Toward Structurally Novel and Metabolically Stable HIV-1 Capsid-Targeting Small Molecules

 , ,
, ,
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Biology Cells
2.2. Method Details
2.2.1. Thermal Shift Assays (TSAs) to Screen Compounds for Effect on HIV-1 CA Hexamer Stability
2.2.2. Virus Production
2.2.3. Anti-HIV-1 and Cytotoxicity Assays
2.3. Microsomal Stability Assay
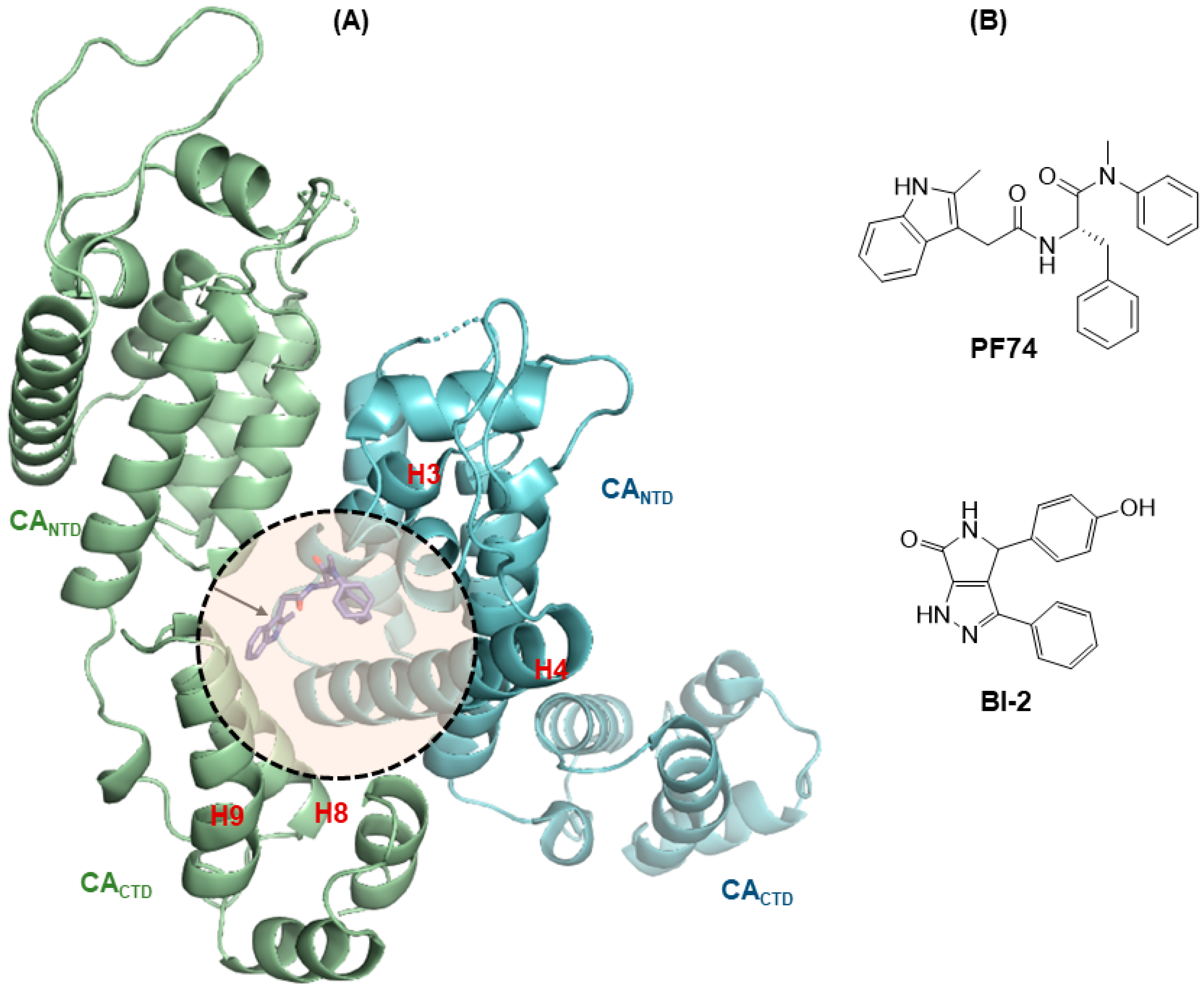
2.4. Molecular Modeling
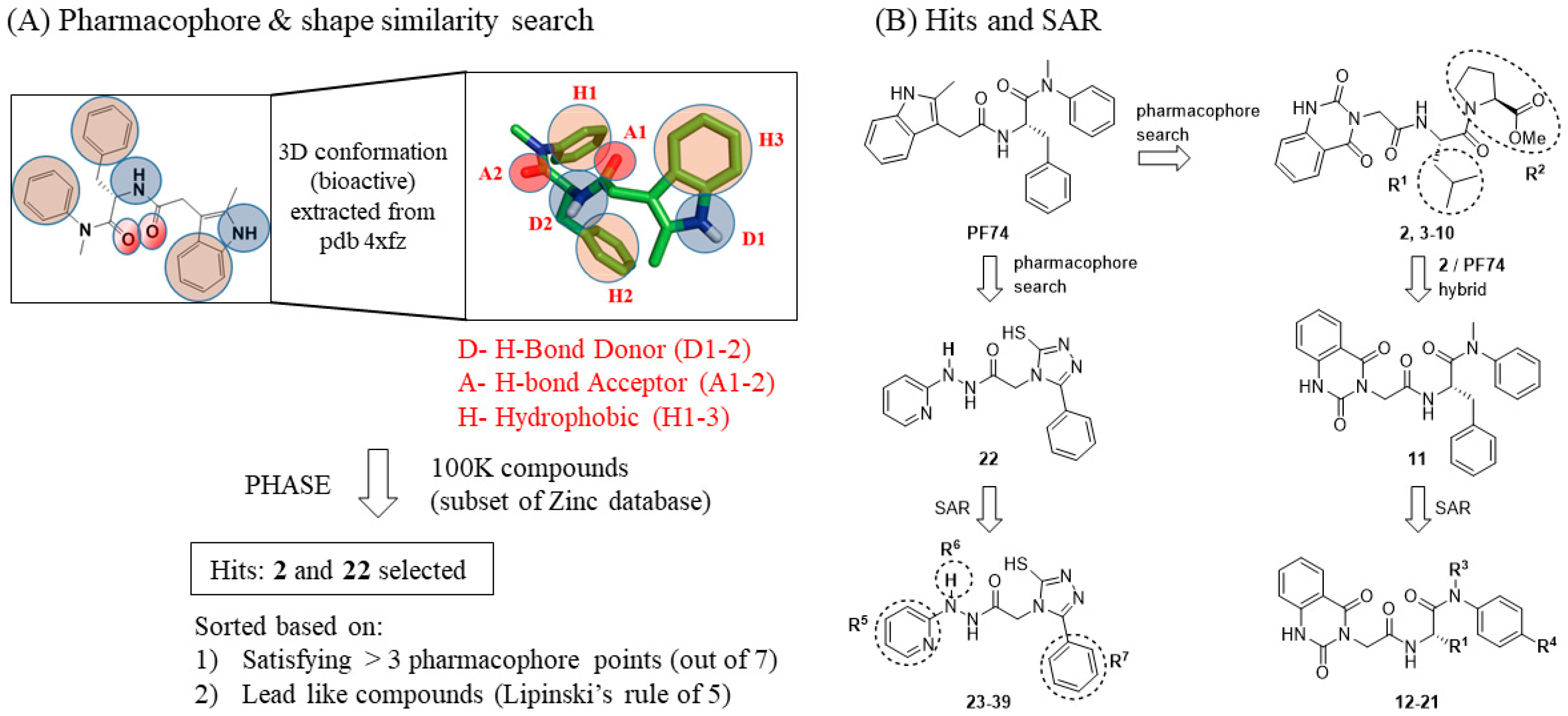
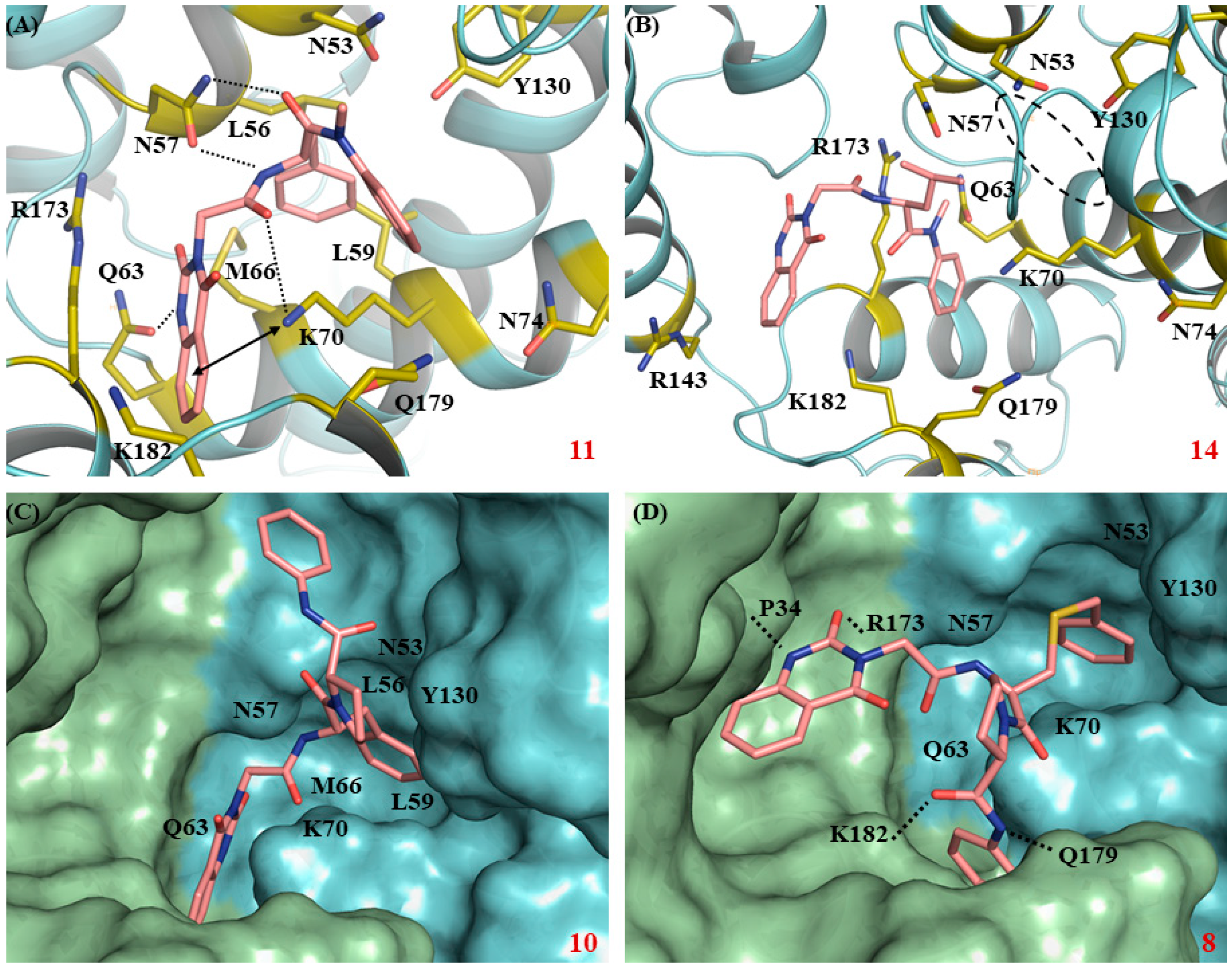
3. Results
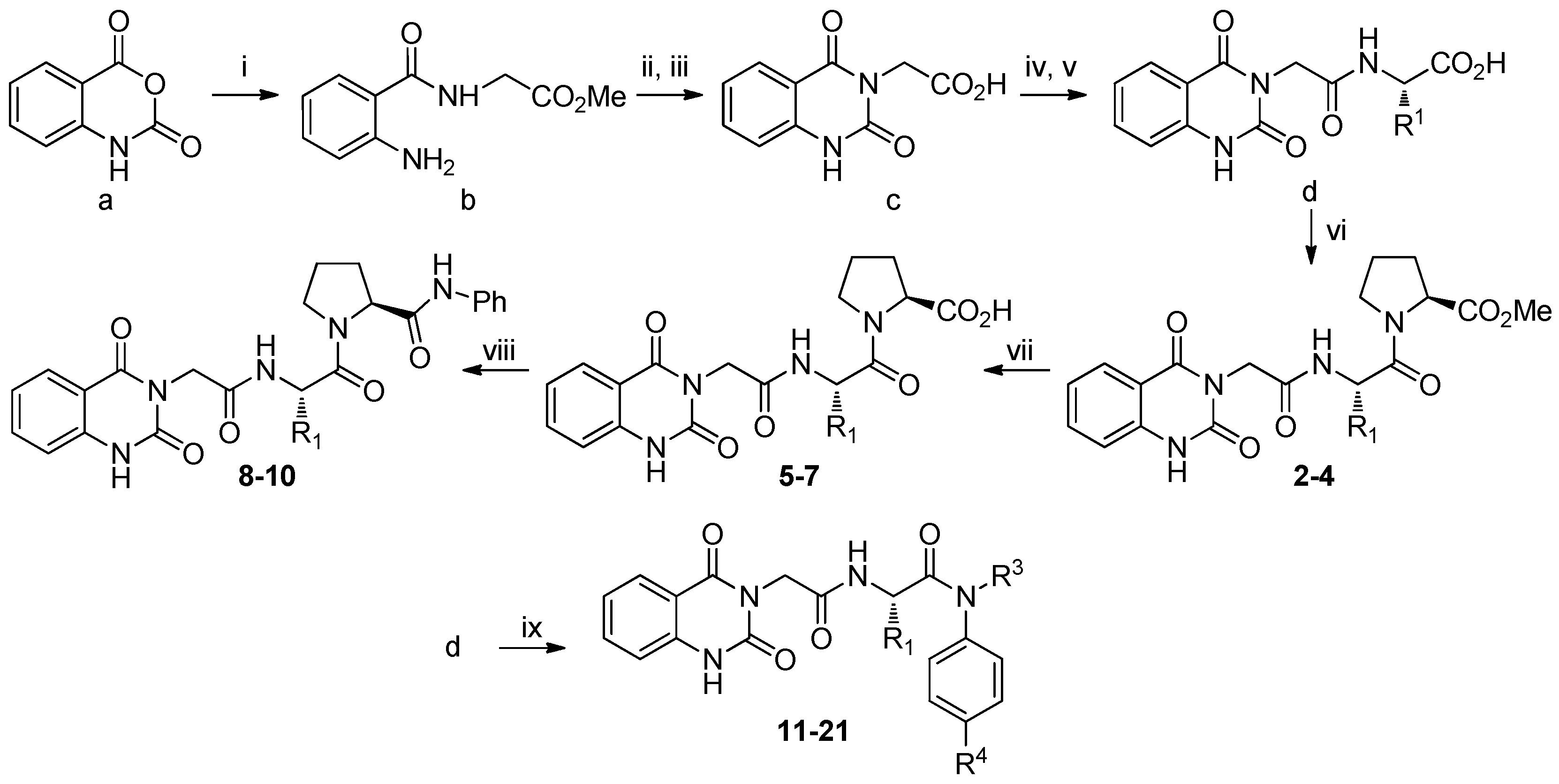
3.1. SAR of Hit 2 (R1 and R2)
3.2. SAR of Hit 11 (R1, R3, and R4)
3.3. SAR of Hit 22 (R5, R6, and R7)
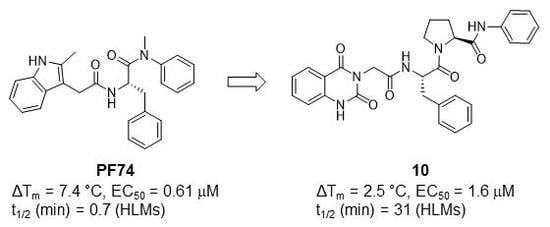
3.4. Metabolic Stability
3.5. Molecular Modeling
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Freed, E.O. HIV-1 assembly, release and maturation. Nat. Rev. Microbiol. 2015, 13, 484–496. [Google Scholar] [CrossRef]
- Ganser, B.K.; Li, S.; Klishko, V.Y.; Finch, J.T.; Sundquist, W.I. Assembly and analysis of conical models for the HIV-1 core. Science 1999, 283, 80–83. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Hill, C.P.; Sundquist, W.I.; Finch, J.T. Image reconstructions of helical assemblies of the HIV-1 CA protein. Nature 2000, 407, 409–413. [Google Scholar] [CrossRef] [PubMed]
- Campbell, E.M.; Hope, T.J. HIV-1 capsid: The multifaceted key player in HIV-1 infection. Nat. Rev. Microbiol. 2015, 13, 471–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sundquist, W.I.; Krausslich, H.G. HIV-1 assembly, budding, and maturation. Cold Spring Harb. Perspect. Med. 2012, 2, a006924. [Google Scholar] [CrossRef]
- Le Sage, V.; Mouland, A.J.; Valiente-Echeverria, F. Roles of HIV-1 capsid in viral replication and immune evasion. Virus Res. 2014, 193, 116–129. [Google Scholar] [CrossRef] [PubMed]
- Hilditch, L.; Towers, G.J. A model for cofactor use during HIV-1 reverse transcription and nuclear entry. Curr. Opin. Virol. 2014, 4, 32–36. [Google Scholar] [CrossRef] [Green Version]
- Ambrose, Z.; Aiken, C. HIV-1 uncoating: Connection to nuclear entry and regulation by host proteins. Virology 2014, 454–455, 371–379. [Google Scholar] [CrossRef] [Green Version]
- Stremlau, M.; Owens, C.M.; Perron, M.J.; Kiessling, M.; Autissier, P.; Sodroski, J. The cytoplasmic body component TRIM5alpha restricts HIV-1 infection in Old World monkeys. Nature 2004, 427, 848–853. [Google Scholar] [CrossRef]
- Sayah, D.M.; Sokolskaja, E.; Berthoux, L.; Luban, J. Cyclophilin A retrotransposition into TRIM5 explains owl monkey resistance to HIV-1. Nature 2004, 430, 569–573. [Google Scholar] [CrossRef]
- Achuthan, V.; Perreira, J.M.; Sowd, G.A.; Puray-Chavez, M.; McDougall, W.M.; Paulucci-Holthauzen, A.; Wu, X.; Fadel, H.J.; Poeschla, E.M.; Multani, A.S.; et al. Capsid-CPSF6 Interaction Licenses Nuclear HIV-1 Trafficking to Sites of Viral DNA Integration. Cell Host Microbe 2018, 24, 392–404.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bejarano, D.A.; Peng, K.; Laketa, V.; Borner, K.; Jost, K.L.; Lucic, B.; Glass, B.; Lusic, M.; Mueller, B.; Krausslich, H.G. HIV-1 nuclear import in macrophages is regulated by CPSF6-capsid interactions at the nuclear pore complex. Elife 2019, 8, e41800. [Google Scholar] [CrossRef] [PubMed]
- Woodward, C.L.; Prakobwanakit, S.; Mosessian, S.; Chow, S.A. Integrase interacts with nucleoporin NUP153 to mediate the nuclear import of human immunodeficiency virus type 1. J. Virol. 2009, 83, 6522–6533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matreyek, K.A.; Yucel, S.S.; Li, X.; Engelman, A. Nucleoporin NUP153 phenylalanine-glycine motifs engage a common binding pocket within the HIV-1 capsid protein to mediate lentiviral infectivity. PLoS Pathog. 2013, 9, e1003693. [Google Scholar] [CrossRef] [Green Version]
- Buffone, C.; Martinez-Lopez, A.; Fricke, T.; Opp, S.; Severgnini, M.; Cifola, I.; Petiti, L.; Frabetti, S.; Skorupka, K.; Zadrozny, K.K.; et al. Nup153 Unlocks the Nuclear Pore Complex for HIV-1 Nuclear Translocation in Nondividing Cells. J. Virol. 2018, 92, e00648-18. [Google Scholar] [CrossRef] [Green Version]
- Meehan, A.M.; Saenz, D.T.; Guevera, R.; Morrison, J.H.; Peretz, M.; Fadel, H.J.; Hamada, M.; van Deursen, J.; Poeschla, E.M. A cyclophilin homology domain-independent role for Nup358 in HIV-1 infection. PLoS Pathog. 2014, 10, e1003969. [Google Scholar] [CrossRef]
- Dharan, A.; Talley, S.; Tripathi, A.; Mamede, J.I.; Majetschak, M.; Hope, T.J.; Campbell, E.M. KIF5B and Nup358 Cooperatively Mediate the Nuclear Import of HIV-1 during Infection. PLoS Pathog. 2016, 12, e1005700. [Google Scholar] [CrossRef] [Green Version]
- Mamede, J.I.; Damond, F.; Bernardo, A.; Matheron, S.; Descamps, D.; Battini, J.L.; Sitbon, M.; Courgnaud, V. Cyclophilins and nucleoporins are required for infection mediated by capsids from circulating HIV-2 primary isolates. Sci. Rep. 2017, 7, 45214. [Google Scholar] [CrossRef] [Green Version]
- Fricke, T.; White, T.E.; Schulte, B.; de Souza Aranha Vieira, D.A.; Dharan, A.; Campbell, E.M.; Brandariz-Nunez, A.; Diaz-Griffero, F. MxB binds to the HIV-1 core and prevents the uncoating process of HIV-1. Retrovirology 2014, 11, 68. [Google Scholar] [CrossRef]
- Xu, B.; Pan, Q.; Liang, C. Role of MxB in Alpha Interferon-Mediated Inhibition of HIV-1 Infection. J. Virol. 2018, 92, e00422-18. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.; Dauphin, A.; Komurlu, S.; McCauley, S.M.; Yurkovetskiy, L.; Carbone, C.; Diehl, W.E.; Strambio-De-Castillia, C.; Campbell, E.M.; Luban, J. Cyclophilin A protects HIV-1 from restriction by human TRIM5alpha. Nat. Microbiol. 2019, 4, 2044–2051. [Google Scholar] [CrossRef]
- Franke, E.K.; Yuan, H.E.; Luban, J. Specific incorporation of cyclophilin A into HIV-1 virions. Nature 1994, 372, 359–362. [Google Scholar] [CrossRef] [PubMed]
- Thali, M.; Bukovsky, A.; Kondo, E.; Rosenwirth, B.; Walsh, C.T.; Sodroski, J.; Gottlinger, H.G. Functional association of cyclophilin A with HIV-1 virions. Nature 1994, 372, 363–365. [Google Scholar] [CrossRef] [PubMed]
- Novikova, M.; Zhang, Y.; Freed, E.O.; Peng, K. Multiple Roles of HIV-1 Capsid during the Virus Replication Cycle. Virol. Sin. 2019, 34, 119–134. [Google Scholar] [CrossRef] [Green Version]
- Gres, A.T.; Kirby, K.A.; KewalRamani, V.N.; Tanner, J.J.; Pornillos, O.; Sarafianos, S.G. STRUCTURAL VIROLOGY. X-ray crystal structures of native HIV-1 capsid protein reveal conformational variability. Science 2015, 349, 99–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pornillos, O.; Ganser-Pornillos, B.K.; Kelly, B.N.; Hua, Y.; Whitby, F.G.; Stout, C.D.; Sundquist, W.I.; Hill, C.P.; Yeager, M. X-ray structures of the hexameric building block of the HIV capsid. Cell 2009, 137, 1282–1292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, G.; Perilla, J.R.; Yufenyuy, E.L.; Meng, X.; Chen, B.; Ning, J.; Ahn, J.; Gronenborn, A.M.; Schulten, K.; Aiken, C.; et al. Mature HIV-1 capsid structure by cryo-electron microscopy and all-atom molecular dynamics. Nature 2013, 497, 643–646. [Google Scholar] [CrossRef]
- Goudreau, N.; Lemke, C.T.; Faucher, A.M.; Grand-Maitre, C.; Goulet, S.; Lacoste, J.E.; Rancourt, J.; Malenfant, E.; Mercier, J.F.; Titolo, S.; et al. Novel inhibitor binding site discovery on HIV-1 capsid N-terminal domain by NMR and X-ray crystallography. ACS Chem. Biol. 2013, 8, 1074–1082. [Google Scholar] [CrossRef]
- Price, A.J.; Jacques, D.A.; McEwan, W.A.; Fletcher, A.J.; Essig, S.; Chin, J.W.; Halambage, U.D.; Aiken, C.; James, L.C. Host cofactors and pharmacologic ligands share an essential interface in HIV-1 capsid that is lost upon disassembly. PLoS Pathog. 2014, 10, e1004459. [Google Scholar] [CrossRef]
- Bhattacharya, A.; Alam, S.L.; Fricke, T.; Zadrozny, K.; Sedzicki, J.; Taylor, A.B.; Demeler, B.; Pornillos, O.; Ganser-Pornillos, B.K.; Diaz-Griffero, F.; et al. Structural basis of HIV-1 capsid recognition by PF74 and CPSF6. Proc. Natl. Acad. Sci. USA 2014, 111, 18625–18630. [Google Scholar] [CrossRef] [Green Version]
- Sowd, G.A.; Serrao, E.; Wang, H.; Wang, W.; Fadel, H.J.; Poeschla, E.M.; Engelman, A.N. A critical role for alternative polyadenylation factor CPSF6 in targeting HIV-1 integration to transcriptionally active chromatin. Proc. Natl. Acad. Sci. USA 2016, 113, E1054–E1063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, J.; Zhou, J.; Shah, V.B.; Aiken, C.; Whitby, K. Small-molecule inhibition of human immunodeficiency virus type 1 infection by virus capsid destabilization. J. Virol. 2011, 85, 542–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saito, A.; Ferhadian, D.; Sowd, G.A.; Serrao, E.; Shi, J.; Halambage, U.D.; Teng, S.; Soto, J.; Siddiqui, M.A.; Engelman, A.N.; et al. Roles of Capsid-Interacting Host Factors in Multimodal Inhibition of HIV-1 by PF74. J. Virol. 2016, 90, 5808–5823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.P.; Francis, A.C.; Meuser, M.E.; Mankowski, M.; Ptak, R.G.; Rashad, A.A.; Melikyan, G.B.; Cocklin, S. Exploring Modifications of an HIV-1 Capsid Inhibitor: Design, Synthesis, and Mechanism of Action. J. Drug Des. Res. 2018, 5, 1070. [Google Scholar] [PubMed]
- Sun, L.; Huang, T.; Dick, A.; Meuser, M.E.; Zalloum, W.A.; Chen, C.H.; Ding, X.; Gao, P.; Cocklin, S.; Lee, K.H.; et al. Design, synthesis and structure-activity relationships of 4-phenyl-1H-1,2,3-triazole phenylalanine derivatives as novel HIV-1 capsid inhibitors with promising antiviral activities. Eur. J. Med. Chem. 2020, 190, 112085. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Zalloum, W.A.; Meuser, M.E.; Jing, L.; Kang, D.; Chen, C.H.; Tian, Y.; Zhang, F.; Cocklin, S.; Lee, K.H.; et al. Discovery of phenylalanine derivatives as potent HIV-1 capsid inhibitors from click chemistry-based compound library. Eur. J. Med. Chem. 2018, 158, 478–492. [Google Scholar] [CrossRef]
- Kubinyi, H. Similarity and dissimilarity: A medicinal chemist’s view. Perspect. Drug Discov. 1998, 9–11, 225–252. [Google Scholar] [CrossRef]
- Maggiora, G.; Vogt, M.; Stumpfe, D.; Bajorath, J. Molecular similarity in medicinal chemistry. J. Med. Chem. 2014, 57, 3186–3204. [Google Scholar] [CrossRef]
- Sterling, T.; Irwin, J.J. ZINC 15—Ligand Discovery for Everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef]
- Dixon, S.L.; Smondyrev, A.M.; Rao, S.N. PHASE: A novel approach to pharmacophore modeling and 3D database searching. Chem. Biol. Drug Des. 2006, 67, 370–372. [Google Scholar] [CrossRef]
- Dixon, S.L.; Smondyrev, A.M.; Knoll, E.H.; Rao, S.N.; Shaw, D.E.; Friesner, R.A. PHASE: A new engine for pharmacophore perception, 3D QSAR model development, and 3D database screening: 1. Methodology and preliminary results. J. Comput. Aided Mol. Des. 2006, 20, 647–671. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A. Lead- and drug-like compounds: The rule-of-five revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Harrison, J.R.; Brand, S.; Smith, V.; Robinson, D.A.; Thompson, S.; Smith, A.; Davies, K.; Mok, N.; Torrie, L.S.; Collie, I.; et al. A Molecular Hybridization Approach for the Design of Potent, Highly Selective, and Brain-Penetrant N-Myristoyltransferase Inhibitors. J. Med. Chem. 2018, 61, 8374–8389. [Google Scholar] [CrossRef] [PubMed]
- Lange, M.J.; Lyddon, T.D.; Johnson, M.C. Diphtheria Toxin A-Resistant Cell Lines Enable Robust Production and Evaluation of DTA-Encoding Lentiviruses. Sci. Rep. 2019, 9, 8985. [Google Scholar] [CrossRef] [Green Version]
- Rosa, A.; Chande, A.; Ziglio, S.; De Sanctis, V.; Bertorelli, R.; Goh, S.L.; McCauley, S.M.; Nowosielska, A.; Antonarakis, S.E.; Luban, J.; et al. HIV-1 Nef promotes infection by excluding SERINC5 from virion incorporation. Nature 2015, 526, 212–217. [Google Scholar] [CrossRef] [Green Version]
- Lo, M.C.; Aulabaugh, A.; Jin, G.; Cowling, R.; Bard, J.; Malamas, M.; Ellestad, G. Evaluation of fluorescence-based thermal shift assays for hit identification in drug discovery. Anal. Biochem. 2004, 332, 153–159. [Google Scholar] [CrossRef]
- Miyazaki, Y.; Doi, N.; Koma, T.; Adachi, A.; Nomaguchi, M. Novel In Vitro Screening System Based on Differential Scanning Fluorimetry to Search for Small Molecules against the Disassembly or Assembly of HIV-1 Capsid Protein. Front. Microbiol. 2017, 8, 1413. [Google Scholar] [CrossRef]
- Pantoliano, M.W.; Petrella, E.C.; Kwasnoski, J.D.; Lobanov, V.S.; Myslik, J.; Graf, E.; Carver, T.; Asel, E.; Springer, B.A.; Lane, P.; et al. High-density miniaturized thermal shift assays as a general strategy for drug discovery. J. Biomol. Screen. 2001, 6, 429–440. [Google Scholar] [CrossRef]
- Adachi, A.; Gendelman, H.E.; Koenig, S.; Folks, T.; Willey, R.; Rabson, A.; Martin, M.A. Production of acquired immunodeficiency syndrome-associated retrovirus in human and nonhuman cells transfected with an infectious molecular clone. J. Virol. 1986, 59, 284–291. [Google Scholar] [CrossRef] [Green Version]
- Schrodinger. Schrödinger Small-Molecule Drug Discovery Suite 2015-4, Schrödinger; LLC: New York, NY, USA, 2015. [Google Scholar]
- Schrodinger. Schrödinger Release 2020-1: Maestro, Schrödinger; LLC: New York, NY, USA, 2020. [Google Scholar]
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aid Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Maxwell, D.S.; TiradoRives, J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meanwell, N.A. Synopsis of some recent tactical application of bioisosteres in drug design. J. Med. Chem. 2011, 54, 2529–2591. [Google Scholar] [CrossRef] [PubMed]
- Masimirembwa, C.M.; Bredberg, U.; Andersson, T.B. Metabolic stability for drug discovery and development: Pharmacokinetic and biochemical challenges. Clin. Pharmacokinet. 2003, 42, 515–528. [Google Scholar] [CrossRef] [PubMed]
- Wacher, V.J.; Silverman, J.A.; Zhang, Y.; Benet, L.Z. Role of P-glycoprotein and cytochrome P450 3A in limiting oral absorption of peptides and peptidomimetics. J. Pharm. Sci. 1998, 87, 1322–1330. [Google Scholar] [CrossRef] [PubMed]
- Eichelbaum, M.; Burk, O. CYP3A genetics in drug metabolism. Nat. Med. 2001, 7, 285–287. [Google Scholar] [CrossRef]
- Xu, L.H.; Liu, H.T.; Murray, B.P.; Callebaut, C.; Lee, M.S.; Hong, A.; Strickley, R.G.; Tsai, L.K.; Stray, K.M.; Wang, Y.J.; et al. Cobicistat (GS-9350): A Potent and Selective Inhibitor of Human CYP3A as a Novel Pharmacoenhancer. Acs Med. Chem. Lett. 2010, 1, 209–213. [Google Scholar] [CrossRef] [Green Version]
- AIDSinfo FDA-Approved HIV Medicines. Available online: https://aidsinfo.nih.gov/understanding-hiv-aids/fact-sheets/21/58/fda-approved-hiv-medicines (accessed on 30 March 2020).






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
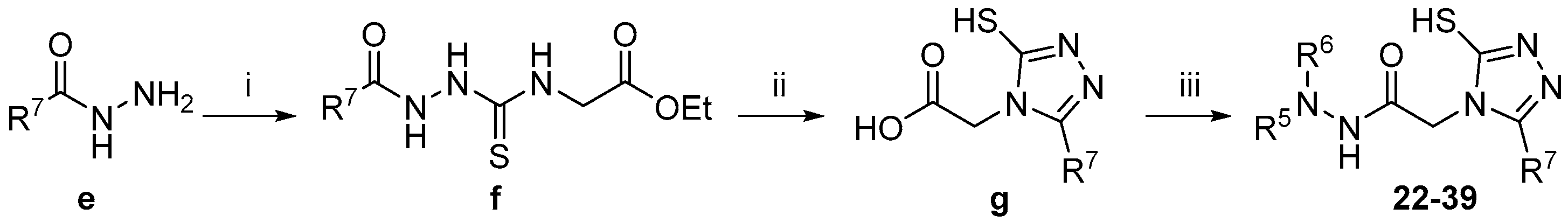{kind=link}

| Compd | R1 | R2 | EC50 (μM) [a] | CC50 (μM) [b] | TSA ΔTm (°C) [c] |
|---|---|---|---|---|---|
| 1 (PF74) | -- | -- | 0.61 ± 0.2 | 76 ± 9 | 7.4 |
| 2 |  |  | >20 | >100 | 0.5 |
| 3 | H |  | >20 | >100 | 0 |
| 4 |  |  | >20 | <100 | 0.5 |
| 5 |  |  | >20 | >100 | 0.8 |
| 6 |  |  | >20 | >100 | 0 |
| 7 |  |  | >20 | >100 | 0 |
| 8 |  |  | >20 | 44 ± 5 | 0.5 |
| 9 |  |  | >20 | >100 | 0.5 |
| 10 |  |  | 1.6 ± 0.1 | >100 | 2.5 |
| Compd | R1 | R3 | R4 | EC50 (μM) [a] | CC50 (μM) [b] | TSA ΔTm (°C) [c] |
|---|---|---|---|---|---|---|
| 1 (PF74) | -- | -- | -- | 0.61 ± 0.2 | 76 ± 9 | 7.4 |
| 11 |  | Me | H | 6.9 ± 0.8 | >100 | 2.7 |
| 12 |  | H | H | >20 | <100 | 0 |
| 13 |  | Me | F | 8.0 ± 1.3 | >100 | 2.4 |
| 14 |  | Me | H | >20 | >100 | 0 |
| 15 |  | H | H | >20 | >100 | 0.7 |
| 16 |  | Me | Me | >20 | >100 | 0 |
| 17 |  | H | H | >20 | >100 | 0 |
| 18 |  | Me | H | >20 | >100 | 0.5 |
| 19 |  | H | H | >20 | >100 | 0.6 |
| 20 |  | Me | Cl | >20 | >100 | 0.5 |
| 21 |  | H |  | >20 | >100 | 0.6 |

| Compd | R5 | R6 | R7 | EC50 (μM) [a] | CC50 (μM) [b] | TSA ΔTm (°C) [c] |
|---|---|---|---|---|---|---|
| 22 |  | H |  | >20 | >100 | −0.5 |
| 23 |  | H |  | >20 | >100 | 0 |
| 24 |  | H |  | >20 | >100 | 0.7 |
| 25 |  | H |  | >20 | >100 | 0.6 |
| 26 |  | H |  | >20 | >100 | 0.7 |
| 27 |  | H |  | >20 | ~100 | 0.7 |
| 28 |  | H |  | >20 | >100 | 0.7 |
| 29 |  | H |  | >20 | >100 | 0 |
| 30 |  | H |  | >20 | >100 | 0.8 |
| 31 |  | H |  | >20 | >100 | −1.2 |
| 32 |  | H |  | >20 | >100 | 0 |
| 33 |  | H |  | >20 | >100 | 0 |
| 34 |  | H |  | >20 | >100 | 0.6 |
| 35 |  | H |  | >20 | >100 | 0.5 |
| 36 |  | H |  | >20 | >100 | 0.8 |
| 37 |  | H |  | >20 | >100 | 1.0 |
| 38 |  | H |  | >20 | >100 | 0.9 |
| 39 |  | Me |  | >20 | >50 | 1.2 |
| Compound | HLM a | HLM a (+Cobi c) | MLM b | MLM b (+Cobi c) |
|---|---|---|---|---|
| PF74 | 0.7 | 91 | 0.6 | 34 |
| 10 | 31 | >120 | 2.9 | 85 |
| Verapamil | 15 | -- | 4.2 | -- |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vernekar, S.K.V.; Sahani, R.L.; Casey, M.C.; Kankanala, J.; Wang, L.; Kirby, K.A.; Du, H.; Zhang, H.; Tedbury, P.R.; Xie, J.; et al. Toward Structurally Novel and Metabolically Stable HIV-1 Capsid-Targeting Small Molecules. Viruses 2020, 12, 452. https://doi.org/10.3390/v12040452
Vernekar SKV, Sahani RL, Casey MC, Kankanala J, Wang L, Kirby KA, Du H, Zhang H, Tedbury PR, Xie J, et al. Toward Structurally Novel and Metabolically Stable HIV-1 Capsid-Targeting Small Molecules. Viruses. 2020; 12(4):452. https://doi.org/10.3390/v12040452
Chicago/Turabian StyleVernekar, Sanjeev Kumar V., Rajkumar Lalji Sahani, Mary C. Casey, Jayakanth Kankanala, Lei Wang, Karen A. Kirby, Haijuan Du, Huanchun Zhang, Philip R. Tedbury, Jiashu Xie, and et al. 2020. "Toward Structurally Novel and Metabolically Stable HIV-1 Capsid-Targeting Small Molecules" Viruses 12, no. 4: 452. https://doi.org/10.3390/v12040452