |
| CRB | >128 | 64 | >128 | 0.5 | 0.5 | 0.06 | 0.5 | 0.03 | 128 |
| CRB∆pbp4 | 0.25 | 1 | 0.25 | 0.25 | 0.25 | 0.06 | 0.5 | 0.03 | 128 |
| Compound | CRB | CRB∆pbp4 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Alone | Combination | Alone | Combination | |||||||
| BPR 1 | Cmpd 2 | BPR 1 | Cmpd 2 | Fold 3 | BPR 1 | Cmpd 2 | BPR 1 | Cmpd 2 | Fold 3 | |
| 5784306 | 128 | 100 | 4 | 12.5 | 32 | 0.25 | 400 | 0.25 | 12.5 | 0 |
| 7611906 | 128 | 400 | 16 | 25 | 8 | 0.25 | 400 | 0.25 | 25 | 0 |
| 7729837 | 128 | 200 | 64 | 50 | 2 | 0.25 | 400 | 0.25 | 50 | 0 |
| 9009498 | 128 | 100 | 8 | 25 | 16 | 0.25 | 100 | 0.06 | 25 | 4 |
| 7974147 | 128 | 400 | 8 | 25 | 16 | 0.25 | 400 | 0.25 | 25 | 0 |
| 9314848 | 128 | 400 | 8 | 25 | 16 | 0.25 | 400 | 0.125 | 25 | 2 |
| Strain | Relevant Characteristics | Source |
|---|---|---|
| USA300 | Community-associated MRSA | [42] |
| USA300∆pbp4 | USA300 pbp4 deletion | [32] |
| USA300∆pbp4 pPBP4 | USA300∆pbp4 harboring pCN40-pbp4 | [32] |
| COL | Hospital-associated MRSA | [43] |
| COLn | TetS COL derivative | [33] |
| COLnex | COLn ∆SCCmec | [33] |
| CRB | COLnex PBP4 over-expresser derivative | [19] |
| CRB∆pbp4 | CRB pbp4 deletion | [24] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Young, M.; Walsh, D.J.; Masters, E.; Gondil, V.S.; Laskey, E.; Klaczko, M.; Awad, H.; McGrath, J.; Schwarz, E.M.; Melander, C.; et al. Identification of Staphylococcus aureus Penicillin Binding Protein 4 (PBP4) Inhibitors. Antibiotics 2022, 11, 1351. https://doi.org/10.3390/antibiotics11101351
Young M, Walsh DJ, Masters E, Gondil VS, Laskey E, Klaczko M, Awad H, McGrath J, Schwarz EM, Melander C, et al. Identification of Staphylococcus aureus Penicillin Binding Protein 4 (PBP4) Inhibitors. Antibiotics. 2022; 11(10):1351. https://doi.org/10.3390/antibiotics11101351
Chicago/Turabian StyleYoung, Mikaeel, Danica J. Walsh, Elysia Masters, Vijay Singh Gondil, Emily Laskey, Michael Klaczko, Hani Awad, James McGrath, Edward M. Schwarz, Christian Melander, and et al. 2022. "Identification of Staphylococcus aureus Penicillin Binding Protein 4 (PBP4) Inhibitors" Antibiotics 11, no. 10: 1351. https://doi.org/10.3390/antibiotics11101351