
Comprehensive Analysis of miRNAs and Target mRNAs between Immature and Mature Testis Tissue in Chinese Red Steppes Cattle

 , , ,
, , ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Animal and Samples
2.3. RNA Extraction and Quality Analysis
2.4. Library Preparation of mRNA and miRNA and Quantification
2.5. Analysis of Differentially Expressed Genes
2.6. GO and KEGG Enrichment Analysis of DEGs
2.7. Analysis of miRNA Profiling and Prediction of Novel miRNA
2.8. miRNA Target Gene Prediction and Annotation Analyses
2.9. Real-Time PCR of DEGs and DERs
3. Results
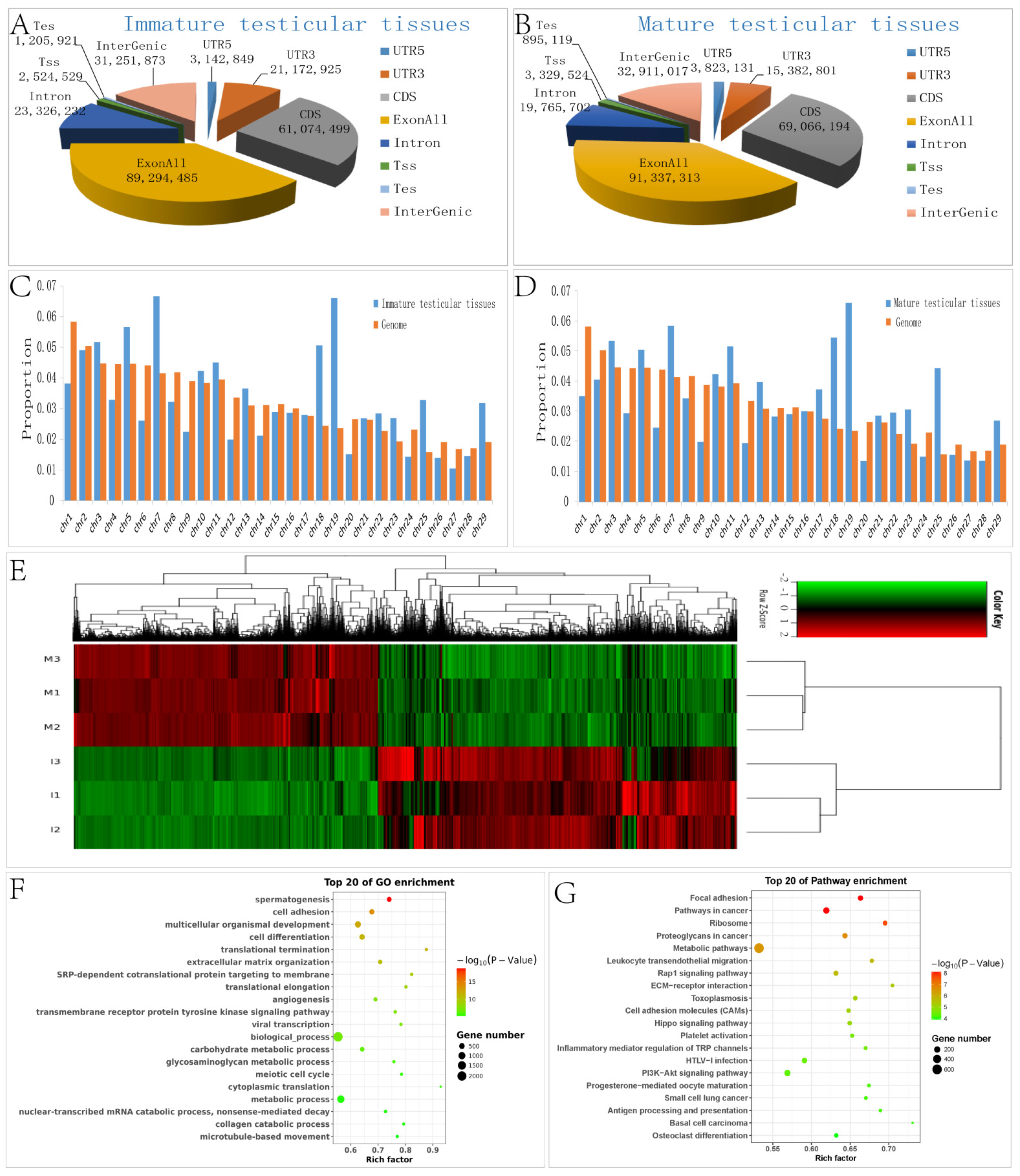
3.1. Differential Expression of mRNAs in Chinese Red Steppes between Immature and Mature Testicular Tissues
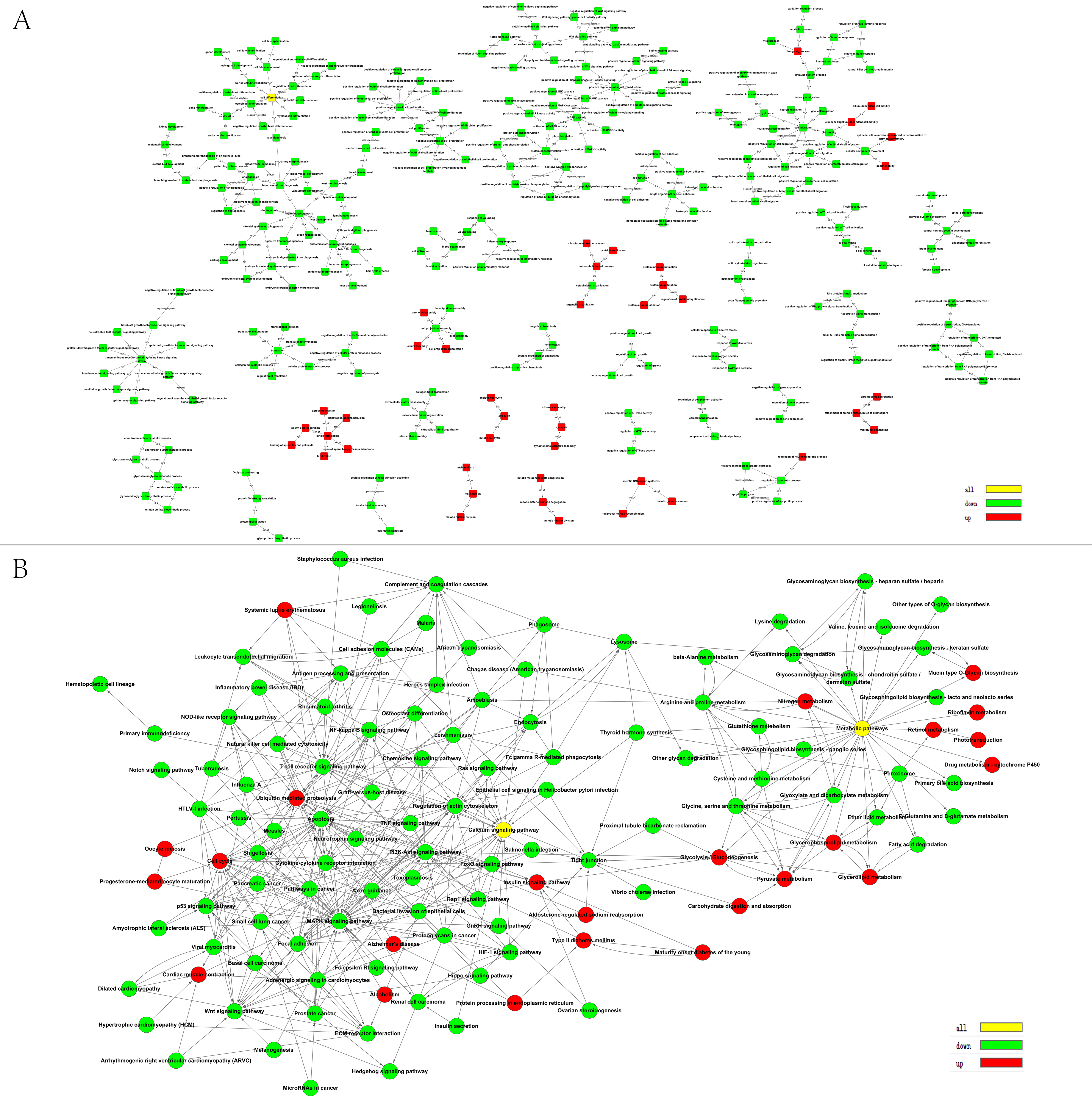
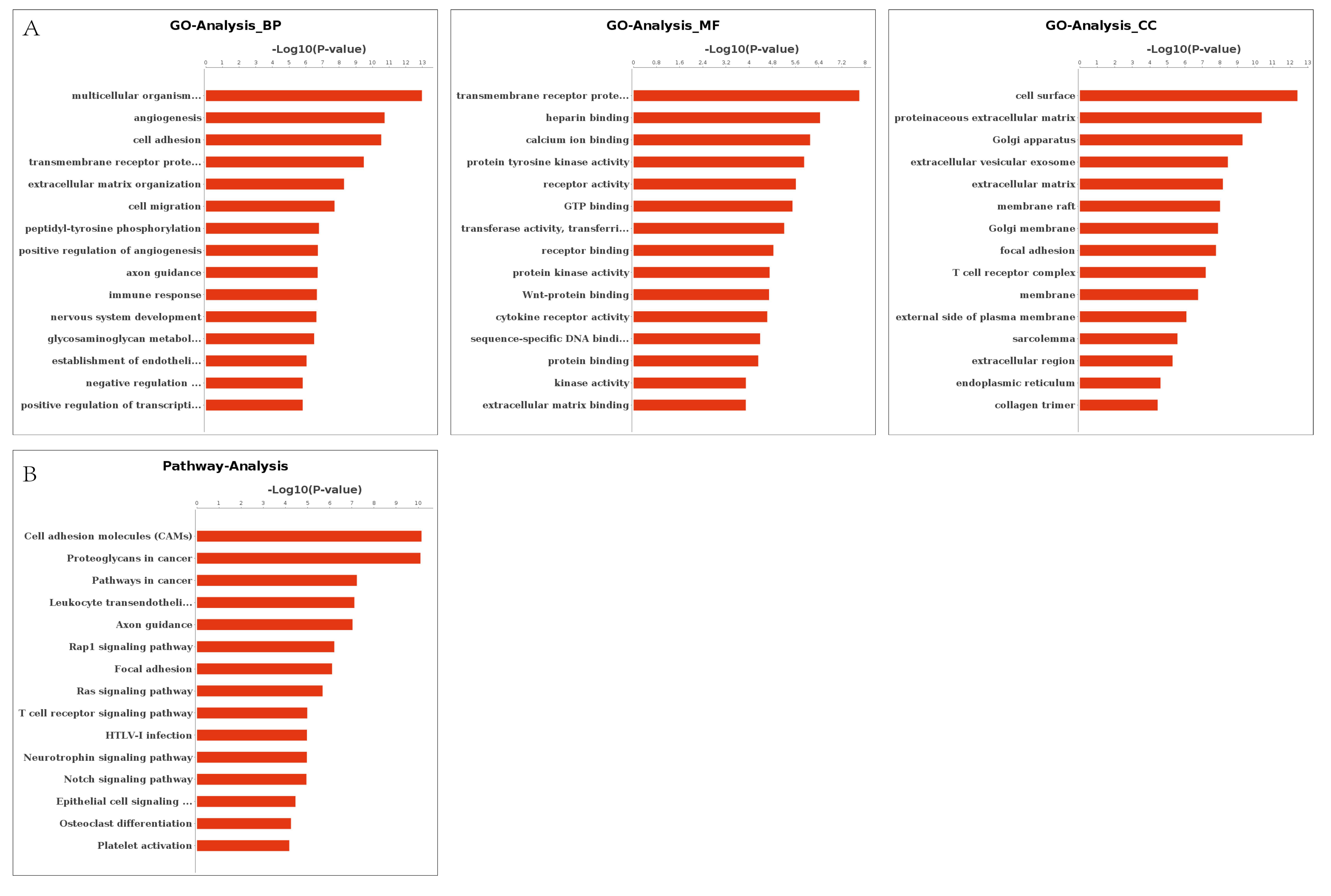
3.2. GO and KEGG Pathway Analyses of DEGs
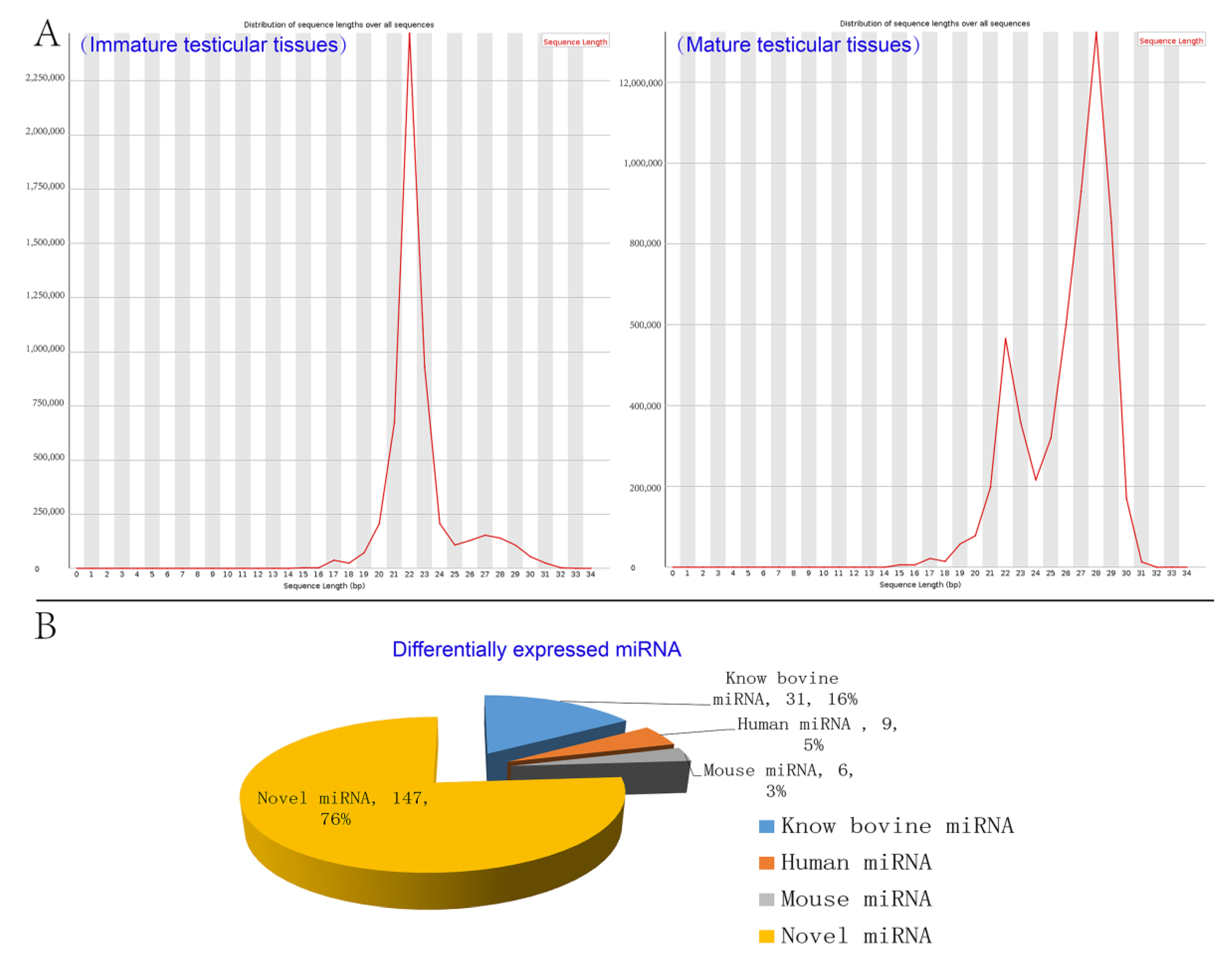
3.3. Analysis of miRNAs Expression Patterns in Testicular Tissues
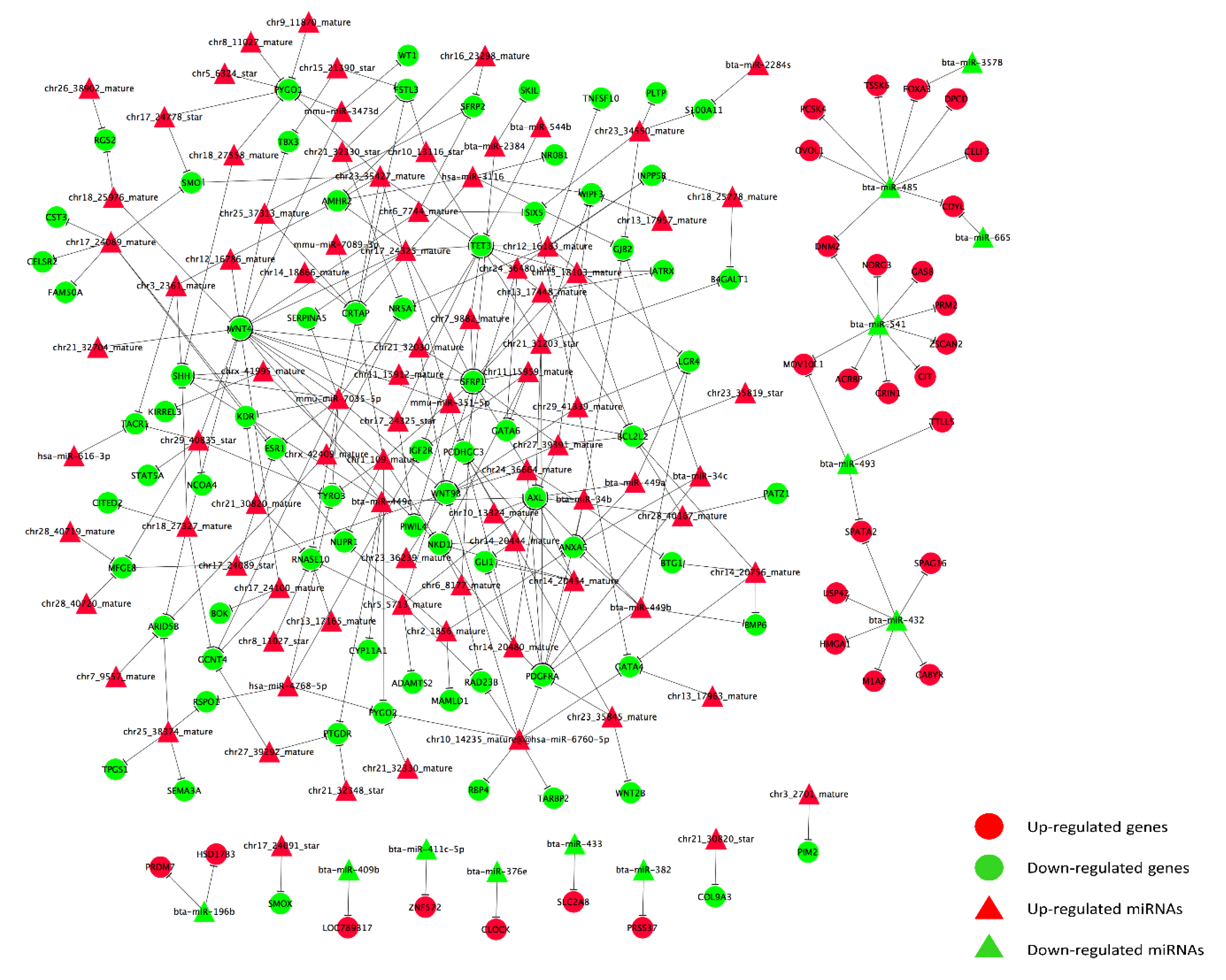
3.4. Prediction and Construction of Target Network between DERs and DEGs
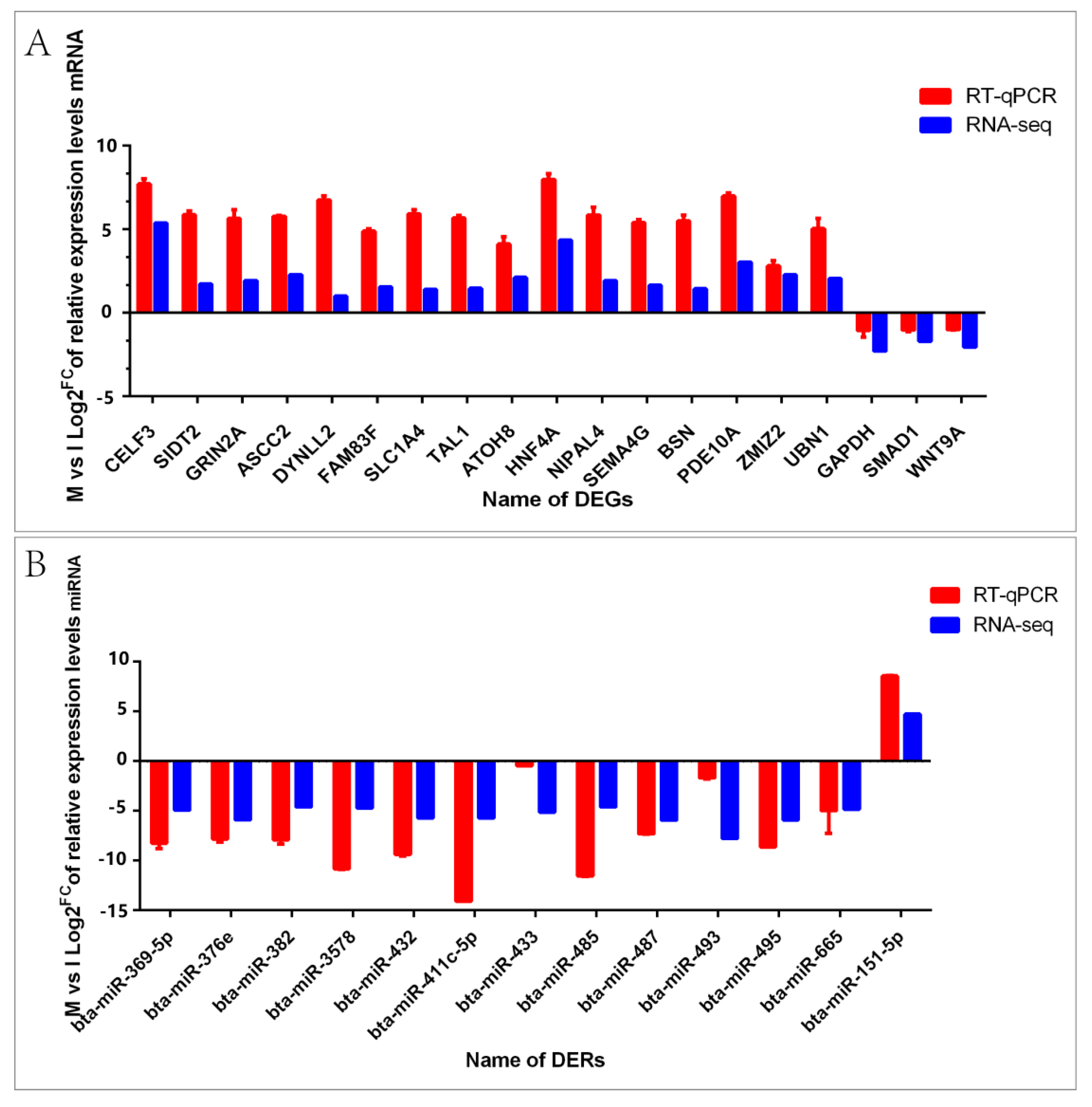
3.5. The Comparative Analysis and Verification of the Expression Levels between Real-Time PCR Result and Sequencing Data
4. Discussion
4.1. Mapping and Length Distributions of the miRNAs Sequences
4.2. Gene Expression Patterns and Roles in Testicular Tissues of Cattle
4.3. Target Predictions of DERs and Target Network
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Griswold, M.D. Spermatogenesis: The Commitment to Meiosis. Physiol. Rev. 2016, 96, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Hecht, N.B. Molecular mechanisms of male germ cell differentiation. BioEssays 1998, 20, 555–561. [Google Scholar] [CrossRef]
- De Martino, C.; Francavilla, S.; Fabbrini, A.; Accinni, L. Mammalian spermatogenesis and its disorders in man. Ultrastruct. Hum. Gametog. Early Embryog. 1989, 5, 1–32. [Google Scholar]
- Card, C.J.; Anderson, E.J.; Zamberlan, S.; Krieger, K.E.; Kaproth, M.; Sartini, B.L. Cryopreserved bovine spermatozoal transcript profile as revealed by high-throughput ribonucleic acid sequencing1. Biol. Reprod. 2013, 88, 49. [Google Scholar] [CrossRef] [PubMed]
- Djureinovic, D.; Fagerberg, L.; Hallström, B.; Danielsson, A.; Lindskog, C.; Uhlén, M.; Pontén, F. The human testis-specific proteome defined by transcriptomics and antibody-based profiling. Mol. Hum. Reprod. 2014, 20, 476–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soumillon, M.; Necsulea, A.; Weier, M.; Brawand, D.; Zhang, X.; Gu, H.; Barthès, P.; Kokkinaki, M.; Nef, S.; Gnirke, A.; et al. Cellular Source and Mechanisms of High Transcriptome Complexity in the Mammalian Testis. Cell Rep. 2013, 3, 2179–2190. [Google Scholar] [CrossRef]
- Ramsköld, D.; Wang, E.T.; Burge, C.B.; Sandberg, R. An Abundance of Ubiquitously Expressed Genes Revealed by Tissue Transcriptome Sequence Data. PLoS Comput. Biol. 2009, 5, e1000598. [Google Scholar] [CrossRef]
- Steuernagel, L.; Meckbach, C.; Heinrich, F.; Zeidler, S.; Schmitt, A.O.; Gültas, M. Computational identification of tissue-specific transcription factor cooperation in ten cattle tissues. PLoS ONE 2019, 14, e0216475. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, A.; Koli, S.; Reddy, K.V.R. Regulatory non-coding transcripts in spermatogenesis: Shedding light on ‘dark matter’. Andrology 2014, 2, 360–369. [Google Scholar] [CrossRef]
- Robles, V.; Herráez, P.; Labbé, C.; Cabrita, E.; Pšenička, M.; Valcarce, D.G.; Riesco, M.F. Molecular basis of spermatogenesis and sperm quality. Gen. Comp. Endocrinol. 2017, 245, 5–9. [Google Scholar] [CrossRef]
- Gao, Y.; Wu, M.; Fan, Y.; Li, S.; Lai, Z.; Huang, Y.; Lan, X.; Lei, C.; Chen, H.; Dang, R. Identification and characterization of circular RNAs in Qinchuan cattle testis. R. Soc. Open Sci. 2018, 5, 180413. [Google Scholar] [CrossRef] [Green Version]
- Qiu, G.-H.; Huang, C.; Zheng, X.; Yang, X. The protective function of noncoding DNA in genome defense of eukaryotic male germ cells. Epigenomics 2018, 10, 499–517. [Google Scholar] [CrossRef]
- Griswold, M.D. The central role of Sertoli cells in spermatogenesis. Semin. Cell Dev. Biol. 1998, 9, 411–416. [Google Scholar] [CrossRef] [Green Version]
- Dufau, M.L. Endocrine regulation and communicating functions of the leydig cell. Annu. Rev. Physiol. 1988, 50, 483–508. [Google Scholar] [CrossRef]
- Braundmeier, A.G.; Miller, D.J. The Search is on: Finding Accurate Molecular Markers of Male Fertility. J. Dairy Sci. 2001, 84, 1915–1925. [Google Scholar] [CrossRef]
- Overstreet, J.W.; Cooper, G.W. Sperm Transport in the Reproductive Tract of the Female Rabbit: II. The Sustained Phase of Transport1. Biol. Reprod. 1978, 19, 115–132. [Google Scholar] [CrossRef]
- Evenson, D.P. Loss of livestock breeding efficiency due to uncompensable sperm nuclear defects. Reprod. Fertil. Dev. 1999, 11, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Januskauskas, A.; Johannisson, A.; Söderquist, L.; Rodriguez-Martinez, H. Assessment of sperm characteristics post-thaw and response to calcium ionophore in relation to fertility in Swedish dairy AI bulls. Theriogenology 2000, 53, 859–875. [Google Scholar] [CrossRef]
- Coetzee, K.; Olmedo, J.; Lombard, C.J. Induced acrosome reactions as fertility predictor. J. Assist. Reprod. Genet. 1994, 11, 470–473. [Google Scholar] [CrossRef] [PubMed]
- Gadea, J.; Matás, C.; Lucas, X. Prediction of porcine semen fertility by homologous in vitro penetration (hIVP) assay. Anim. Reprod. Sci. 1998, 54, 95–108. [Google Scholar] [CrossRef]
- Harrison, R.A.P.; Vickers, S.E. Use of fluorescent probes to assess membrane integrity in mammalian spermatozoa. J. Reprod. Fertil. 1990, 88, 343–352. [Google Scholar] [CrossRef] [PubMed]
- Amann, R.P.; Seidel, G.E.; Brink, Z.A., Jr. Exposure of thawed frozen bull sperm to a synthetic peptide before artificial insemination increases fertility. J. Androl. 1999, 20, 42–46. [Google Scholar] [PubMed]
- Harayama, H.; Minami, K.; Kishida, K.; Noda, T. Protein biomarkers for male artificial insemination subfertility in bovine spermatozoa. Reprod. Med. Biol. 2017, 16, 89–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Son, M.; Tremoen, N.H.; Gaustad, A.H.; Myromslien, F.D.; Våge, D.I.; Stenseth, E.-B.; Zeremichael, T.T.; Grindflek, E. RNA sequencing reveals candidate genes and polymorphisms related to sperm DNA integrity in testis tissue from boars. BMC Vet. Res. 2017, 13, 362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hermann, B.P.; Mutoji, K.N.; Velte, E.K.; Ko, D.; Oatley, J.M.; Geyer, C.B.; McCarrey, J.R. Transcriptional and Translational Heterogeneity among Neonatal Mouse Spermatogonia1. Biol. Reprod. 2015, 92, 54. [Google Scholar] [CrossRef] [PubMed]
- Hu, F.; Xu, K.; Zhou, Y.; Wu, C.; Wang, S.; Xiao, J.; Wen, M.; Zhao, R.; Luo, K.; Tao, M.; et al. Different expression patterns of sperm motility-related genes in testis of diploid and tetraploid cyprinid fish. Biol. Reprod. 2017, 96, 907–920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Wang, F.; Li, F.; Ren, C.; Pang, J.; Wan, Y.; Wang, Z.; Feng, X.; Zhang, Y. Comprehensive analysis of long noncoding RNA and mRNA expression patterns in sheep testicular maturation. Biol. Reprod. 2018, 99, 650–661. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.L.; Ponsuksili, S.; Tholen, E.; Jennen, D.G.; Schellander, K.; Wimmers, K. Candidate gene markers for sperm quality and fertility of boar. Anim. Reprod. Sci. 2005, 92, 349–363. [Google Scholar] [CrossRef] [PubMed]
- Feugang, J.M.; Kaya, A.; Page, G.P.; Chen, L.; Mehta, T.; Hirani, K.; Nazareth, L.; Topper, E.; Gibbs, R.; Memili, E. Two-stage genome-wide association study identifies integrin beta 5 as having potential role in bull fertility. BMC Genom. 2009, 10, 176. [Google Scholar] [CrossRef] [Green Version]
- He, L.; Wang, S.; Deng, H.; Dong, H.; Chen, J. Solexa Profiling Identifies Differentially Expressed MiRNAs Between Sexually Immature and Mature Equine Testis. Braz. Arch. Biol. Technol. 2018, 61, 61. [Google Scholar] [CrossRef]
- He, Z.; Kokkinaki, M.; Pant, D.; Gallicano, G.I.; Dym, M. Small RNA molecules in the regulation of spermatogenesis. Reproduction 2009, 137, 901–911. [Google Scholar] [CrossRef] [Green Version]
- Benjamini, Y.; Drai, D.; Elmer, G.; Kafkafi, N.; Golani, I. Controlling the false discovery rate in behavior genetics research. Behav. Brain Res. 2001, 125, 279–284. [Google Scholar] [CrossRef] [Green Version]
- Leng, N.; Dawson, J.A.; Thomson, J.A.; Ruotti, V.; Rissman, A.I.; Smits, B.M.G.; Haag, J.D.; Gould, M.N.; Stewart, R.M.; Kendziorski, C. EBSeq: An empirical Bayes hierarchical model for inference in RNA-seq experiments. Bioinformatics 2013, 29, 2073. [Google Scholar] [CrossRef] [Green Version]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef] [Green Version]
- Aokikinoshita, K.F.; Kanehisa, M. Gene annotation and pathway mapping in kegg. Methods Mol. Biol. 2007, 396, 71. [Google Scholar]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Ogata, H.; Goto, S.; Fujibuchi, W.; Kanehisa, M. Computation with the KEGG pathway database. Biosystems 1998, 47, 119–128. [Google Scholar] [CrossRef]
- Zhang, J.D.; Wiemann, S. KEGGgraph: A graph approach to KEGG PATHWAY in R and bioconductor. Bioinformatics 2009, 25, 1470–1471. [Google Scholar] [CrossRef] [PubMed]
- Kozomara, A.; Birgaoanu, M.; Griffiths-Jones, S. miRBase: From microRNA sequences to function. Nucleic Acids Res. 2019, 47, D155–D162. [Google Scholar] [CrossRef] [PubMed]
- Rehmsmeier, M.; Steffen, P.; Höchsmann, M.; Giegerich, R. Fast and effective prediction of microRNA/target duplexes. RNA 2004, 10, 1507–1517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Betel, D.; Wilson, M.; Gabow, A.; Marks, D.S.; Sander, C. The microRNA.org resource: Targets and expression. Nucleic Acids Res. 2008, 36, D149–D153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kramer, M.F. Stem-Loop RT-qPCR for miRNAs. Curr. Protoc. Mol. Biol. 2011, 95, 15.10.1–15.10.15. [Google Scholar] [CrossRef] [PubMed]
- Fu, M.; Xu, K.-H.; Xu, W.-M. Research advances of Dicer in regulating reproductive function. Hereditas 2016, 38, 612–622. [Google Scholar] [PubMed]
- Korhonen, H. The Essential Role of DICER in Spermatogenesis and male Fertility. Ph.D. Thesis, University of Turku, Turku, Finland, 2016. [Google Scholar]
- Yadav, R.P.; Kotaja, N. Small RNAs in spermatogenesis. Mol. Cell. Endocrinol. 2014, 382, 498–508. [Google Scholar] [CrossRef] [PubMed]
- Kotaja, N. MicroRNAs and spermatogenesis. Fertil. Steril. 2014, 101, 1552–1562. [Google Scholar] [CrossRef]
- Lau, N.C.; Seto, A.G.; Jinkuk, K.; Satomi, K.M.; Toru, N.; Bartel, D.P.; Kingston, R.E. Characterization of the pirna complex from rat testes. Science 2006, 313, 363–367. [Google Scholar] [CrossRef] [Green Version]
- Han, B.W.; Zamore, P.D. piRNAs. Curr. Biol. 2014, 24, 730–733. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Ju, Z.; Li, Q.; Hou, Q.; Wang, C.; Li, J.; Li, R.; Wang, L.; Sun, T.; Hang, S.; et al. Solexa Sequencing of Novel and Differentially Expressed MicroRNAs in Testicular and Ovarian Tissues in Holstein Cattle. Int. J. Biol. Sci. 2011, 7, 1016–1026. [Google Scholar] [CrossRef]
- Sharma, D.D.; Neerja, W.; Neetu, K.; Kanchan, S.; Shankar, P.B.; Majumdar, S.S. Dickkopf homolog 3 (dkk3) plays a crucial role upstream of WNT/β-catenin signaling for sertoli cell mediated regulation of spermatogenesis. PLoS ONE 2013, 8, e63603. [Google Scholar]
- Tanwar, P.S.; Tomoko, K.T.; Lihua, Z.; Poonam, R.; Taketo, M.M.; Jose, T. Constitutive WNT/beta-catenin signaling in murine sertoli cells disrupts their differentiation and ability to support spermatogenesis. Biol. Reprod. 2010, 82, 422. [Google Scholar] [CrossRef] [Green Version]
- Rappaport, M.S.; Smith, E.P. Insulin-like growth factor I inhibits aromatization induced by follice-stimulating hormone in rat sertoli cell culture. Biol. Reprod. 1996, 54, 446–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borland, K.; Mita, M.; Oppenheimer, C.L.; Blinderman, L.A.; Massague, J.; Hall, P.F.; Czech, M.P. The actions of insulin-like growth factors I and II on cultured sertoli cells. Endocrinology 1984, 114, 240. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.A.; Lilianne, N.; Lauren, P.; Spicer, L.J.; Davis, J.S. Follicle-stimulating hormone amplifies insulin-like growth factor I-mediated activation of AKT/protein kinase b signaling in immature rat sertoli cells. Endocrinology 2002, 143, 2259–2267. [Google Scholar] [CrossRef] [PubMed]
- Mullaney, B.P.; Skinner, M.K. Transforming growth factor-beta (beta 1, beta 2, and beta 3) gene expression and action during pubertal development of the seminiferous tubule: Potential role at the onset of spermatogenesis. Mol. Endocrinol. 1993, 7, 67–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Catherine, I.; Loveland, K.L. Smads and cell fate: Distinct roles in specification, development, and tumorigenesis in the testis. IUBMB Life 2013, 65, 85–97. [Google Scholar]
- Capra, E.; Turri, F.; Lazzari, B.; Cremonesi, P.; Gliozzi, T.M.; Fojadelli, I.; Stella, A.; Pizzi, F. Small RNA sequencing of cryopreserved semen from single bull revealed altered miRNAs and piRNAs expression between High- and Low-motile sperm populations. BMC Genom. 2017, 18, 14. [Google Scholar] [CrossRef] [Green Version]
- Kasturi, S.S.; Tannir, J.; Brannigan, R.E. The metabolic syndrome and male infertility. J. Androl. 2008, 29, 251–259. [Google Scholar] [CrossRef]
- Pasquali, R.; Casimirri, F.; Vicennati, V. Weight control and its beneficial effect on fertility in women with obesity and polycystic ovary syndrome. Hum. Reprod. 1997, 12, 82–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelson, S.M.; Fleming, R. Obesity and reproduction. Proc. Nutr. Soc. 2008, 28, 1–6. [Google Scholar] [CrossRef]
- Butler, W.R.; Smith, R.D. Interrelationships Between Energy Balance and Postpartum Reproductive Function in Dairy Cattle. J. Dairy Sci. 1989, 72, 767–783. [Google Scholar] [CrossRef]
- Spicer, L.J.; Francisco, C.C. The Adipose Obese Gene Product, Leptin: Evidence of a Direct Inhibitory Role in Ovarian Function. Endocrinology 1997, 138, 3374–3379. [Google Scholar] [CrossRef] [PubMed]
- Arnett, D.W.; Holland, G.L.; Totusek, R. Some Effects of Obesity in Beef Females1. J. Anim. Sci. 1971, 33, 1129–1136. [Google Scholar] [CrossRef] [PubMed]
- Vick, M.M.; Sessions, D.R.; Murphy, B.A.; Kennedy, E.L.; Reedy, S.E.; Fitzgerald, B.P. Obesity is associated with altered metabolic and reproductive activity in the mare: Effects of metformin on insulin sensitivity and reproductive cyclicity. Reprod. Fertil. Dev. 2006, 18, 609–617. [Google Scholar] [CrossRef]
- Leroy, J.L.; Sturmey, R.G.; Van Hoeck, V.; De Bie, J.; McKeegan, P.J.; Bols, P. Dietary Fat Supplementation and the Consequences for Oocyte and Embryo Quality: Hype or Significant Benefit for Dairy Cow Reproduction? Reprod. Domest. Anim. 2014, 49, 353–361. [Google Scholar] [CrossRef]
- Gülüm, K.; Scott, N.M.; Craig, N.; Prins, G.S.; Carole, O. Genome-wide association study identifies candidate genes for male fertility traits in humans. Am. J. Hum. Genet. 2012, 90, 950–961. [Google Scholar]
- Yao, C.; Sun, M.; Yuan, Q.; Niu, M.; Chen, Z.; Hou, J.; Wang, H.; Wen, L.; Liu, Y.; Li, Z.; et al. MiRNA-133b promotes the proliferation of human Sertoli cells through targeting GLI3. Oncotarget 2016, 7, 2201–2219. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.; Qin, L.; Yang, Y.; Dong, X.; Zhao, Z.; Zhang, G.; Zhao, Z. PDPN gene promotes the proliferation of immature Bovine Sertoli cells in vitro. Anim. Reprod. Sci. 2017, 179, 35–43. [Google Scholar] [CrossRef]
- Cai, X.; Yu, S.; Mipam, T.D.; Yang, F.; Zhao, W.; Liu, W.; Cao, S.Z.; Shen, L.; Zhao, F.; Sun, L.; et al. Comparative analysis of testis transcriptomes associated with male infertility in cattleyak. Theriogenology 2017, 88, 28–42. [Google Scholar] [CrossRef]
- Katsuhiko, H.; Lopes, S.M.; De Sousa, C.; Masahiro, K.; Fuchou, T.; Petra, H.; Kaiqin, L.; Donal, O.C.; Das, P.P.; Alexander, T.; et al. Microrna biogenesis is required for mouse primordial germ cell development and spermatogenesis. PLoS ONE 2008, 3, e1738. [Google Scholar]
- Comazzetto, S.; Giacomo, M.D.; Rasmussen, K.D.; Much, C.; Azzi, C.; Perlas, E.; Morgan, M.; O’Carroll, D. Oligoasthenoteratozoospermia and Infertility in Mice Deficient for miR-34b/c and miR-449 Loci. PLoS Genet. 2014, 10, e1004597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, S.; Tang, C.; Zhang, Y.; Wu, J.; Bao, J.; Zheng, H.; Xu, C.; Yan, W. mir-34b/c and mir-449a/b/c are required for spermatogenesis, but not for the first cleavage division in mice. Biol. Open 2015, 4, 212–223. [Google Scholar] [CrossRef] [Green Version]
- Abu-Halima, M.; Backes, C.; Leidinger, P.; Keller, A.; Lubbad, A.M.; Hammadeh, M.; Meese, E. MicroRNA expression profiles in human testicular tissues of infertile men with different histopathologic patterns. Fertil. Steril. 2014, 101, 78–86. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, X.; Mata, A.; Bassas, L.; Larriba, S. Altered miRNA Signature of Developing Germ-cells in Infertile Patients Relates to the Severity of Spermatogenic Failure and Persists in Spermatozoa. Sci. Rep. 2015, 5, 17991. [Google Scholar] [CrossRef] [Green Version]
- Huszar, J.M.; Payne, C.J. Microrna 146 (mir146) modulates spermatogonial differentiation by retinoic acid in mice. Biol. Reprod. 2013, 88, 15. [Google Scholar] [CrossRef] [PubMed]
- Qin, L.; Zhang, G.; Zhu, C.; Wu, J.; Zhao, Z.; Zhao, Y. The predicted target genes of mir-122/449a validation and effects on sertoli cells proliferation. Pak. J. Zool. 2018, 50, 273–281. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| AccID | Mature | Immature | Log2FC | FDR | Regulate |
|---|---|---|---|---|---|
| TRIM42 | 1754.37 | 0.00 | 20 | 0.00 | up |
| SPACA4 | 879.07 | 0.00 | 20 | 0.00 | up |
| FAM187B | 776.40 | 0.00 | 20 | 0.00 | up |
| LOC781895 | 693.90 | 0.00 | 20 | 0.00 | up |
| PRPS1L1 | 634.28 | 0.00 | 20 | 0.00 | up |
| LOC100335845 | 557.42 | 0.00 | 20 | 0.00 | up |
| TMIGD3 | 423.84 | 0.00 | 20 | 0.00 | up |
| LOC784318 | 405.71 | 0.00 | 20 | 0.00 | up |
| CCDC182 | 396.56 | 0.00 | 20 | 0.00 | up |
| LOC101904646 | 362.72 | 0.00 | 20 | 0.00 | up |
| TMEM239 | 352.87 | 0.00 | 20 | 0.00 | up |
| LOC101907601 | 339.97 | 0.00 | 20 | 0.00 | up |
| LOC615451 | 280.92 | 0.00 | 20 | 0.00 | up |
| LOC516636 | 232.22 | 0.00 | 20 | 0.00 | up |
| LOC101906055 | 216.52 | 0.00 | 20 | 0.00 | up |
| LOC104976276 | 0.00 | 68.67 | −20 | 0.00 | down |
| LOR | 0.00 | 37.29 | −20 | 0.00 | down |
| LOC781553 | 0.00 | 34.92 | −20 | 0.00 | down |
| PDIA2 | 0.00 | 105.11 | −20 | 0.00 | down |
| CHRNA1 | 0.00 | 64.93 | −20 | 0.00 | down |
| LOC100847574 | 0.00 | 51.13 | −20 | 0.00 | down |
| MMP12 | 0.00 | 29.71 | −20 | 0.00 | down |
| LOC107133052 | 0.00 | 28.63 | −20 | 0.00 | down |
| MMP7 | 0.00 | 22.75 | −20 | 0.00 | down |
| LOC101904303 | 0.00 | 22.03 | −20 | 0.00 | down |
| LOC104970370 | 0.00 | 21.73 | −20 | 0.00 | down |
| GABRG3 | 0.00 | 16.98 | −20 | 0.00 | down |
| LOC104970773 | 0.00 | 16.71 | −20 | 0.00 | down |
| CSMD3 | 0.00 | 16.21 | −20 | 0.00 | down |
| LOC100139712 | 0.00 | 15.90 | −20 | 0.00 | down |
| AccID | Mature | Immature | Log2FC | FDR | Style |
|---|---|---|---|---|---|
| bta-miR-449a | 10,427.15 | 14.20 | 9.52 | 0.00 | up |
| bta-miR-449b | 326.43 | 1.49 | 7.77 | 0.00 | up |
| bta-miR-34b | 4001.49 | 35.13 | 6.83 | 0.00 | up |
| bta-miR-34c | 75,119.87 | 561.35 | 7.06 | 0.00 | up |
| bta-miR-449c | 151.18 | 1.49 | 6.66 | 0.00 | up |
| bta-miR-146a | 379.95 | 8.22 | 5.53 | 0.00 | up |
| bta-miR-375 | 671.60 | 20.18 | 5.06 | 0.00 | up |
| bta-miR-544b | 28.09 | 0.00 | 20.00 | 0.01 | up |
| bta-miR-2384 | 64.22 | 2.24 | 4.84 | 0.02 | up |
| bta-miR-151-5p | 13,120.22 | 488.10 | 4.75 | 0.02 | up |
| bta-miR-2284s | 17.39 | 0.00 | 20.00 | 0.04 | up |
| bta-miR-493 | 1.34 | 290.77 | −7.76 | 0.00 | down |
| bta-miR-409b | 2.68 | 224.24 | −6.39 | 0.01 | down |
| bta-miR-409a | 0.00 | 94.93 | −20.00 | 0.01 | down |
| bta-miR-495 | 16.05 | 964.99 | −5.91 | 0.01 | down |
| bta-miR-487b | 5.35 | 327.39 | −5.94 | 0.01 | down |
| bta-miR-376e | 4.01 | 238.44 | −5.89 | 0.01 | down |
| bta-miR-411c-5p | 6.69 | 346.08 | −5.69 | 0.01 | down |
| bta-miR-196b | 0.00 | 69.51 | −20.00 | 0.02 | down |
| bta-miR-376b | 1.34 | 92.69 | −6.11 | 0.02 | down |
| bta-miR-433 | 13.38 | 469.41 | −5.13 | 0.03 | down |
| bta-miR-3956 | 0.00 | 50.08 | −20.00 | 0.03 | down |
| bta-miR-432 | 255.53 | 13391.71 | −5.71 | 0.03 | down |
| bta-miR-541 | 0.00 | 44.85 | −20.00 | 0.03 | down |
| bta-miR-369-3p | 9.36 | 284.79 | −4.93 | 0.03 | down |
| bta-miR-665 | 10.70 | 307.21 | −4.84 | 0.04 | down |
| bta-miR-496 | 0.00 | 38.87 | −20.00 | 0.04 | down |
| bta-miR-3578 | 16.05 | 417.84 | −4.70 | 0.04 | down |
| bta-miR-543 | 20.07 | 504.54 | −4.65 | 0.05 | down |
| bta-miR-382 | 17.39 | 419.33 | −4.59 | 0.05 | down |
| bta-miR-485 | 17.39 | 418.58 | −4.59 | 0.05 | down |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fang, X.; Qin, L.; Yu, H.; Jiang, P.; Xia, L.; Gao, Z.; Yang, R.; Zhao, Y.; Yu, X.; Zhao, Z. Comprehensive Analysis of miRNAs and Target mRNAs between Immature and Mature Testis Tissue in Chinese Red Steppes Cattle. Animals 2021, 11, 3024. https://doi.org/10.3390/ani11113024
Fang X, Qin L, Yu H, Jiang P, Xia L, Gao Z, Yang R, Zhao Y, Yu X, Zhao Z. Comprehensive Analysis of miRNAs and Target mRNAs between Immature and Mature Testis Tissue in Chinese Red Steppes Cattle. Animals. 2021; 11(11):3024. https://doi.org/10.3390/ani11113024
Chicago/Turabian StyleFang, Xibi, Lihong Qin, Haibin Yu, Ping Jiang, Lixin Xia, Zhen Gao, Runjun Yang, Yumin Zhao, Xianzhong Yu, and Zhihui Zhao. 2021. "Comprehensive Analysis of miRNAs and Target mRNAs between Immature and Mature Testis Tissue in Chinese Red Steppes Cattle" Animals 11, no. 11: 3024. https://doi.org/10.3390/ani11113024