
Prediction of Hanwoo Cattle Phenotypes from Genotypes Using Machine Learning Methods

 ,
,  , , and
, , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Data
2.2. Statistical Methods
2.2.1. Genomic Best Linear Unbiased Prediction (GBLUP)
2.2.2. Random Forest (RF)
2.2.3. Extreme Gradient Boosting Method (XGB)
2.2.4. Support Vector Machine (SVM)
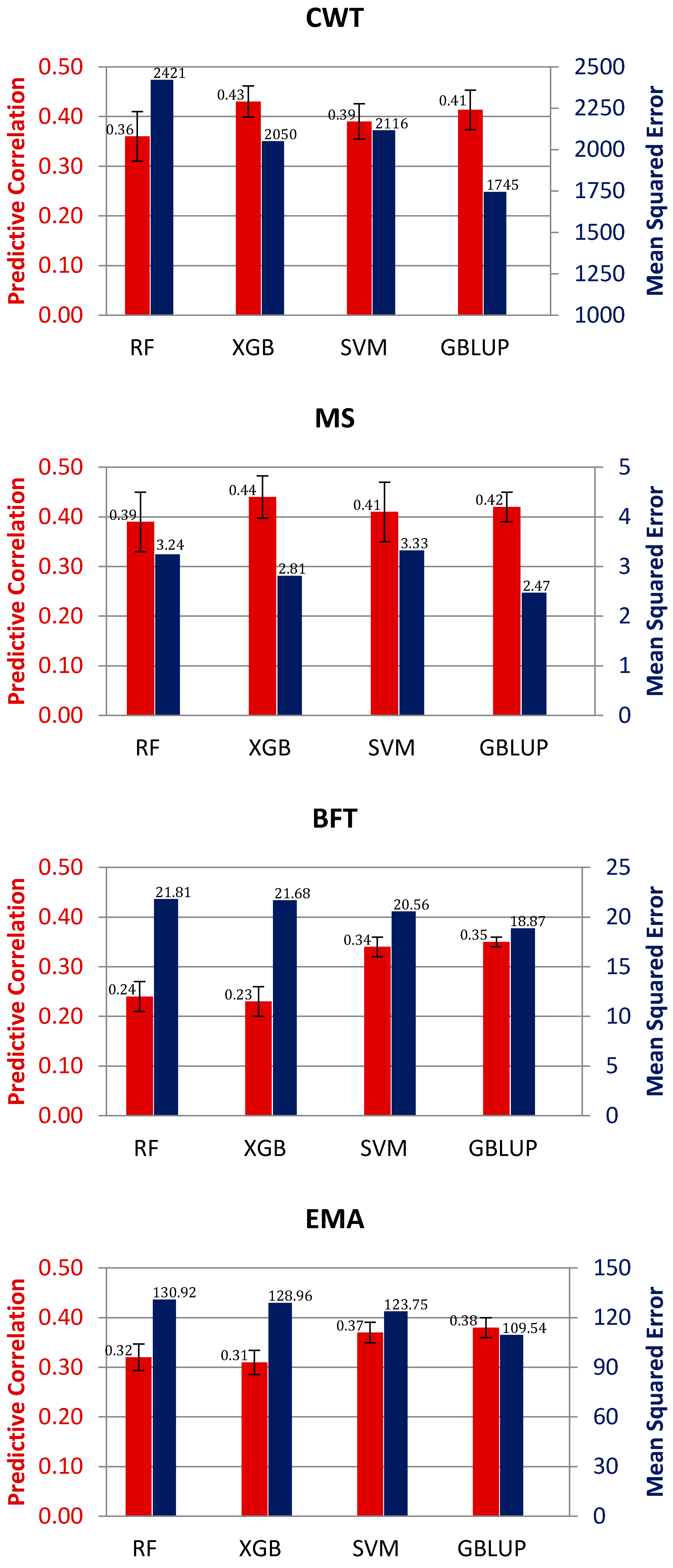
3. Results and Discussion
3.1. Genetic Parameters
3.2. Genomic Prediction
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Lee, S.-H.; Park, B.-H.; Sharma, A.; Dang, C.-G.; Lee, S.-S.; Choi, T.-J.; Choy, Y.-H.; Kim, H.-C.; Jeon, K.-J.; Kim, S.-D.; et al. Hanwoo cattle: Origin, domestication, breeding strategies and genomic selection. J. Anim. Sci. Technol. 2015, 56, 2. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Alam, M.; Park, M.N. Breeding initiatives for Hanwoo cattle to thrive as a beef industry—A review study. J. Anim. Breed. Genom. 2017, 1, 102–124. [Google Scholar] [CrossRef]
- VanRaden, P.M. Efficient methods to compute genomic predictions. J. Dairy Sci. 2008, 91, 4414–4423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aguilar, I.; Misztal, I.; Johnson, D.L.; Legarra, A.; Tsuruta, S.; Lawlor, T.J. Hot topic: A unified approach to utilize phenotypic, full pedigree, and genomic information for genetic evaluation of Holstein final score1. J. Dairy Sci. 2010, 93, 743–752. [Google Scholar] [CrossRef]
- Meuwissen, T.H.; Hayes, B.J.; Goddard, M. Prediction of total genetic value using genome-wide dense marker maps. Genetics 2001, 157, 1819–1829. [Google Scholar] [CrossRef] [PubMed]
- de los Campos, G.; Naya, H.; Gianola, D.; Crossa, J.; Legarra, A.; Manfredi, E.; Weigel, K.; Cotes, J.M. Predicting Quantitative Traits With Regression Models for Dense Molecular Markers and Pedigree. Genetics 2009, 182, 375–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moser, G.; Lee, S.H.; Hayes, B.J.; Goddard, M.E.; Wray, N.R.; Visscher, P.M. Simultaneous Discovery, Estimation and Prediction Analysis of Complex Traits Using a Bayesian Mixture Model. PLoS Genet. 2015, 11, e1004969. [Google Scholar] [CrossRef]
- Endelman, J.B. Ridge Regression and Other Kernels for Genomic Selection with R Package rrBLUP. Plant Genome 2011, 4. [Google Scholar] [CrossRef] [Green Version]
- van Eeuwijk, F.A.; Bink, M.C.A.M.; Chenu, K.; Chapman, S.C. Detection and use of QTL for complex traits in multiple environments. Curr. Opin. Plant Biol. 2010, 13, 193–205. [Google Scholar] [CrossRef]
- de los Campos, G.; Hickey, J.M.; Pong-Wong, R.; Daetwyler, H.D.; Calus, M.P.L. Whole-Genome Regression and Prediction Methods Applied to Plant and Animal Breeding. Genetics 2013, 193, 327–345. [Google Scholar] [CrossRef] [Green Version]
- González-Recio, O.; Forni, S. Genome-wide prediction of discrete traits using bayesian regressions and machine learning. Genet. Sel. Evol. 2011, 43, 7. [Google Scholar] [CrossRef] [Green Version]
- González-Recio, O.; Rosa, G.J.M.; Gianola, D. Machine learning methods and predictive ability metrics for genome-wide prediction of complex traits. Livest. Sci. 2014, 166, 217–231. [Google Scholar] [CrossRef]
- Breiman, L. Random Forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef] [Green Version]
- Friedman, J.H. Greedy function approximation: A gradient boosting machine. Ann. Stat. 2001, 1189–1232. [Google Scholar]
- Aruna, S.; Dr, S. A Novel SVM based CSSFFS Feature Selection Algorithm for Detecting Breast Cancer. Int. J. Comput. Appl. 2011, 31, 14–20. [Google Scholar]
- Chen, T.; Guestrin, C. XgBoost: A Scalable Tree Boosting System. In Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining (KDD ’16); Association for Computing Machinery: New York, NY, USA, 2016; pp. 785–794. [Google Scholar] [CrossRef] [Green Version]
- Zhao, W.; Lai, X.; Liu, D.; Zhang, Z.; Ma, P.; Wang, Q.; Zhang, Z.; Pan, Y. Applications of Support Vector Machine in Genomic Prediction in Pig and Maize Populations. Front. Genet. 2020, 11, 598318. [Google Scholar] [CrossRef]
- Li, B.; Zhang, N.; Wang, Y.-G.; George, A.W.; Reverter, A.; Li, Y. Genomic Prediction of Breeding Values Using a Subset of SNPs Identified by Three Machine Learning Methods. Front. Genet. 2018, 9, 237. [Google Scholar] [CrossRef] [PubMed]
- Misztal, I.; Tsuruta, S.; Lourenco, D.; Aguilar, I.; Legarra, A.; Vitezica, Z. Manual for BLUPF90 family of programs. Athens Univ. GA 2014, 199. [Google Scholar]
- Lopez, B.I.; Lee, S.-H.; Park, J.-E.; Shin, D.-H.; Oh, J.-D.; de las Heras-Saldana, S.; van der Werf, J.; Chai, H.-H.; Park, W.; Lim, D. Weighted Genomic Best Linear Unbiased Prediction for Carcass Traits in Hanwoo Cattle. Genes 2019, 10, 1019. [Google Scholar] [CrossRef] [Green Version]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.H.; Van der Werf, J.H.J. MTG2: An efficient algorithm for multivariate linear mixed model analysis based on genomic information. Bioinformatics 2016, 32, 1420–1422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V. Scikit-learn: Machine learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Smola, A.J.; Schölkopf, B. A tutorial on support vector regression. Stat. Comput. 2004, 14, 199–222. [Google Scholar] [CrossRef] [Green Version]
- Choi, T.J.; Alam, M.; Cho, C.I.; Lee, J.G.; Park, B.; Kim, S.; Koo, Y.; Roh, S.H. Genetic parameters for yearling weight, carcass traits, and primal-cut yields of Hanwoo cattle1. J. Anim. Sci. 2015, 93, 1511–1521. [Google Scholar] [CrossRef]
- Do, C.; Park, B.; Kim, S.; Choi, T.; Yang, B.; Park, S.; Song, H. Genetic Parameter Estimates of Carcass Traits under National Scale Breeding Scheme for Beef Cattle. Asian-Australas. J. Anim. Sci. 2016, 29, 1083–1094. [Google Scholar] [CrossRef] [PubMed]
- Lopez, B.I.M.; An, N.; Srikanth, K.; Lee, S.; Oh, J.-D.; Shin, D.-H.; Park, W.; Chai, H.-H.; Park, J.-E.; Lim, D. Genomic Prediction Based on SNP Functional Annotation Using Imputed Whole-Genome Sequence Data in Korean Hanwoo Cattle. Front. Genet. 2021, 11, 603822. [Google Scholar] [CrossRef] [PubMed]
- Abdollahi-Arpanahi, R.; Gianola, D.; Peñagaricano, F. Deep learning versus parametric and ensemble methods for genomic prediction of complex phenotypes. Genet. Sel. Evol. 2020, 52, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogutu, J.O.; Piepho, H.-P.; Schulz-Streeck, T. A comparison of random forests, boosting and support vector machines for genomic selection. BMC Proc. 2011, 5, S11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moser, G.; Tier, B.; Crump, R.E.; Khatkar, M.S.; Raadsma, H.W. A comparison of five methods to predict genomic breeding values of dairy bulls from genome-wide SNP markers. Genet. Sel. Evol. 2009, 41, 56. [Google Scholar] [CrossRef] [Green Version]
- Long, N.; Gianola, D.; Rosa, G.J.M.; Weigel, K.A. Application of support vector regression to genome-assisted prediction of quantitative traits. Theor. Appl. Genet. 2011, 123, 1065. [Google Scholar] [CrossRef]
- Alves, A.A.C.; da Costa, R.M.; Bresolin, T.; Fernandes Júnior, G.A.; Espigolan, R.; Ribeiro, A.M.F.; Carvalheiro, R.; de Albuquerque, L.G. Genome-wide prediction for complex traits under the presence of dominance effects in simulated populations using GBLUP and machine learning methods. J. Anim. Sci. 2020, 98, skaa179. [Google Scholar] [CrossRef] [PubMed]
- Tusell, L.; Bergsma, R.; Gilbert, H.; Gianola, D.; Piles, M. Machine Learning Prediction of Crossbred Pig Feed Efficiency and Growth Rate From Single Nucleotide Polymorphisms. Front. Genet. 2020, 11, 567818. [Google Scholar] [CrossRef] [PubMed]
- Wolpert, D.H. The lack of a priori distinctions between learning algorithms. Neural Comput. 1996, 8, 1341–1390. [Google Scholar] [CrossRef]


{kind=link}
| Trait | Mean | SD | Min | Max |
|---|---|---|---|---|
| CWT (in kg) | 439.33 | 49.47 | 159 | 645 |
| MS (1–9) | 5.99 | 1.84 | 1 | 9 |
| BFT (in mm) | 14.24 | 4.78 | 1 | 45 |
| EMA (in cm2) | 96.15 | 11.96 | 35 | 155 |
| Trait | N_Estimator | Criterion | Max_Features | Min_Samples_Leaf | Min_Samples_Split | Max_Depth | Bootstrap |
|---|---|---|---|---|---|---|---|
| CWT | 400 | MSE | auto | 4 | 10 | 70 | TRUE |
| MS | 600 | MSE | auto | 4 | 2 | 40 | TRUE |
| BFT | 2000 | MSE | auto | 2 | 2 | 90 | TRUE |
| EMA | 1400 | MSE | auto | 4 | 2 | 100 | TRUE |
| Trait | σ2a | σ2e | σ2p | h2 |
|---|---|---|---|---|
| CWT | 773.00 | 1266.48 | 2039.48 | 0.38 |
| MS | 1.29 | 1.67 | 2.96 | 0.44 |
| BFT | 7.77 | 14.04 | 21.81 | 0.36 |
| EMA | 43.80 | 81.89 | 125.69 | 0.35 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Srivastava, S.; Lopez, B.I.; Kumar, H.; Jang, M.; Chai, H.-H.; Park, W.; Park, J.-E.; Lim, D. Prediction of Hanwoo Cattle Phenotypes from Genotypes Using Machine Learning Methods. Animals 2021, 11, 2066. https://doi.org/10.3390/ani11072066
Srivastava S, Lopez BI, Kumar H, Jang M, Chai H-H, Park W, Park J-E, Lim D. Prediction of Hanwoo Cattle Phenotypes from Genotypes Using Machine Learning Methods. Animals. 2021; 11(7):2066. https://doi.org/10.3390/ani11072066
Chicago/Turabian StyleSrivastava, Swati, Bryan Irvine Lopez, Himansu Kumar, Myoungjin Jang, Han-Ha Chai, Woncheoul Park, Jong-Eun Park, and Dajeong Lim. 2021. "Prediction of Hanwoo Cattle Phenotypes from Genotypes Using Machine Learning Methods" Animals 11, no. 7: 2066. https://doi.org/10.3390/ani11072066