
New Statistical Models for Copolymerization

Abstract
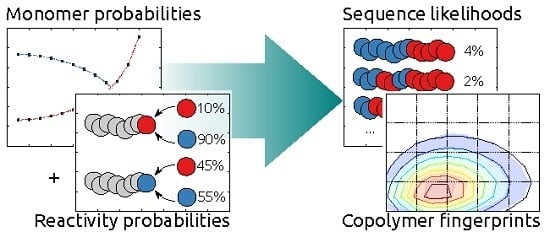
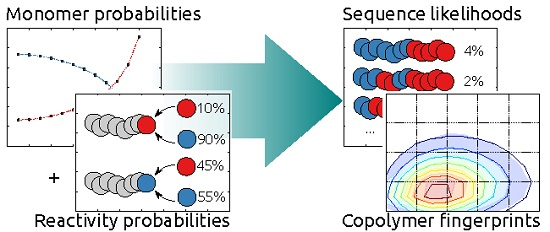
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
3. Results and Discussion
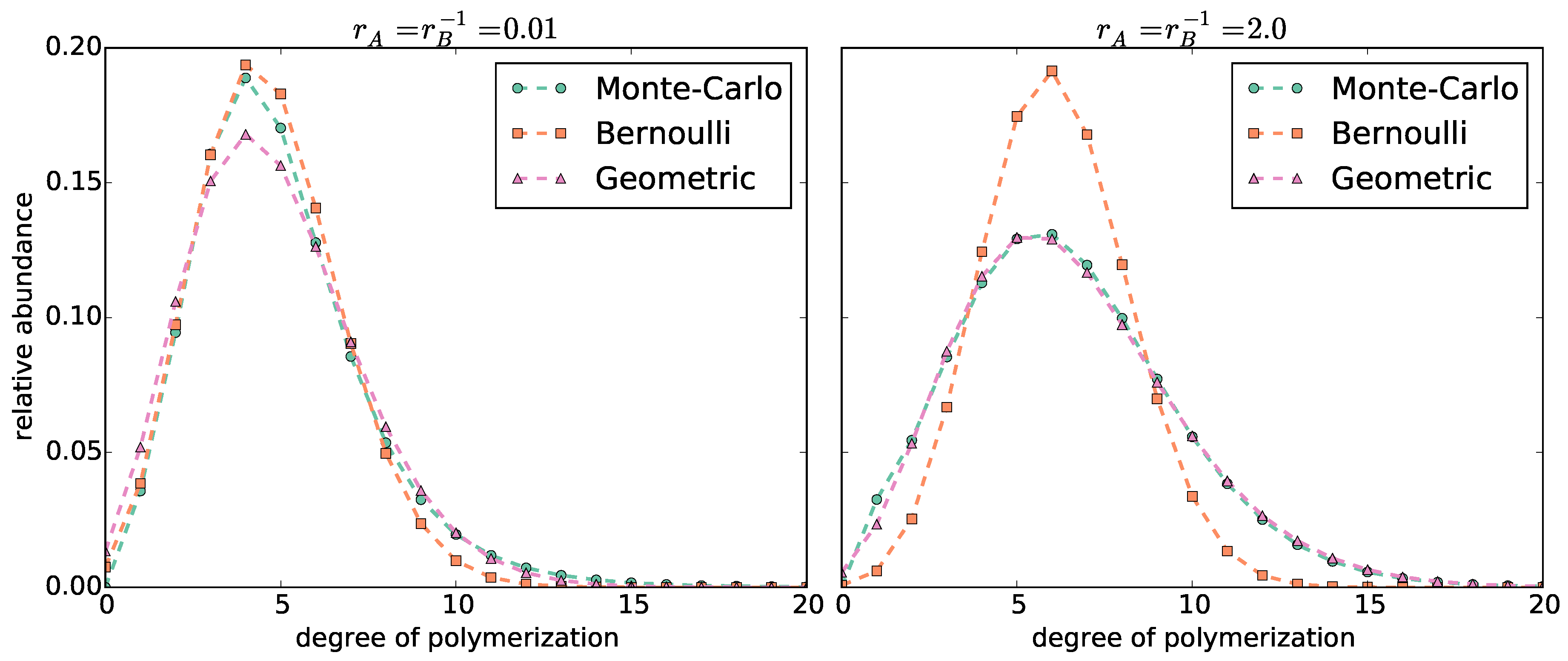
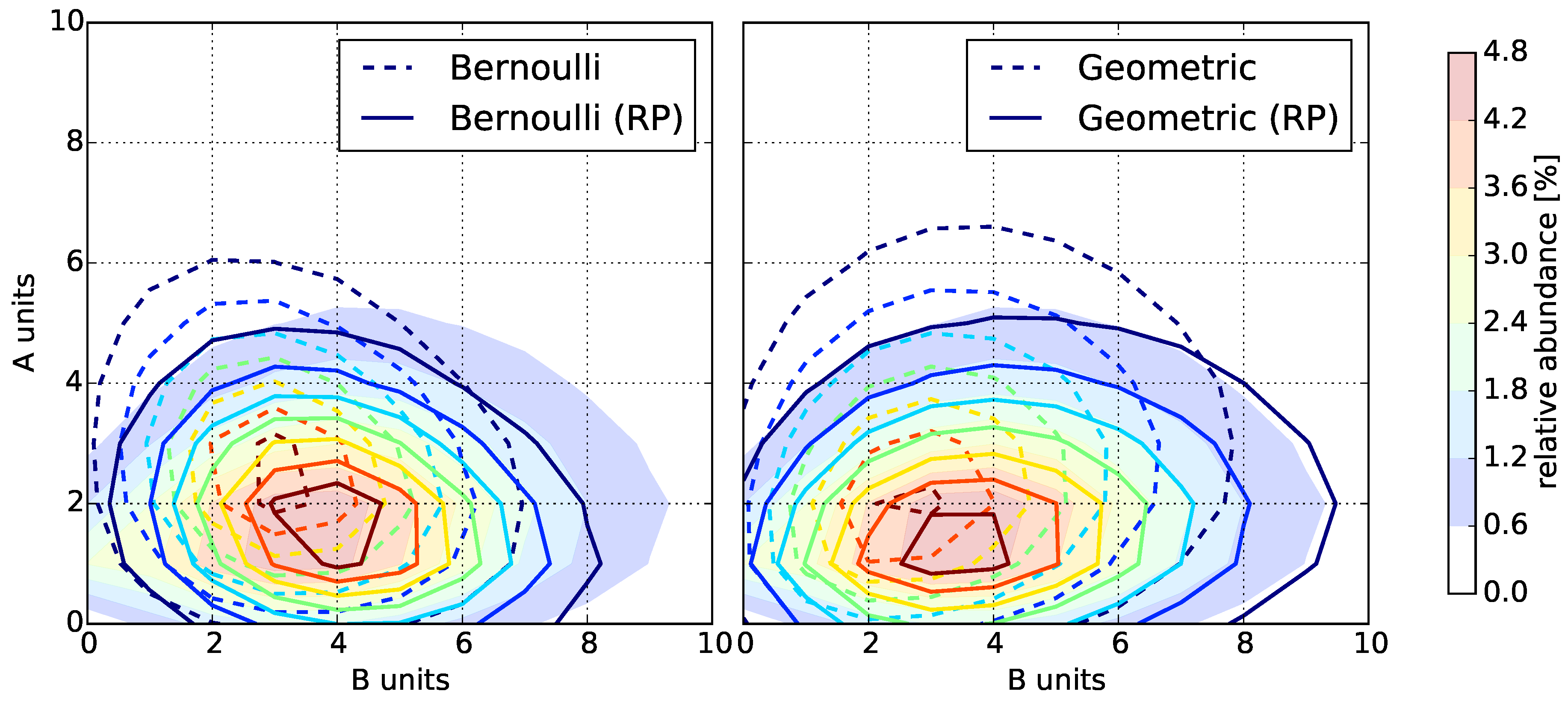
3.1. Bernoulli Model
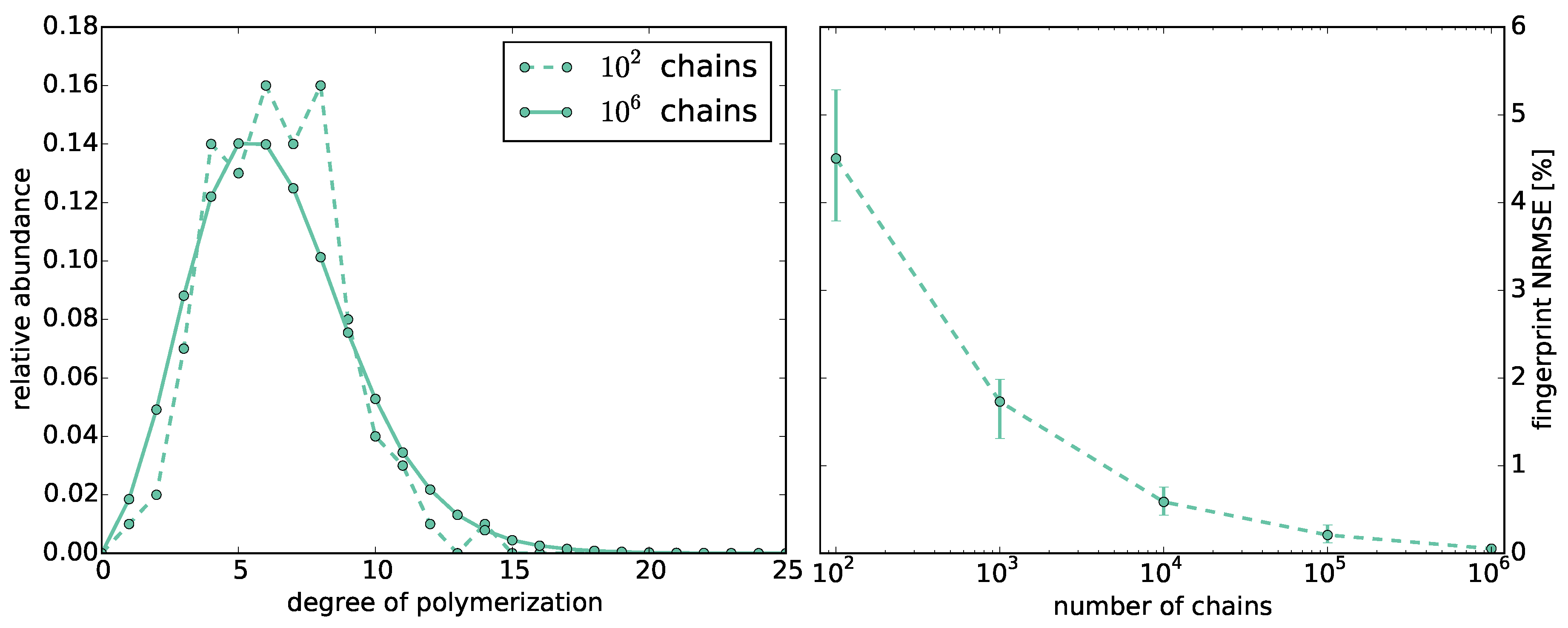
3.1.1. Chain Lengths
3.1.2. Fingerprint Model
3.1.3. Reactivity Ratios
3.2. Geometric Model
3.2.1. Chain Length
3.2.2. Fingerprint Model
3.2.3. Reactivity Ratios
3.3. Single Chain Models
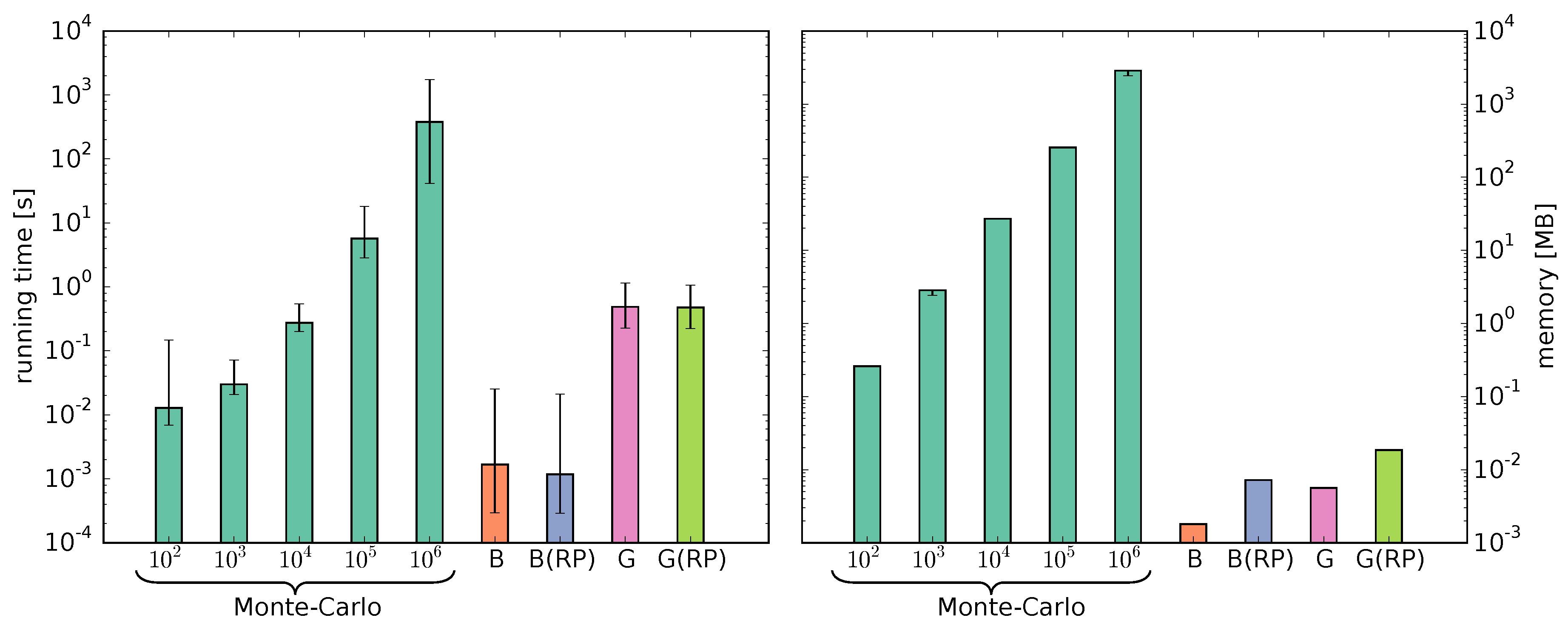
3.4. Parameter Estimation
3.5. Model Evaluation
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| ODE | Ordinary differential equation |
| MS | Mass spectrometry |
| MALDI-TOF MS | Matrix-assisted laser desorption/ionization time-of-flight mass spectrometry |
| NRMSE | Normalized root mean square error |
References
- Mayo, F.R.; Lewis, F.M. Copolymerization. I. A basis for comparing the behavior of monomers in copolymerization; the copolymerization of styrene and methyl methacrylate. J. Am. Chem. Soc. 1944, 66, 1594–1601. [Google Scholar] [CrossRef]
- Kryven, I.; Iedema, P.D. Deterministic modeling of copolymer microstructure: Composition drift and sequence patterns. Macromol. React. Eng. 2015, 9, 285–306. [Google Scholar] [CrossRef]
- Fischer, B.; Roth, V.; Roos, F.; Grossmann, J.; Baginsky, S.; Widmayer, P.; Gruissem, W.; Buhmann, J.M. NovoHMM: A hidden Markov model for de novo peptide sequencing. Anal. Chem. 2005, 77, 7265–7273. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Caffo, B.; Jaffee, H.A.; Irizarry, R.A.; Feinberg, A.P. Redefining CpG islands using hidden Markov models. Biostatistics 2010, 11, 499–514. [Google Scholar] [CrossRef] [PubMed]
- González Díaz, H.; Molina, R.; Uriarte, E. Stochastic molecular descriptors for polymers. 1. Modelling the properties of icosahedral viruses with 3D-Markovian negentropies. Polymer 2004, 45, 3845–3853. [Google Scholar] [CrossRef]
- González-Díaz, H.; Pérez-Bello, A.; Uriarte, E. Stochastic molecular descriptors for polymers. 3. Markov electrostatic moments as polymer 2D-folding descriptors: RNA-QSAR for mycobacterial promoters. Polymer 2005, 46, 6461–6473. [Google Scholar] [CrossRef]
- González-Díaz, H.; Saíz-Urra, L.; Molina, R.; Uriarte, E. Stochastic molecular descriptors for polymers. 2. Spherical truncation of electrostatic interactions on entropy based polymers 3D-QSAR. Polymer 2005, 46, 2791–2798. [Google Scholar] [CrossRef]
- Cruz-Monteagudo, M.; Munteanu, C.R.; Borges, F.; Cordeiro, M.N.D.S.; Uriarte, E.; Chou, K.C.; González-Díaz, H. Stochastic molecular descriptors for polymers. 4. Study of complex mixtures with topological indices of mass spectra spiral and star networks: The blood proteome case. Polymer 2008, 49, 5575–5587. [Google Scholar] [CrossRef]
- González-Díaz, H.; Uriarte, E. Biopolymer stochastic moments. I. Modeling human rhinovirus cellular recognition with protein surface electrostatic moments. Biopolymers 2005, 77, 296–303. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Montoto, L.G.; Dea-Ayuela, M.A.; Prado-Prado, F.J.; Bolas-Fernández, F.; Ubeira, F.M.; González-Díaz, H. Study of peptide fingerprints of parasite proteins and drug-DNA interactions with Markov-Mean-Energy invariants of biopolymer molecular-dynamic lattice networks. Polymer 2009, 50, 3857–3870. [Google Scholar] [CrossRef]
- Rodriguez-Soca, Y.; Munteanu, C.R.; Dorado, J.; Rabuñal, J.; Pazos, A.; González-Díaz, H. Plasmod-PPI: A web-server predicting complex biopolymer targets in plasmodium with entropy measures of protein-protein interactions. Polymer 2010, 51, 264–273. [Google Scholar] [CrossRef]
- Brandrup, J.; Immergut, E.H. Polymer Handbook, 4th ed.; Grulke, E.A., Ed.; Wiley: Hoboken, NJ, USA, 1999. [Google Scholar]
- Gillespie, D.T. Exact stochastic simulation of coupled chemical reactions. J. Phys. Chem. 1977, 81, 2340–2361. [Google Scholar] [CrossRef]
- Meimaroglou, D.; Kiparissides, C. Review of Monte Carlo Methods for the Prediction of Distributed Molecular and Morphological Polymer Properties. Ind. Eng. Chem. Res. 2014, 53, 8963–8979. [Google Scholar] [CrossRef]
- D’hooge, D.R.; Van Steenberge, P.H.; Derboven, P.; Reyniers, M.F.; Marin, G.B. Model-based design of the polymer microstructure: Bridging the gap between polymer chemistry and engineering. Polym. Chem. 2015, 6, 7081–7096. [Google Scholar] [CrossRef]
- Brandão, A.L.T.; Soares, J.B.P.; Pinto, J.C.; Alberton, A.L. When polymer reaction engineers play dice: Applications of Monte Carlo Models in PRE. Macromol. React. Eng. 2015, 9, 141–185. [Google Scholar] [CrossRef]
- Willemse, R.X.E. New insights into free-radical (co)polymerization kinetics. Ph.D. Thesis, University of Technology Eindhoven, Eindhoven, The Netherlands, 2005. [Google Scholar]
- Drache, M.; Schmidt-Naake, G.; Buback, M.; Vana, P. Modeling RAFT polymerization kinetics via Monte Carlo methods: Cumyl dithiobenzoate mediated methyl acrylate polymerization. Polymer 2005, 46, 8483–8493. [Google Scholar] [CrossRef]
- Drache, M. Modeling the product composition during controlled radical polymerizations with mono- and bifunctional alkoxyamines. Macromol. Symp. 2009, 275–276, 52–58. [Google Scholar] [CrossRef]
- Szymanski, R. On the determination of the ratios of the propagation rate constants on the basis of the MWD of copolymer chains: A new Monte Carlo algorithm. e-Polymers 2009, 9, 538–552. [Google Scholar] [CrossRef]
- Van Steenberge, P.H.M.; D’hooge, D.R.; Wang, Y.; Zhong, M.; Reyniers, M.F.; Konkolewicz, D.; Matyjaszewski, K.; Marin, G.B. Linear gradient quality of ATRP copolymers. Macromolecules 2012, 45, 8519–8531. [Google Scholar] [CrossRef]
- Drache, M.; Drache, G. Simulating controlled radical polymerizations with mcPolymer—A Monte Carlo approach. Polymers 2012, 4, 1416–1442. [Google Scholar] [CrossRef]
- Engler, M.S.; Crotty, S.; Barthel, M.J.; Pietsch, C.; Knop, K.; Schubert, U.S.; Böcker, S. COCONUT—An efficient tool for estimating copolymer compositions from mass spectra. Anal. Chem. 2015, 87, 5223–5231. [Google Scholar] [CrossRef] [PubMed]
- Montaudo, M.S. Mass spectra of copolymers. Mass Spectrom. Rev. 2002, 21, 108–144. [Google Scholar] [CrossRef] [PubMed]
- Pasch, H. MALDI-TOF Mass Spectrometry of Synthetic Polymers; Schrepp, W., Ed.; Springer: Berlin, Germany, 2003. [Google Scholar]
- Vivó-Truyols, G.; Staal, B.; Schoenmakers, P.J. Strip-based regression: A method to obtain comprehensive co-polymer architectures from matrix-assisted laser desorption ionisation-mass spectrometry data. J. Chromatogr. A 2010, 1217, 4150–4159. [Google Scholar] [CrossRef] [PubMed]
- Weidner, S.M.; Falkenhagen, J.; Bressler, I. Copolymer composition determined by LC-MALDI-TOF MS coupling and MassChrom2D data analysis. Macromol. Chem. Phys. 2012, 213, 2404–2411. [Google Scholar] [CrossRef]
- Horský, J.; Walterová, Z. Fingerprint multiplicity in MALDI-TOF mass spectrometry of copolymers. Macromol. Symp. 2014, 339, 9–16. [Google Scholar] [CrossRef]
- Engler, M.S.; Crotty, S.; Barthel, M.J.; Pietsch, C.; Schubert, U.S.; Böcker, S. Abundance correction for mass discrimination effects in polymer mass spectra. Rapid Commun. Mass Spectrom. 2016, 30, 1233–1241. [Google Scholar] [CrossRef]
- Raeder, H.; Schrepp, W. MALDI-TOF mass spectrometry in the analysis of synthetic polymers. Acta Polym. 1998, 49, 272–293. [Google Scholar] [CrossRef]
- Schriemer, D.C.; Li, L. Mass discrimination in the analysis of polydisperse polymers by MALDI time-of-flight mass spectrometry. 1. Sample preparation and desorption/ionization issues. Anal. Chem. 1997, 69, 4169–4175. [Google Scholar] [CrossRef]
- Schriemer, D.C.; Li, L. Mass discrimination in the analysis of polydisperse polymers by MALDI time-of-flight mass spectrometry. 2. Instrumental issues. Anal. Chem. 1997, 69, 4176–4183. [Google Scholar] [CrossRef]
- Hoteling, A.J.; Erb, W.J.; Tyson, R.J.; Owens, K.G. Exploring the importance of the relative solubility of matrix and analyte in MALDI sample preparation using HPLC. Anal. Chem. 2004, 76, 5157–5164. [Google Scholar] [CrossRef] [PubMed]
- Wilczek-Vera, G.; Danis, P.O.; Eisenberg, A. Individual block length distributions of block copolymers of polystyrene-block-poly(R-methylstyrene) by MALDI/TOF mass spectrometry. Macromolecules 1996, 29, 4036–4044. [Google Scholar] [CrossRef]
- Teraoka, I. Polymer Solutions; Wiley: New York, NY, USA, 2002. [Google Scholar]






© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Engler, M.S.; Scheubert, K.; Schubert, U.S.; Böcker, S. New Statistical Models for Copolymerization. Polymers 2016, 8, 240. https://doi.org/10.3390/polym8060240
Engler MS, Scheubert K, Schubert US, Böcker S. New Statistical Models for Copolymerization. Polymers. 2016; 8(6):240. https://doi.org/10.3390/polym8060240
Chicago/Turabian StyleEngler, Martin S., Kerstin Scheubert, Ulrich S. Schubert, and Sebastian Böcker. 2016. "New Statistical Models for Copolymerization" Polymers 8, no. 6: 240. https://doi.org/10.3390/polym8060240