Hybrid Versus Autochthonous Turkey Populations: Homozygous Genomic Regions Occurrences Due to Artificial and Natural Selection


Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sampling and Population Genetic Diversity
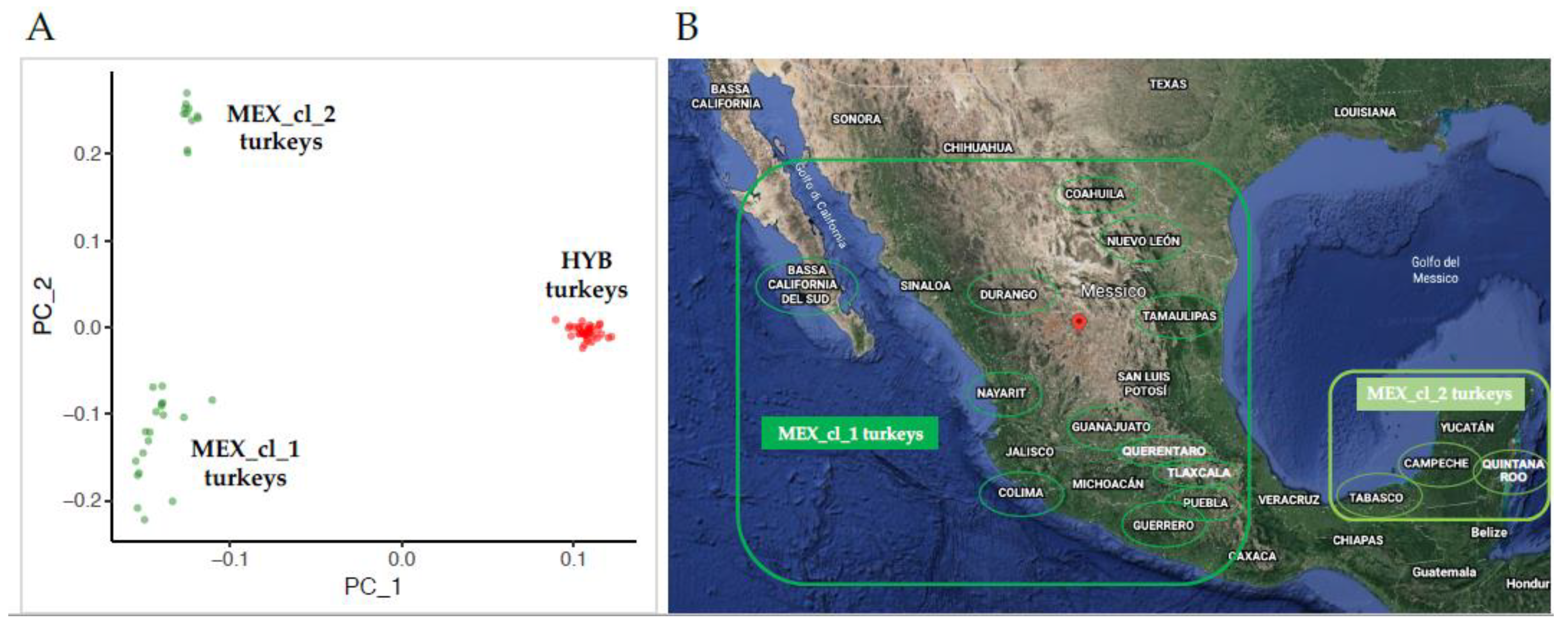
- Principal components analysis (PCA) based on allele genotypes using the SVS software (SVS) version 8.8.3 (Golden Helix Inc., Bozeman, MT, USA). The graphical visualization of PCA was obtained by the ggplot2 R package (https://CRAN.R-project.org/package=ggplot2) [16].
- Estimation of pairwise Fixation Index (i.e., Wright’s F-statistic FST) using the dedicated module of SVS.
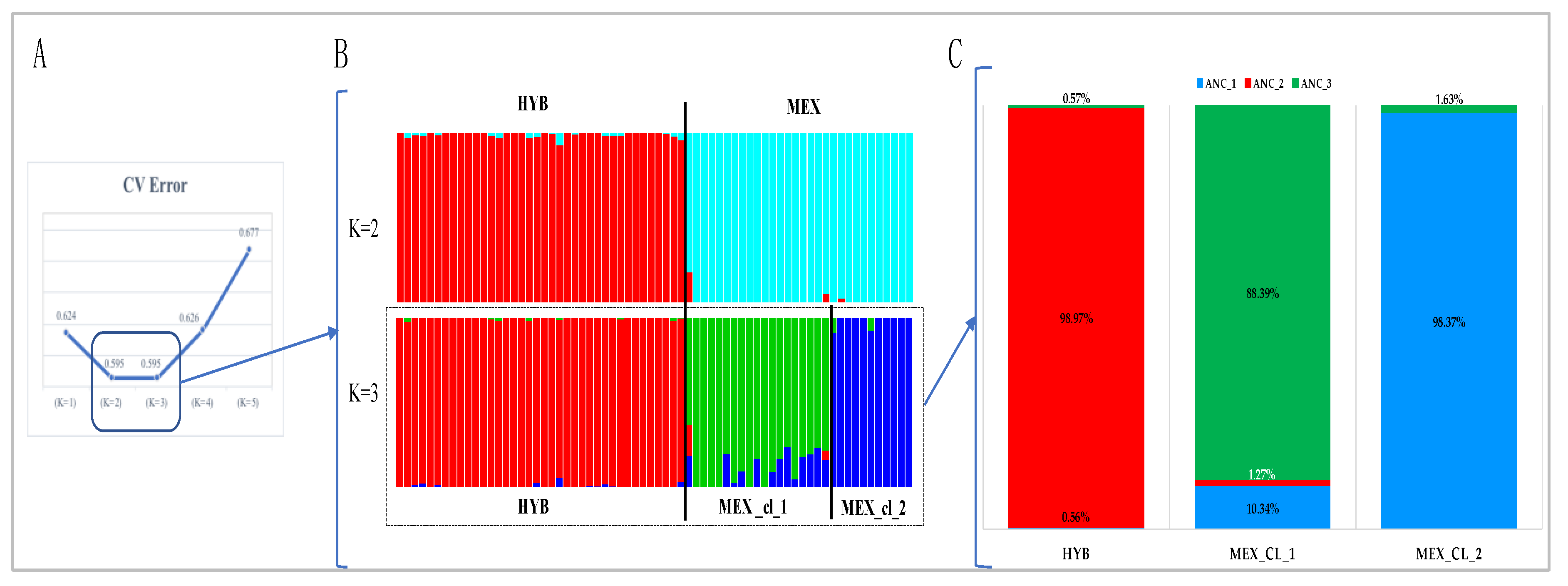
- Determination of the most probable number of ancestral populations with the ADMIXTURE v.1.3.0 software [17]. ADMIXTURE was run from K = 2 to K = 5, and the optimal number of clusters (K-value) was determined as the one having the lowest cross-validation error (CV). Data input files were generated for ADMIXTURE using PLINK software version 1.07 [18]. The R script suggested by ADMIXTURE procedure, was used to perform a graphical representation of ADMIXTURE results.
2.2. Identification of Runs of Homozygosity (ROH)
2.3. Inbreeding Coefficients
3. Results
3.1. Population Genetic Diversity
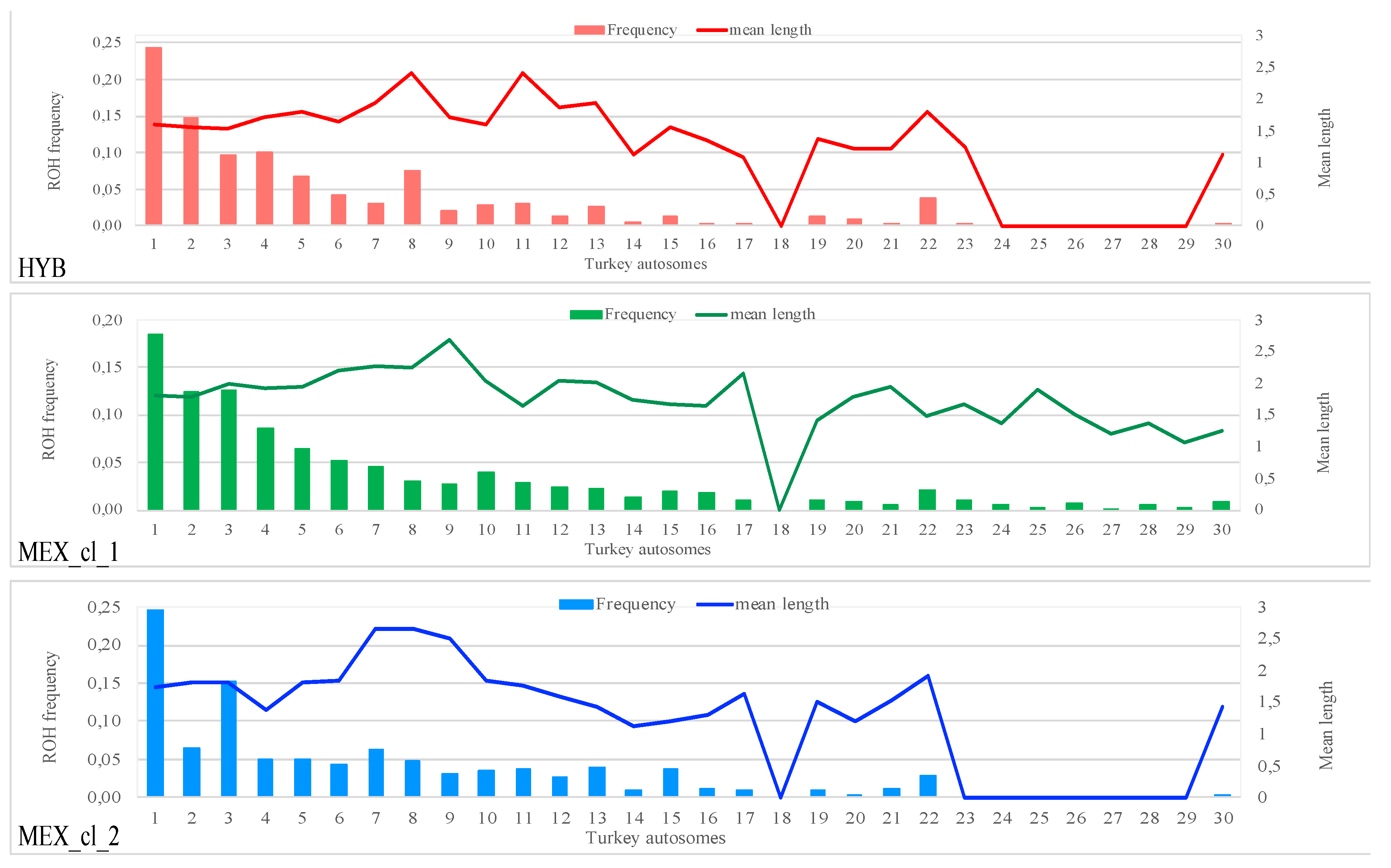
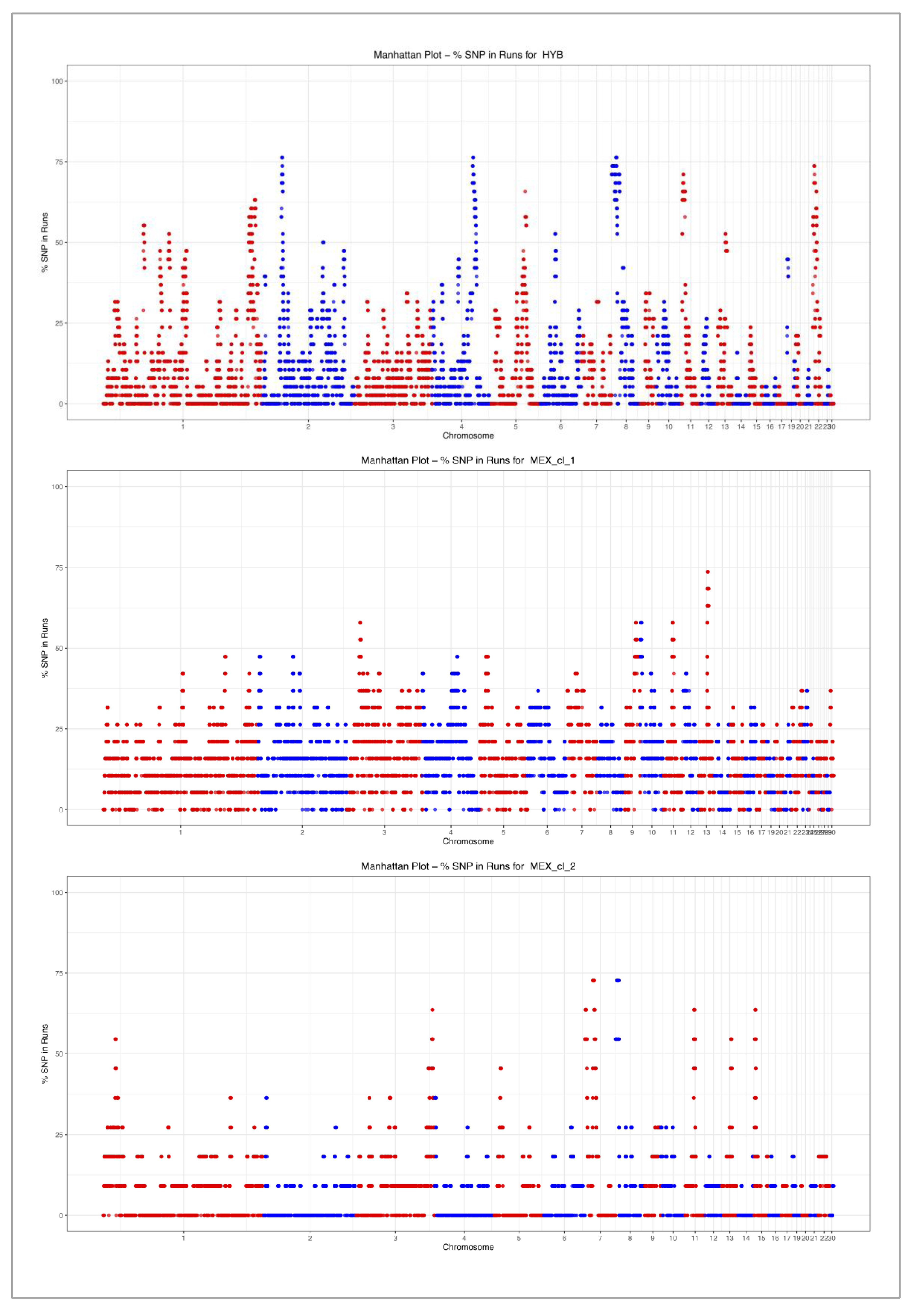
3.2. Runs of Homozygosity (ROH)
3.3. Inbreeding Coefficients (F-FROH)
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Thornton, E.K.; Emery, K.F. The uncertain origins of Mesoamerican turkey domestication. J. Archaeol. Method Theory 2017, 24, 328–351. [Google Scholar] [CrossRef]
- Schorger, A.W. The Wild Turkey. Its History and Domestication; University of Ottawa Press: Ottawa, ON, Canada, 1996. [Google Scholar]
- Crawford, R.D. Introduction to Europe and diffusion of domesticated turkeys from the America. Arch. Zootec. 1992, 41, 2. [Google Scholar]
- FAO. The Second Report on the State of the World’s Animal Genetic Resources for Food and Agriculture; FAO: Rome, Italy, 2015. [Google Scholar]
- Rios Utrera, A.; Roman Ponce, S.I.; Velez Izquierdo, A.; Cabrera Torre, E.; Cantu Covarrubias, A.; De la Cruz Colin, L.; Durán Aguilar, M.; Maldonado Jaquez, J.A.; Martínez Silva, F.E.; Martínez Velázquez, G.; et al. Analysis of morphological variables in Mexican backyard turkeys (Meleagris gallopavo gallopavo). Rev. Mex. Cienc. Pecu. 2016, 7, 377–389. [Google Scholar] [CrossRef] [Green Version]
- Dalloul, R.A.; Long, J.A.; Zimin, A.V.; Aslam, L.; Beal, K.; Ann Blomberg, L.; Bouffard, P.; Burt, D.W.; Crasta, O.; Crooijmans, R.P.M.A.; et al. Multi-Platform Next-Generation Sequencing of the Domestic Turkey (Meleagris gallopavo): Genome Assembly and Analysis. PLoS Biol. 2010, 8, e1000475. [Google Scholar] [CrossRef]
- Strillacci, M.G.; Gorla, E.; Ríos-Utrera, A.; Vega-Murillo, V.E.; Montaño-Bermudez, M.; Garcia-Ruiz, A.; Cerolini, S.; Román-Ponce, S.I.; Bagnato, A. Copy Number Variation Mapping and Genomic Variation of Autochthonous and Commercial Turkey Populations. Front. Genet. 2019, 10, 982. [Google Scholar] [CrossRef]
- Marras, G.; Wood, B.J.; Makanjuola, B.; Malchiodi, F.; Peeters, K.; van As, P.; Baes, C.F.; Biscarini, F. Characterization of runs of homozygosity and heterozygosity-rich regions in a commercial turkey (Meleagris gallopavo) population. In Proceedings of the 11th World Congress on Genetics Applied to Livestock Production, Auckland, New Zealand, 10–16 February 2018; p. 763. [Google Scholar]
- Aslam, M.L.; Bastiaansen, J.W.M.; Elferink, M.G.; Megens, H.-J.; Crooijmans, R.P.M.A.; Blomberg, L.A.; Fleischer, R.C.; Van Tassell, C.P.; Sonstegard, T.S.; Schroeder, S.G.; et al. Whole genome SNP discovery and analysis of genetic diversity in Turkey (Meleagris gallopavo). BMC Genom. 2012, 13, 391. [Google Scholar] [CrossRef] [Green Version]
- Guan, X.; Silva, P.; Gyenai, K.; Xu, J.; Geng, T.; Smith, E. Mitochondrial DNA-based analyses of relatedness among turkeys, Meleagris gallopavo. Biochem. Genet. 2015, 53, 29–41. [Google Scholar] [CrossRef]
- Canales Vergara, A.M.; Landi, V.; Delgado Bermejo, J.V.; Martínez, A.; Cervantes Acosta, P.; Pons Barro, Á.; Bigi, D.; Sponenberg, P.; Helal, M.; Hossein Banabazi, M.; et al. Tracing Worldwide Turkey Genetic Diversity Using D-loop Sequence Mitochondrial DNA Analysis. Animals 2019, 9, 897. [Google Scholar] [CrossRef] [Green Version]
- Ferenčaković, M.; Sölkner, J.; Curik, I. Estimating autozygosity from high-throughput information: Effects of SNP density and genotyping errors. Genet. Sel. Evol. 2013, 45, 42. [Google Scholar] [CrossRef] [Green Version]
- Ceballos, F.C.; Joshi, P.K.; Clark, D.W.; Ramsay, M.; Wilson, J.F. Runs of homozygosity: Windows into population history and trait architecture. Nat. Rev. Genet. 2018, 19, 220. [Google Scholar] [CrossRef]
- Johnson, E.C.; Evans, L.M.; Keller, M.C. Relationships between estimated autozygosity and complex traits in the UK Biobank. PLoS Genet. 2018, 14, e1007556. [Google Scholar] [CrossRef]
- Sams, A.J.; Boyko, A.R. Fine-Scale Resolution of Runs of Homozygosity Reveal Patterns of Inbreeding and Substantial Overlap with Recessive Disease Genotypes in Domestic Dogs. G3 Genes Genomes Genet. 2019, 9, 117–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016; ISBN 978-3-319-24277-4. [Google Scholar]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [Green Version]
- Biscarini, F.; Cozzi, P.; Gaspa, G.; Marras, G. detectRUNS: An R Package to Detect Runs of Homozygosity and Heterozygosity in Diploid Genomes Genomes. R Package Version 0.9.5. Available online: https://CRAN.R-project.org/package=detectRUNS (accessed on 1 July 2020).
- Purfield, D.C.; Berry, D.P.; McParland, S.; Bradley, D.G. Runs of homozygosity and population history in cattle. BMC Genet. 2012, 13, 70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- NCBI Online Database. Available online: https://ftp.ncbi.nlm.nih.gov/genomes/all/annotation_releases/9103/103/GCF_000146605.3_Turkey_5.1 (accessed on 1 July 2020).
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DAVID Online Database. Available online: http://david.abcc.ncifcrf.gov/summary.jsp (accessed on 1 July 2020).
- Chicken Quantitative Trait Loci (QTL) Database (Chicken QTLdb). Available online: https://www.animalgenome.org/cgi-bin/QTLdb/GG/genesrch (accessed on 1 July 2020).
- Google Earth. Available online: https://earth.google.com/web/search/México/@23.55407672,-102.62049061,571.6762355a,7328746.89000249d,35y,0h,0t,0r/data=CigiJgokCfVcOQAj8zhAEfVcOQAj8zjAGdmNrjOG80pAIdmNrjOG80rA (accessed on 27 July 2020).
- Reed, K.M.; Bauer, M.M.; Monson, M.S.; Benoit, B.; Chaves, L.D.; O’Hare, T.H.; Delany, M.E. Defining the Turkey MHC: Identification of expressed class I- and class IIB-like genes independent of the MHC-B. Immunogenetics 2011, 63, 753. [Google Scholar] [CrossRef]
- Ai, H.; Xiao, S.; Zhang, Z.; Yang, B.; Li, L.; Guo, Y.; Lin, G.; Ren, J.; Huang, L. Three novel quantitative trait loci for skin thickness in swine identified by linkage and genome-wide association studies. Anim. Genet. 2014, 45, 524–533. [Google Scholar] [CrossRef] [PubMed]
- Karisa, B.K.; Thomson, J.; Wang, Z.; Stothard, P.; Moore, S.S.; Plastow, G.S. Candidate genes and single nucleotide polymorphisms associated with variation in residual feed intake in beef cattle. J. Anim. Sci. 2013, 91, 3502–3513. [Google Scholar] [CrossRef] [Green Version]
- Cichocki, F.; Wu, C.Y.; Zhang, B.; Felices, M.; Tesi, B.; Tuininga, K.; Dougherty, P.; Taras, E.; Hinderlie, P.; Blazar, B.R.; et al. ARID5B regulates metabolic programming in human adaptive NK cells. J. Exp. Med. 2018, 215, 2379–2395. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Qiao, Y.; Di, Q.; Le, X.; Zhang, L.; Zhang, X.; Zhang, C.; Cheng, J.; Zong, S.; Koide, S.S.; et al. Interaction of SH3P13 and DYDC1 protein: A germ cell component that regulates acrosome biogenesis during spermiogenesis. Eur. J. Cell Biol. 2009, 88, 509–520. [Google Scholar] [CrossRef] [PubMed]
- Recla, J.M.; Robledo, R.F.; Gatti, D.M.; Bult, C.J.; Churchill, G.A.; Chesler, E.J. Precise genetic mapping and integrative bioinformatics in Diversity Outbred mice reveals Hydin as a novel pain gene. Mamm. Genome 2014, 25, 211–222. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Gao, Y.; Kim, Y.S.; Iqbal, A.; Kim, J.J. A whole genome association study to detect additive and dominant single nucleotide polymorphisms for growth and carcass traits in Korean native cattle, Hanwoo. Asian-Australas. J. Anim. Sci. 2017, 30, 8–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leal-Gutiérrez, J.D.; Elzo, M.A.; Johnson, D.D.; Hamblen, H.; Mateescu, R.G. Genome wide association and gene enrichment analysis reveal membrane anchoring and structural proteins associated with meat quality in beef. BMC Genom. 2019, 20, 151. [Google Scholar] [CrossRef] [PubMed]
- Castro, L.M.; Rosa, G.; Lopes, F.B.; Regitano, L.; Rosa, A.; Magnabosco, C.U. Genomewide association mapping and pathway analysis of meat tenderness in Polled Nellore cattle. J. Anim. Sci. 2017, 95, 1945–1956. [Google Scholar] [CrossRef]
- Packialakshmi, B.; Liyanage, R.; Lay Jr, J.; Okimoto, R.; Rath, N. Prednisolone-induced predisposition to femoral head separation and the accompanying plasma protein changes in chickens. Biomark. Insights 2015, 10, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Sell-Kubiak, E.; Wimmers, K.; Reyer, H.; Szwaczkowski, T. Genetic aspects of feed efficiency and reduction of environmental footprint in broilers: A review. J. Appl. Genet. 2017, 58, 487–498. [Google Scholar] [CrossRef] [Green Version]
- McGovern, S.P.; Purfield, D.C.; Ring, S.C.; Carthy, T.R.; Graham, D.A.; Berry, D.P. Candidate genes associated with the heritable humoral response to Mycobacterium avium ssp. paratuberculosis in dairy cows have factors in common with gastrointestinal diseases in humans. J. Dairy Sci. 2019, 102, 4249–4263. [Google Scholar] [CrossRef] [Green Version]
- Choi, I.; Bates, R.O.; Raney, N.E.; Steibel, J.P.; Ernst, C.W. Evaluation of QTL for carcass merit and meat quality traits in a US commercial Duroc population. Meat Sci. 2012, 92, 132–138. [Google Scholar] [CrossRef]
- Hu, Y.; Sun, Q.; Liu, J.; Jia, Y.; Cai, D.; Idriss, A.A.; Omer, N.A.; Zhao, R. In ovo injection of betaine alleviates corticosterone-induced fatty liver in chickens through epigenetic modifications. Sci. Rep. 2017, 7, 40251. [Google Scholar] [CrossRef]
- Wang, Y.; Xu, H.Y.; Gilbert, E.R.; Peng, X.; Zhao, X.L.; Liu, Y.P.; Zhu, Q. Detection of SNPs in the TBC1D1 gene and their association with carcass traits in chicken. Gene 2014, 547, 288–294. [Google Scholar] [CrossRef] [PubMed]
- Manjula, P.; Cho, S.; Suh, K.J.; Seo, D.; Lee, J.H. Single Nucleotide Polymorphism of TBC1D1 Gene Association with Growth Traits and Serum Clinical-Chemical Traits in Chicken. Korean J. Poult. Sci. 2018, 45, 291–298. [Google Scholar] [CrossRef]
- Yang, Z.J.; Fu, L.; Zhang, G.W.; Yang, Y.; Chen, S.Y.; Wang, J.; Lai, S.J. Identification and Association of SNPs in TBC1D1 Gene with Growth Traits in Two Rabbit Breeds. Asian-Australas. J. Anim Sci 2013, 26, 1529–1535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubin, C.-J.; Zody, M.C.; Eriksson, J.; Meadows, J.R.S.; Sherwood, E.; Webster, M.T.; Jiang, L.; Ingman, M.; Sharpe, T.; Ka, S.; et al. Whole-genome resequencing reveals loci under selection during chicken domestication. Nature 2010, 464, 587–591. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
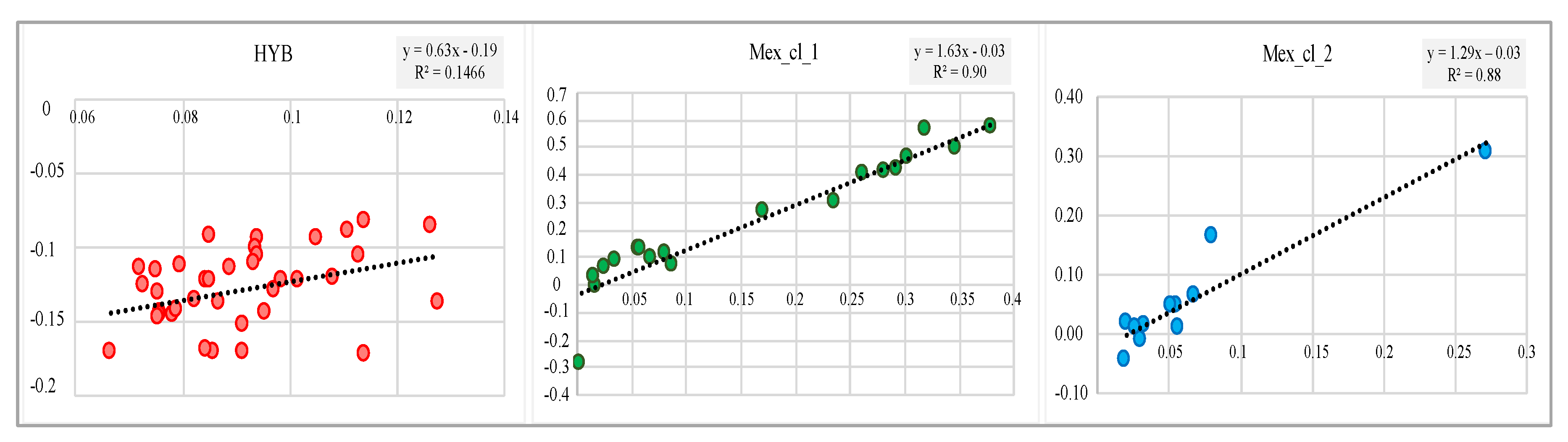{kind=link}
| Pop | Samples | N. ROH | Min–Max (Mean) N. ROH per Sample | Min–Max (Mean) ROH Length per Sample |
|---|---|---|---|---|
| HYB | 38 | 1809 | 36–66 (47) | 1–6.970 (1.717) |
| MEX_cl_1 | 19 | 1438 | 2–170 (75) | 1–7.652 (1.911) |
| MEX_cl_2 | 11 | 355 | 11–120 (32) | 1–7.999 (1.825) |
| Chr | Start ROH Position | End ROH Position | ROH Length | Gene 1 |
|---|---|---|---|---|
| ROH_island in HYB | ||||
| 2 | 23,019,543 | 23,992,770 | 973,227 | MTA3, HAAO |
| 4 | 48,508,445 | 49,752,492 | 124,4047 | RBM47, CHRNA9, RHOH, N4BP2, PDS5A, UBE2K, SMIM14, UGDH, LIAS, RPL9, KLB, RFC1, WDR19, KLHL5, TMEM156, FAM114A1, KLF3, TBC1D1, PGM2, RELL1, C4H4orf19, NWD2 |
| 8 | 35,534 | 3,564,550 | 3,529,016 | GHITM, NRG3, SH2D4B, TSPAN14, FAM213A, EXOSC3, DYDC1, MAT1A, RASGEF1A, CSGALNACT2, RET, BMS1, PLAC9, ANXA11, ECD, FAM149B1, DNAJC9, TFAM, UBE2D1, CISD1, IPMK, PCDH15 |
| 8 | 4,968,066 | 5,916,410 | 948,344 | CHAT, OGDHL, PARG, NCOA4, GPRIN2, SYT15, FAM35A, GLUD1, ADIRF, SNCG, MMRN2, BMPR1A, LDB3, OPN4, WAPL |
| 8 | 8,483,294 | 9,342,515 | 859,221 | RTKN2, ARID5B, TMEM26, RHOBTB1, CDK1 |
| 11 | 3,058,621 | 3,097,354 | 38,733 | - |
| 22 | 1,595,252 | 1,986,192 | 390,940 | ASIP, EIF2S2, RALY |
| ROH_island in MEX_cl_1 | ||||
| 13 | 11,244,459 | 11,371,128 | 126,669 | HYDIN, MTSS1L, SF3B3 |
| ROH_island in MEX_cl_2 | ||||
| 7 | 9,145,051 | 10,872,877 | 1,727,826 | DNAH7, BIRC5, STK17B, HECW2, GTF3C3, C7H2orf66, PGAP1, ANKRD44, SF3B1, COQ10B, HSPD1, RFTN2, BOLL, PLCL1, SATB2, C7H2orf69, TYW5, MAIP1, SPATS2L, KCTD18, SGO2, AOX1, BZW1, CLK1, PPIL3, NIF3L1, ORC2, FAM126B |
| 8 | 1,401,303 | 3,748,153 | 2,346,850 | EXOSC3, DYDC1, MAT1A, RASGEF1A, CSGALNACT2, RET, BMS1, PLAC9, ANXA11, ECD, FAM149B1, DNAJC9, TFAM, UBE2D1, CISD1, IPMK, PCDH15, PRKG1 |
| Pop. | Gene | Phenotype | Species | References |
|---|---|---|---|---|
| MEX_cl_2 | ANKRD44 | Skin thickness | Swine | [27] |
| MEX_cl_2 | AOX1 | Residual feed intake | Bovine | [28] |
| HYB | ARID5B | Adaptive immunity | Human | [29] |
| HYB/MEX_cl_2 | DYDC1 | Acrosome biogenesis; Spermiogenesis | Mouse | [30] |
| MEX_cl_1 | HYDIN | Thermal pain response | Mice | [31] |
| MEX_cl_1 | HYDIN | Marbling score | Bovine | [32] |
| HYB | MMRN2 | Meat juiciness | Bovine | [33] |
| HYB | MMRN2 | Meat tenderness | Bovine | [34] |
| MEX_cl_2 | NIF3L1 | Skin thickness | Swine | [27] |
| MEX_cl_2 | ORC2 | Marbling score | Bovine | [32] |
| HYB/MEX_cl_2 | PCDH15 | Femoral head separation | Chicken | [35] |
| HYB | PGM2 | Feed efficiency; Reduction of environmental footprint | Chicken | [36] |
| MEX_cl_2 | PLCL1 | Skin thickness | Swine | [27] |
| MEX_cl_2 | PRKG1 | Humoral response to Mycobacterium avium ssp. Paratuberculosis | Bovine | [37] |
| MEX_cl_2 | SF3B1 | Carcass merit and meat quality | Swine | [38] |
| HYB/MEX_cl_2 | TFAM | CORT-induced fatty liver protection | Chicken | [39] |
| HYB | TBC1D1 | Carcass | Chicken | [40] |
| HYB | TBC1D1 | Growth and Serum Clinical-Chemical | Chicken | [41] |
| HYB | TBC1D1 | Growth | Rabbit | [42] |
| POP | Obs Hom | Exp Hom | Obs Het | Exp Het | F Mean (SD) | FROH Mean (SD) |
|---|---|---|---|---|---|---|
| HYB | 58.5 | 63.3 | 41.5 | 36.2 | −0.129 (0.025) | 0.130 (0.015) |
| MEX_cl_1 | 76.6 | 69.8 | 23.4 | 29.4 | 0.227 (0.23) | 0.161 (0.13) |
| MEX_cl_2 | 72.7 | 71.1 | 27.3 | 27.6 | 0.056 (0.10) | 0.065 (0.07) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Strillacci, M.G.; Marelli, S.P.; Martinez-Velazquez, G. Hybrid Versus Autochthonous Turkey Populations: Homozygous Genomic Regions Occurrences Due to Artificial and Natural Selection. Animals 2020, 10, 1318. https://doi.org/10.3390/ani10081318
Strillacci MG, Marelli SP, Martinez-Velazquez G. Hybrid Versus Autochthonous Turkey Populations: Homozygous Genomic Regions Occurrences Due to Artificial and Natural Selection. Animals. 2020; 10(8):1318. https://doi.org/10.3390/ani10081318
Chicago/Turabian StyleStrillacci, Maria Giuseppina, Stefano Paolo Marelli, and Guillermo Martinez-Velazquez. 2020. "Hybrid Versus Autochthonous Turkey Populations: Homozygous Genomic Regions Occurrences Due to Artificial and Natural Selection" Animals 10, no. 8: 1318. https://doi.org/10.3390/ani10081318