
Genetic and Chemical Engineering of Phages for Controlling Multidrug-Resistant Bacteria


Abstract
:1. Introduction
2. Phage Genetic Modification
2.1. Virulence Gene Overexpression
2.2. MDR System Circumvention
2.2.1. Pathogen-Specific Gene
2.2.2. Biofilm
2.2.3. SOS System
2.3. Host Range Expansion
Tail Fiber Protein
3. Phage Chemical Modification
3.1. Phage–Chemical Crosslink
3.2. Phage Immobilization
4. Challenges in Phage Therapy
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ventola, C.L. The antibiotic resistance crisis: Part 1: Causes and threats. P T 2015, 40, 277–283. [Google Scholar]
- Wright, G.D. The antibiotic resistome: The nexus of chemical and genetic diversity. Nat. Rev. Genet. 2007, 5, 175–186. [Google Scholar] [CrossRef]
- Gu, D.; Dong, N.; Zheng, Z.; Lingbin, S.; Huang, M.; Wang, L.; Chan, E.W.-C.; Shu, L.; Yu, J.; Zhang, R.; et al. A fatal outbreak of ST11 carbapenem-resistant hypervirulent Klebsiella pneumoniae in a Chinese hospital: A molecular epidemiological study. Lancet Infect. Dis. 2018, 18, 37–46. [Google Scholar] [CrossRef]
- Yong, D.; Toleman, M.A.; Giske, C.G.; Cho, H.S.; Sundman, K.; Lee, K.; Walsh, T.R. Characterization of a New Metallo-β-Lactamase Gene, blaNDM-1, and a Novel Erythromycin Esterase Gene Carried on a Unique Genetic Structure in Klebsiella pneumoniae Sequence Type 14 from India. Antimicrob. Agents Chemother. 2009, 53, 5046–5054. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Pop, M. ARDB--Antibiotic Resistance Genes Database. Nucleic Acids Res. 2008, 37, D443–D447. [Google Scholar] [CrossRef] [Green Version]
- Mohr, K.I. History of Antibiotics Research. Curr. Top. Microbiol. Immunol. 2016, 398, 237–272. [Google Scholar] [CrossRef] [PubMed]
- Chan, B.K.; Abedon, S.T.; Loc-Carrillo, C. Phage cocktails and the future of phage therapy. Future Microbiol. 2013, 8, 769–783. [Google Scholar] [CrossRef] [PubMed]
- Kortright, K.E.; Chan, B.K.; Koff, J.L.; Turner, P.E. Phage Therapy: A Renewed Approach to Combat Antibiotic-Resistant Bacteria. Cell Host Microbe 2019, 25, 219–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Twort, F.W. Further Investigations on the Nature of Ultra-Microscopic Viruses and their Cultivation. J. Hyg. 1936, 36, 204–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pires, D.P.; Cleto, S.; Sillankorva, S.; Azeredo, J.; Lu, T.K. Genetically Engineered Phages: A Review of Advances over the Last Decade. Microbiol. Mol. Biol. Rev. 2016, 80, 523–543. [Google Scholar] [CrossRef] [Green Version]
- Domingo-Calap, P.; Delgado-Martínez, J. Bacteriophages: Protagonists of a Post-Antibiotic Era. Antibiotics 2018, 7, 66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemon, D.J.; Kay, M.K.; Titus, J.K.; Ford, A.A.; Chen, W.; Hamlin, N.J.; Hwang, Y.Y. Construction of a genetically modified T7Select phage system to express the antimicrobial peptide 1018. J. Microbiol. 2019, 57, 532–538. [Google Scholar] [CrossRef] [PubMed]
- Nir-Paz, R.; Gelman, D.; Khouri, A.; Sisson, B.M.; Fackler, J.; Alkalay-Oren, S.; Khalifa, L.; Rimon, A.; Yerushalmy, O.; Bader, R.; et al. Successful Treatment of Antibiotic-resistant, Poly-microbial Bone Infection With Bacteriophages and Antibiotics Combination. Clin. Infect. Dis. 2019, 69, 2015–2018. [Google Scholar] [CrossRef] [PubMed]
- Bao, J.; Wu, N.; Zeng, Y.; Chen, L.; Li, L.; Yang, L.; Zhang, Y.; Guo, M.; Li, L.; Li, J.; et al. Non-active antibiotic and bacteriophage synergism to successfully treat recurrent urinary tract infection caused by extensively drug-resistant Klebsiella pneumoniae. Emerg. Microbes Infect. 2020, 9, 771–774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schooley, R.T.; Biswas, B.; Gill, J.J.; Hernandez-Morales, A.; Lancaster, J.; Lessor, L.; Barr, J.J.; Reed, S.L.; Rohwer, F.; Benler, S.; et al. Development and Use of Personalized Bacteriophage-Based Therapeutic Cocktails To Treat a Patient with a Dissem-inated Resistant Acinetobacter baumannii Infection. Antimicrob. Agents Chemother. 2017, 61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Citorik, R.J.; Mimee, M.K.; Lu, T.K. Sequence-specific antimicrobials using efficiently delivered RNA-guided nucleases. Nat. Biotechnol. 2014, 32, 1141–1145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bikard, D.; Euler, C.W.; Jiang, W.; Nussenzweig, P.M.; Goldberg, G.W.; Duportet, X.; Fischetti, V.A.; Marraffini, L.A. Exploiting CRISPR-Cas nucleases to produce sequence-specific antimicrobials. Nat. Biotechnol. 2014, 32, 1146–1150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hagens, S.; Blasi, U. Genetically modified filamentous phage as bactericidal agents: A pilot study. Lett. Appl. Microbiol. 2003, 37, 318–323. [Google Scholar] [CrossRef]
- Westwater, C.; Kasman, L.M.; Schofield, D.A.; Werner, P.A.; Dolan, J.W.; Schmidt, M.G.; Norris, J.S. Use of Genetically Engineered Phage To Deliver Antimicrobial Agents to Bacteria: An Alternative Therapy for Treatment of Bacterial Infections. Antimicrob. Agents Chemother. 2003, 47, 1301–1307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, T.K.; Collins, J.J. Engineered bacteriophage targeting gene networks as adjuvants for antibiotic therapy. Proc. Natl. Acad. Sci. USA 2009, 106, 4629–4634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, T.K.; Collins, J.J. Dispersing biofilms with engineered enzymatic bacteriophage. Proc. Natl. Acad. Sci. USA 2007, 104, 11197–11202. [Google Scholar] [CrossRef] [Green Version]
- Pei, R.; Lamas-Samanamud, G.R. Inhibition of Biofilm Formation by T7 Bacteriophages Producing Quorum-Quenching Enzymes. Appl. Environ. Microbiol. 2014, 80, 5340–5348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yosef, I.; Manor, M.; Kiro, R.; Qimron, U. Temperate and lytic bacteriophages programmed to sensitize and kill antibiotic-resistant bacteria. Proc. Natl. Acad. Sci. USA 2015, 112, 7267–7272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.Y.; Moon, B.Y.; Park, J.W.; Thornton, J.A.; Park, Y.H.; Seo, K.S. Genetic engineering of a temperate phage-based delivery system for CRISPR/Cas9 antimicrobials against Staphylococcus aureus. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahichi, F.; Synnott, A.J.; Yamamichi, K.; Osada, T.; Tanji, Y. Site-specific recombination of T2 phage using IP008 long tail fiber genes provides a targeted method for expanding host range while retaining lytic activity. FEMS Microbiol. Lett. 2009, 295, 211–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, T.-Y.; Lo, Y.-H.; Tseng, P.-W.; Chang, S.-F.; Lin, Y.-T.; Chen, T.-S. A T3 and T7 Recombinant Phage Acquires Efficient Adsorption and a Broader Host Range. PLoS ONE 2012, 7, e30954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marzari, R.; Sblattero, D.; Righi, M.; Bradbury, A. Extending filamentous phage host range by the grafting of a heterologous receptor binding domain. Gene 1997, 185, 27–33. [Google Scholar] [CrossRef]
- Yehl, K.; Lemire, S.; Yang, A.C.; Ando, H.; Mimee, M.; Torres, M.D.T.; De La Fuente-Nunez, C.; Lu, T.K. Engineering Phage Host-Range and Suppressing Bacterial Resistance through Phage Tail Fiber Mutagenesis. Cell 2019, 179, 459–469.e9. [Google Scholar] [CrossRef] [PubMed]
- Kutateladze, M.; Adamia, R. Phage therapy experience at the Eliava Institute. Médecine et Maladies Infectieuses 2008, 38, 426–430. [Google Scholar] [CrossRef]
- Park, T.; Struck, D.K.; Deaton, J.F.; Young, R. Topological dynamics of holins in programmed bacterial lysis. Proc. Natl. Acad. Sci. USA 2006, 103, 19713–19718. [Google Scholar] [CrossRef] [Green Version]
- Pfalzgraff, A.; Brandenburg, K.; Weindl, G. Antimicrobial Peptides and Their Therapeutic Potential for Bacterial Skin Infections and Wounds. Front. Pharmacol. 2018, 9, 281. [Google Scholar] [CrossRef]
- Chan, D.I.; Prenner, E.J.; Vogel, H.J. Tryptophan- and arginine-rich antimicrobial peptides: Structures and mechanisms of action. Biochim. Biophys. Acta (BBA) Biomembr. 2006, 1758, 1184–1202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De La Fuente-Núñez, C.; Reffuveille, F.; Haney, E.F.; Straus, S.K.; Hancock, R.E.W. Broad-Spectrum Anti-biofilm Peptide That Targets a Cellular Stress Response. PLoS Pathog. 2014, 10, e1004152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De La Fuente-Núñez, C.; Korolik, V.; Bains, M.; Nguyen, U.; Breidenstein, E.B.M.; Horsman, S.; Lewenza, S.; Burrows, L.; Hancock, R.E.W. Inhibition of Bacterial Biofilm Formation and Swarming Motility by a Small Synthetic Cationic Peptide. Antimicrob. Agents Chemother. 2012, 56, 2696–2704. [Google Scholar] [CrossRef] [Green Version]
- Nikaido, H. Multidrug Resistance in Bacteria. Annu. Rev. Biochem. 2009, 78, 119–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rafii, F. Effects of treatment with antimicrobial agents on the human colonic microflora. Ther. Clin. Risk Manag. 2008, 4, 1343–1357. [Google Scholar] [CrossRef] [Green Version]
- Yoon, M.Y.; Yoon, S.S. Disruption of the Gut Ecosystem by Antibiotics. Yonsei Med. J. 2018, 59, 4–12. [Google Scholar] [CrossRef] [PubMed]
- Dolan, A.E.; Hou, Z.; Xiao, Y.; Gramelspacher, M.J.; Heo, J.; Howden, S.E.; Freddolino, P.L.; Ke, A.; Zhang, Y. Introducing a Spectrum of Long-Range Genomic Deletions in Human Embryonic Stem Cells Using Type I CRISPR-Cas. Mol. Cell 2019, 74, 936–950.e5. [Google Scholar] [CrossRef] [PubMed]
- Bjarnsholt, T. The Role of Bacterial Biofi lms in Chronic Infections. APMIS Suppl. 2013, 1–51. [Google Scholar] [CrossRef] [PubMed]
- Flemming, H.-C.; Wingender, J. The biofilm matrix. Nat. Rev. Microbiol. 2010, 8, 623–633. [Google Scholar] [CrossRef]
- Elias, S.; Banin, E. Multi-species biofilms: Living with friendly neighbors. FEMS Microbiol. Rev. 2012, 36, 990–1004. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.-M.; Liu, J.; Wang, K.; Zhang, X.; Zhao, T.; Luo, H.-M. Observations of Bacterial Biofilm on Ureteral Stent and Studies on the Distribution of Pathogenic Bacteria and Drug Resistance. Urol. Int. 2018, 101, 320–326. [Google Scholar] [CrossRef] [PubMed]
- Silva, A.J.; Benitez, J.A. Vibrio cholerae Biofilms and Cholera Pathogenesis. PLoS Negl. Trop. Dis. 2016, 10, e0004330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Achermann, Y.; Goldstein, E.J.C.; Coenye, T.; Shirtliff, M.E. Propionibacterium acnes: From Commensal to Opportunistic Biofilm-Associated Implant Pathogen. Clin. Microbiol. Rev. 2014, 27, 419–440. [Google Scholar] [CrossRef] [Green Version]
- Kuang, X.; Chen, V.; Xu, X. Novel Approaches to the Control of Oral Microbial Biofilms. BioMed Res. Int. 2018, 2018, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pang, Z.; Raudonis, R.; Glick, B.R.; Lin, T.-J.; Cheng, Z. Antibiotic resistance in Pseudomonas aeruginosa: Mechanisms and alternative therapeutic strategies. Biotechnol. Adv. 2019, 37, 177–192. [Google Scholar] [CrossRef] [PubMed]
- Hall, C.W.; Mah, T.-F. Molecular mechanisms of biofilm-based antibiotic resistance and tolerance in pathogenic bacteria. FEMS Microbiol. Rev. 2017, 41, 276–301. [Google Scholar] [CrossRef] [PubMed]
- Hathroubi, S.; Mekni, M.A.; Domenico, P.; Nguyen, D.; Jacques, M. Biofilms: Microbial Shelters Against Antibiotics. Microb. Drug Resist. 2017, 23, 147–156. [Google Scholar] [CrossRef]
- Itoh, Y.; Wang, X.; Hinnebusch, B.J.; Preston, J.F.; Romeo, T. Depolymerization of β-1,6-N-Acetyl-d-Glucosamine Disrupts the Integrity of Diverse Bacterial Biofilms. J. Bacteriol. 2005, 187, 382–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waters, C.M.; Bassler, B.L. QUORUM SENSING: Cell-to-Cell Communication in Bacteria. Annu. Rev. Cell Dev. Biol. 2005, 21, 319–346. [Google Scholar] [CrossRef] [Green Version]
- Reading, N.C.; Sperandio, V. Quorum sensing: The many languages of bacteria. FEMS Microbiol. Lett. 2006, 254, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Whiteley, M.; Diggle, S.P.; Greenberg, E.P. Progress in and promise of bacterial quorum sensing research. Nat. Cell Biol. 2017, 551, 313–320. [Google Scholar] [CrossRef]
- Mclean, R.J.; Whiteley, M.; Stickler, D.J.; Fuqua, W.C. Evidence of autoinducer activity in naturally occurring biofilms. FEMS Microbiol. Lett. 1997, 154, 259–263. [Google Scholar] [CrossRef]
- Toyofuku, M.; Nomura, N.; Eberl, L. Types and origins of bacterial membrane vesicles. Nat. Rev. Microbiol. 2019, 17, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Da Re, S.; Garnier, F.; Guérin, E.; Campoy, S.; Denis, F.; Ploy, M.-C. The SOS response promotes qnrB quinolone-resistance determinant expression. EMBO Rep. 2009, 10, 929–933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guerin, É.; Cambray, G.; Sanchez-Alberola, N.; Campoy, S.; Erill, I.; Da Re, S.; Gonzalez-Zorn, B.; Barbé, J.; Ploy, M.-C.; Mazel, D. The SOS Response Controls Integron Recombination. Science 2009, 324, 1034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cambray, G.; Sanchez-Alberola, N.; Campoy, S.; Guerin, É.; Da Re, S.; González-Zorn, B.; Ploy, M.-C.; Barbé, J.; Mazel, D.; Erill, I. Prevalence of SOS-mediated control of integron integrase expression as an adaptive trait of chromosomal and mobile integrons. Mob. DNA 2011, 2, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Bai, H.; Du, J.-F.; Hu, M.; Qi, J.; Cai, Y.-N.; Niu, W.-W.; Liu, Y.-Q. Analysis of mechanisms of resistance and tolerance of Escherichia coli to enrofloxacin. Ann. Microbiol. 2011, 62, 293–298. [Google Scholar] [CrossRef]
- Harms, A.; Maisonneuve, E.; Gerdes, K. Mechanisms of bacterial persistence during stress and antibiotic exposure. Science 2016, 354, aaf4268. [Google Scholar] [CrossRef] [PubMed]
- Kohanski, M.A.; Dwyer, D.J.; Hayete, B.; Lawrence, C.A.; Collins, J.J. A Common Mechanism of Cellular Death Induced by Bactericidal Antibiotics. Cell 2007, 130, 797–810. [Google Scholar] [CrossRef] [Green Version]
- Mapes, A.C.; Trautner, B.W.; Liao, K.S.; Ramig, R.F. Development of expanded host range phage active on biofilms of multi-drug resistantPseudomonas aeruginosa. Bacteriophage 2016, 6, e1096995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Jiang, X. Multiple strategies to activate gold nanoparticles as antibiotics. Nanoscale 2013, 5, 8340–8350. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Pan, P.; Zheng, D.-W.; Bao, P.; Zeng, X.; Zhang, X.-Z. Bioinorganic hybrid bacteriophage for modulation of intestinal microbiota to remodel tumor-immune microenvironment against colorectal cancer. Sci. Adv. 2020, 6, eaba1590. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Yang, Y.; Guo, Y.; Lu, S.; Du, Y.; Li, J.-J.; Zhang, X.; Leung, N.L.C.; Zhao, Z.; Niu, G.; et al. Phage-Guided Targeting, Discriminative Imaging, and Synergistic Killing of Bacteria by AIE Bioconjugates. J. Am. Chem. Soc. 2020, 142, 3959–3969. [Google Scholar] [CrossRef]
- Dong, S.; Shi, H.; Zhang, X.; Chen, X.; Cao, N.; Mao, C.; Gao, X.; Wang, L. Difunctional bacteriophage conjugated with photosensitizers for Candida albicans-targeting photodynamic inactivation. Int. J. Nanomed. 2018, 13, 2199–2216. [Google Scholar] [CrossRef] [Green Version]
- Anany, H.; Chen, W.; Pelton, R.; Griffiths, M.W. Biocontrol of Listeria monocytogenes and Escherichia coli O157:H7 in Meat by Using Phages Immobilized on Modified Cellulose Membranes. Appl. Environ. Microbiol. 2011, 77, 6379–6387. [Google Scholar] [CrossRef] [Green Version]
- Liana, A.E.; Marquis, C.P.; Gunawan, C.; Gooding, J.J.; Amal, R. Antimicrobial activity of T4 bacteriophage conjugated indium tin oxide surfaces. J. Colloid Interface Sci. 2018, 514, 227–233. [Google Scholar] [CrossRef]
- Dougherty, T.J.; Gomer, C.J.; Henderson, B.W.; Jori, G.; Kessel, D.; Korbelik, M.; Moan, J.; Peng, Q. Photodynamic Therapy. J. Natl. Cancer Inst. 1998, 90, 889–905. [Google Scholar] [CrossRef] [Green Version]
- Schafer, M.; Schmitz, C.; Horneck, G. High sensitivity of Deinococcus radiodurans to photodynamically-produced singlet oxygen. Int. J. Radiat. Biol. 1998, 74, 249–253. [Google Scholar] [CrossRef]
- Gouvêa, D.M.; Mendonça, R.C.S.; Soto, M.L.; Cruz, R.S. Acetate cellulose film with bacteriophages for potential antimicrobial use in food packaging. LWT 2015, 63, 85–91. [Google Scholar] [CrossRef] [Green Version]
- Romero-Calle, D.; Benevides, R.G.; Góes-Neto, A.; Billington, C. Bacteriophages as Alternatives to Antibiotics in Clinical Care. Antibiotics 2019, 8, 138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dąbrowska, K.; Świtała-Jeleń, K.; Opolski, A.; Górski, A. Possible association between phages, Hoc protein, and the immune system. Arch. Virol. 2005, 151, 209–215. [Google Scholar] [CrossRef]
- Van Belleghem, J.D.; Clement, F.; Merabishvili, M.; Lavigne, R.; Vaneechoutte, M. Pro- and anti-inflammatory responses of peripheral blood mononuclear cells induced by Staphylococcus aureus and Pseudomonas aeruginosa phages. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geier, M.R.; Trigg, M.E.; Merril, C.R. Fate of Bacteriophage Lambda in Non-immune Germ-free Mice. Nat. Cell Biol. 1973, 246, 221–223. [Google Scholar] [CrossRef]
- Morozova, V.V.; Vlassov, V.V.; Tikunova, N.V. Applications of bacteriophages in the treatment of localized infections in humans. Front. Microbiol. 2018, 9, 1696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Payne, R.J.H. Phage therapy: The peculiar kinetics of self-replicating pharmaceuticals. Clin. Pharmacol. Ther. 2000, 68, 225–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vitiello, C.L.; Merril, C.R.; Adhya, S. An amino acid substitution in a capsid protein enhances phage survival in mouse circulatory system more than a 1000-fold. Virus Res. 2005, 114, 101–103. [Google Scholar] [CrossRef] [PubMed]
- Merril, C.R.; Biswas, B.; Carlton, R.; Jensen, N.C.; Creed, G.J.; Zullo, S.; Adhya, S. Long-circulating bacteriophage as antibacterial agents. Proc. Natl. Acad. Sci. USA 1996, 93, 3188–3192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanji, Y.; Shimada, T.; Fukudomi, H.; Miyanaga, K.; Nakai, Y.; Unno, H. Therapeutic use of phage cocktail for controlling Escherichia coli O157:H7 in gastrointestinal tract of mice. J. Biosci. Bioeng. 2005, 100, 280–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reindel, R.; Fiore, C.R. Phage Therapy: Considerations and Challenges for Development. Clin. Infect. Dis. 2017, 64, 1589–1590. [Google Scholar] [CrossRef] [Green Version]
- Dalpke, A.; Frank, J.; Peter, M.; Heeg, K. Activation of Toll-Like Receptor 9 by DNA from Different Bacterial Species. Infect. Immun. 2006, 74, 940–946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sweere, J.M.; Van Belleghem, J.D.; Ishak, H.; Bach, M.S.; Popescu, M.; Sunkari, V.; Kaber, G.; Manasherob, R.; Suh, G.A.; Cao, X.; et al. Bacteriophage trigger antiviral immunity and prevent clearance of bacterial infection. Science 2019, 363, eaat9691. [Google Scholar] [CrossRef] [PubMed]
- Opal, S.M. Endotoxins and other sepsis triggers. Contrib. Nephrol. 2010, 14–24. [Google Scholar] [CrossRef]
- Brüssow, H. What is needed for phage therapy to become a reality in Western medicine? Virology 2012, 434, 138–142. [Google Scholar] [CrossRef] [Green Version]
- Pirnay, J.-P.; Blasdel, B.G.; Bretaudeau, L.; Buckling, A.; Chanishvili, N.; Clark, J.R.; Corte-Real, S.; Debarbieux, L.; Dublanchet, A.; De Vos, D.; et al. Quality and Safety Requirements for Sustainable Phage Therapy Products. Pharm. Res. 2015, 32, 2173–2179. [Google Scholar] [CrossRef] [Green Version]
- El-Shibiny, A.; El-Sahhar, S. Bacteriophages: The possible solution to treat infections caused by pathogenic bacteria. Can. J. Microbiol. 2017, 63, 865–879. [Google Scholar] [CrossRef] [Green Version]
- Furfaro, L.L.; Payne, M.S.; Chang, B.J. Bacteriophage Therapy: Clinical Trials and Regulatory Hurdles. Front. Cell. Infect. Microbiol. 2018, 8, 376. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
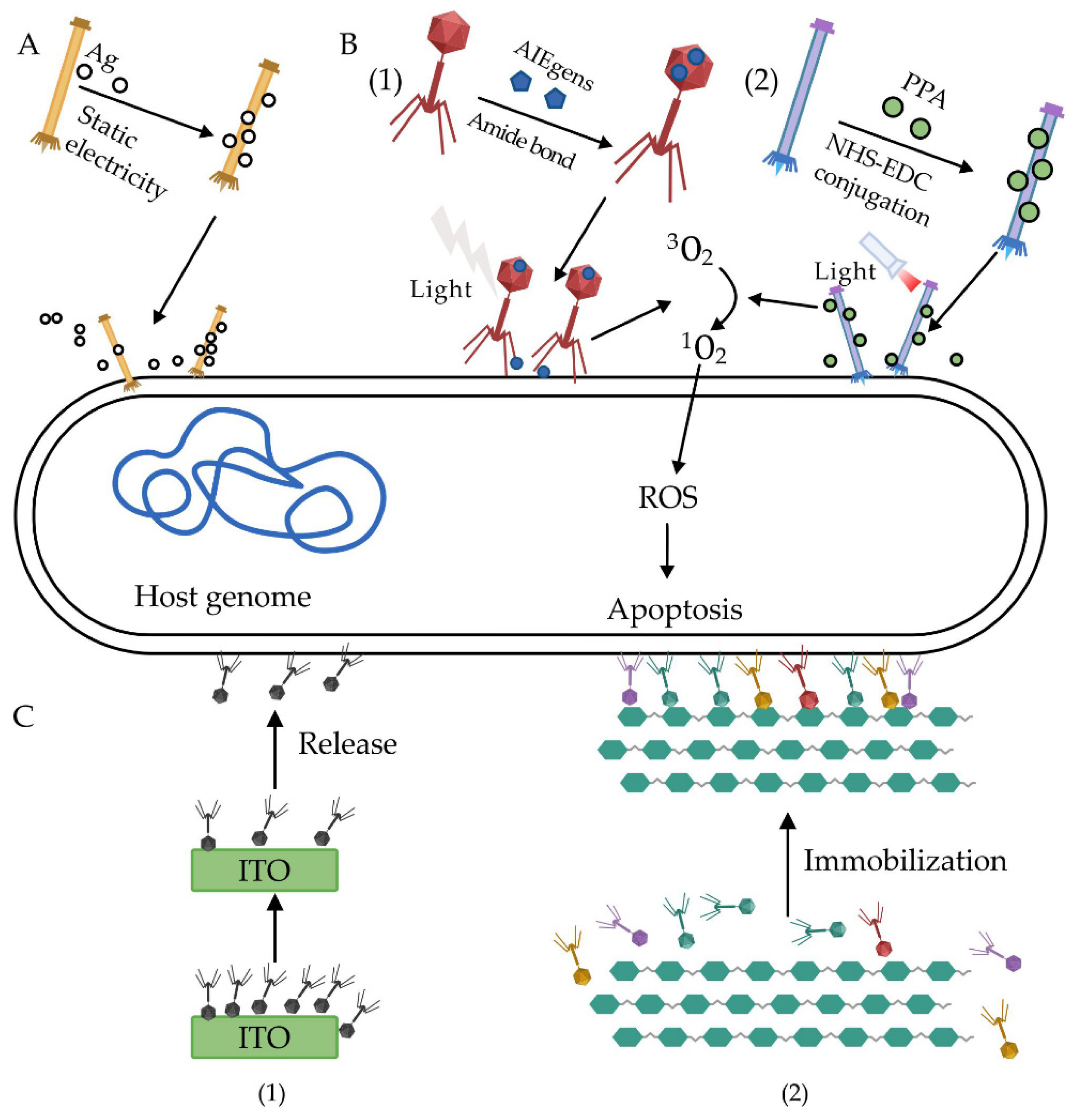{kind=link}
| Phage | Genetic Modification | Mechanism | Goal/Target | Ref. |
|---|---|---|---|---|
| M13 | λS105; Bgl Ⅱ | Membrane damage; DNA breakage | To reduce endotoxin | [18] |
| M13 | Gef; ChpBK | Membrane damage; mRNA degradation | To increase bactericidal efficiency | [19] |
| T7Select | peptide 1018 | Kill cells; inhibit biofilm | Biofilm | [12] |
| M13mp18 | LexA3 | Suppress SOS system | Antibiotic-resistant bacteria | [20] |
| Wild-type T7 | DspB | Hydrolysis β-1,6-N-acetyl-d-glucosamine | Biofilm | [21] |
| T7Select415-1 | AiiA | Inhibit quorum sensing | Biofilm | [22] |
| M13 phagemid | CRISPR-cas9 | Target resistance genes | Antibiotic-resistant bacteria | [17] |
| M13 phagemid | CRISPR-cas9 | Target resistance genes and virulent genes | Antibiotic-resistant bacteria | [16] |
| λ phage | CRISPR-cas3 | Target resistance genes | Antibiotic-resistant bacteria | [23] |
| φ SaBov | CRISPR-cas9 | Target the nuc gene | Antibiotic-resistant bacteria | [24] |
| T2, T3, Fd | Tail fiber genes | Expand the host range | Antiphage bacteria | [25,26,27,28] |
| Phage | Chemical Modification | Binding Force | Ref |
|---|---|---|---|
| M13 | Silver nanoparticles (AgNPs) | Ionic binding | [63] |
| PAP | AIEgens | Amide bond | [64] |
| JM phage | Pheophorbide a (PPA) | EDC/NHS Crosslinking | [65] |
| Bacteriophage T4 (ATCC 11303-B4) | Indium tin oxide (ITO) | Ionic binding | [66] |
| Phage cocktail | Cellulose membrane | Ionic binding | [67] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, D.; Chen, J.; Zhao, X.; Luo, Y.; Jin, M.; Fan, F.; Park, C.; Yang, X.; Sun, C.; Yan, J.; et al. Genetic and Chemical Engineering of Phages for Controlling Multidrug-Resistant Bacteria. Antibiotics 2021, 10, 202. https://doi.org/10.3390/antibiotics10020202
Guo D, Chen J, Zhao X, Luo Y, Jin M, Fan F, Park C, Yang X, Sun C, Yan J, et al. Genetic and Chemical Engineering of Phages for Controlling Multidrug-Resistant Bacteria. Antibiotics. 2021; 10(2):202. https://doi.org/10.3390/antibiotics10020202
Chicago/Turabian StyleGuo, Dingming, Jingchao Chen, Xueyang Zhao, Yanan Luo, Menglu Jin, Fenxia Fan, Chaiwoo Park, Xiaoman Yang, Chuqing Sun, Jin Yan, and et al. 2021. "Genetic and Chemical Engineering of Phages for Controlling Multidrug-Resistant Bacteria" Antibiotics 10, no. 2: 202. https://doi.org/10.3390/antibiotics10020202