Rational Design of Amphiphilic Diblock Copolymer/MWCNT Surface Modifiers and Their Application for Direct Electrochemical Sensing of DNA

 , and
, and
Abstract
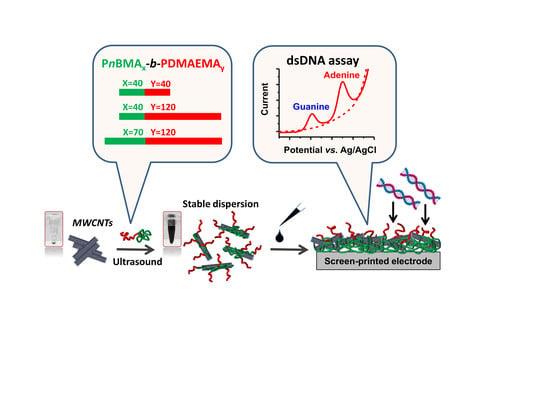
:
1. Introduction
2. Materials and Methods
2.1. Synthesis of PnBMA-Br Macroinitiators
2.2. Synthesis of PnBMA-b-PDMAEMA Diblock Copolymers
2.3. Dispersing of Carbon Nanomaterials
2.4. 1H Nuclear Magnetic Resonance (1H–NMR) Spectroscopy
2.5. Size Exclusion Chromatography (SEC)
2.6. Transmission Electron Microscopy (TEM)
2.7. CryogenicTransmission Electron Microscopy (cryo-TEM)
2.8. Electrochemical Measurements
2.9. Spectrophotometric Measurements
3. Results and Discussion
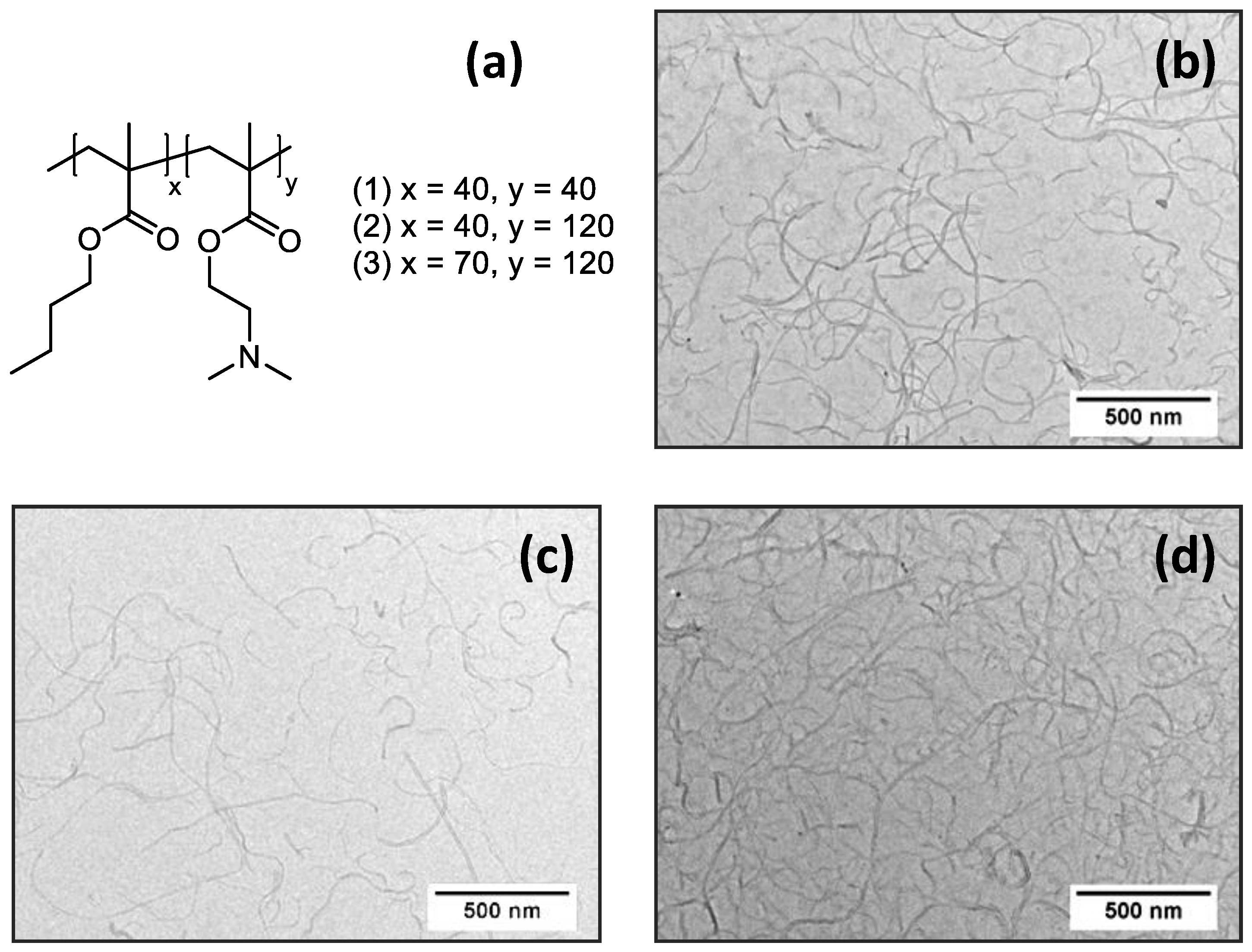
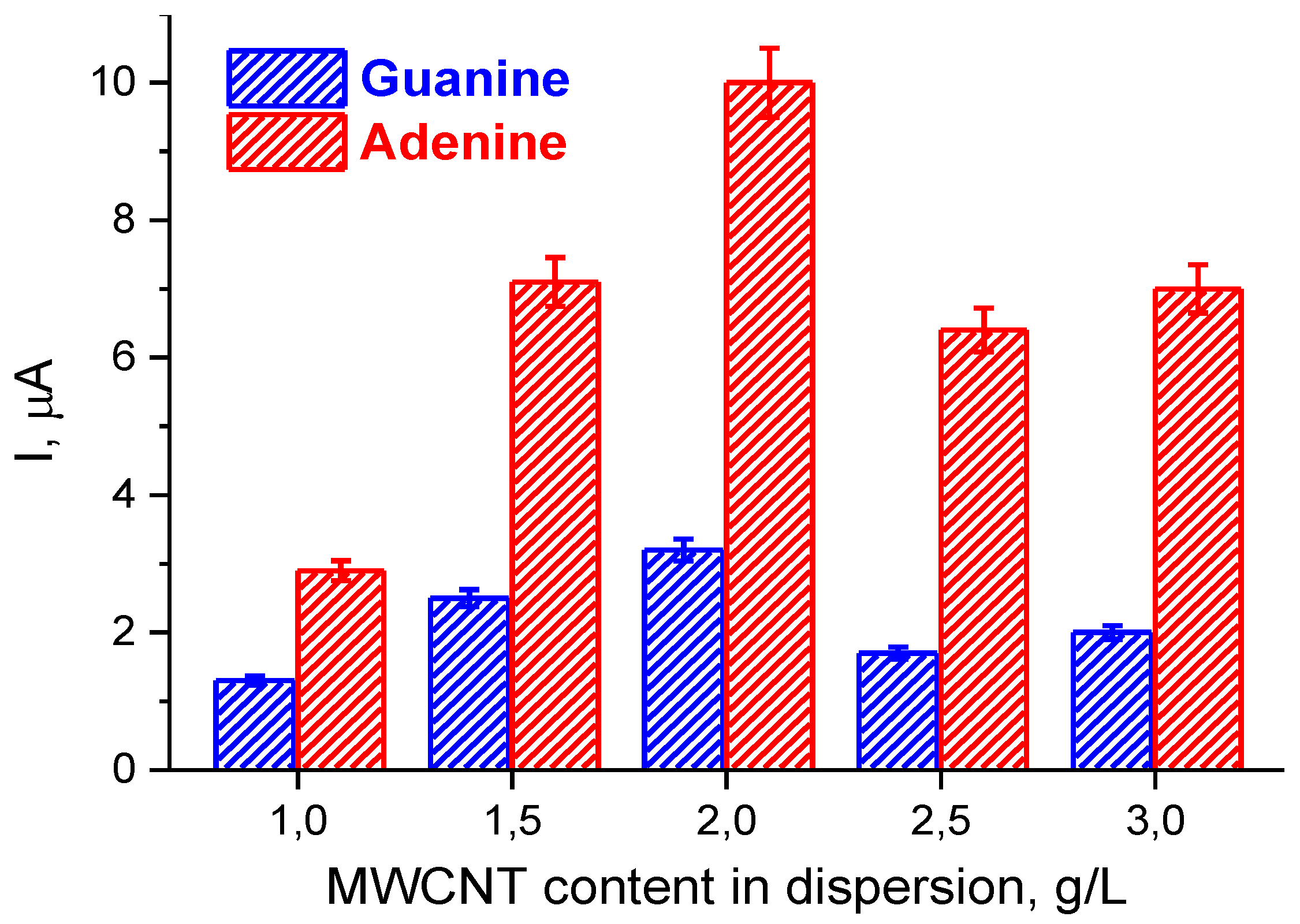
3.1. Preparation and Characterization of (PnBMAx-b-PDMAEMAy + MWCNT) Dispersions
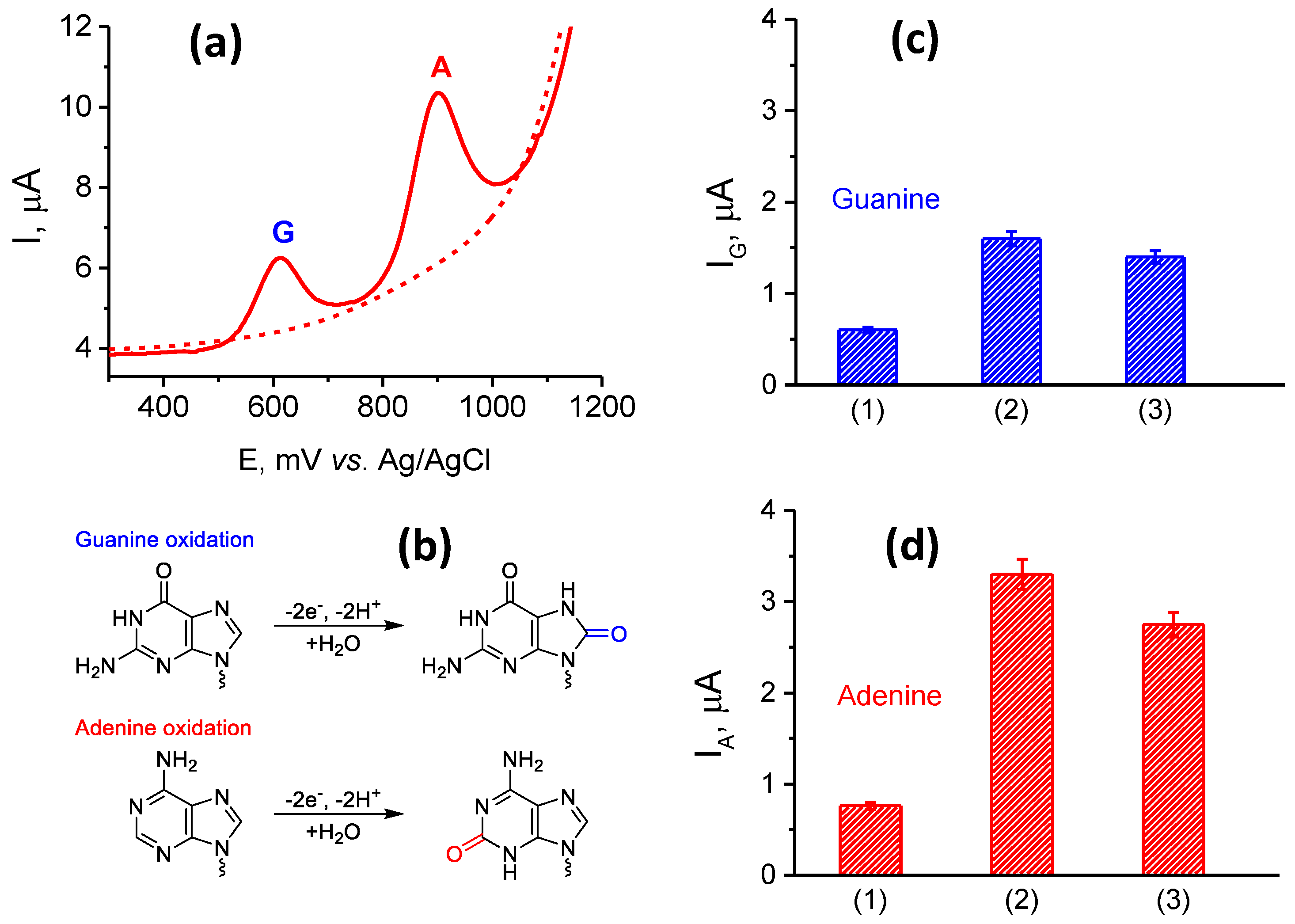
3.2. Electrochemical Characterization of Electrodes Modified by (PnBMAx-b-PDMAEMAy + MWCNT) Dispersions
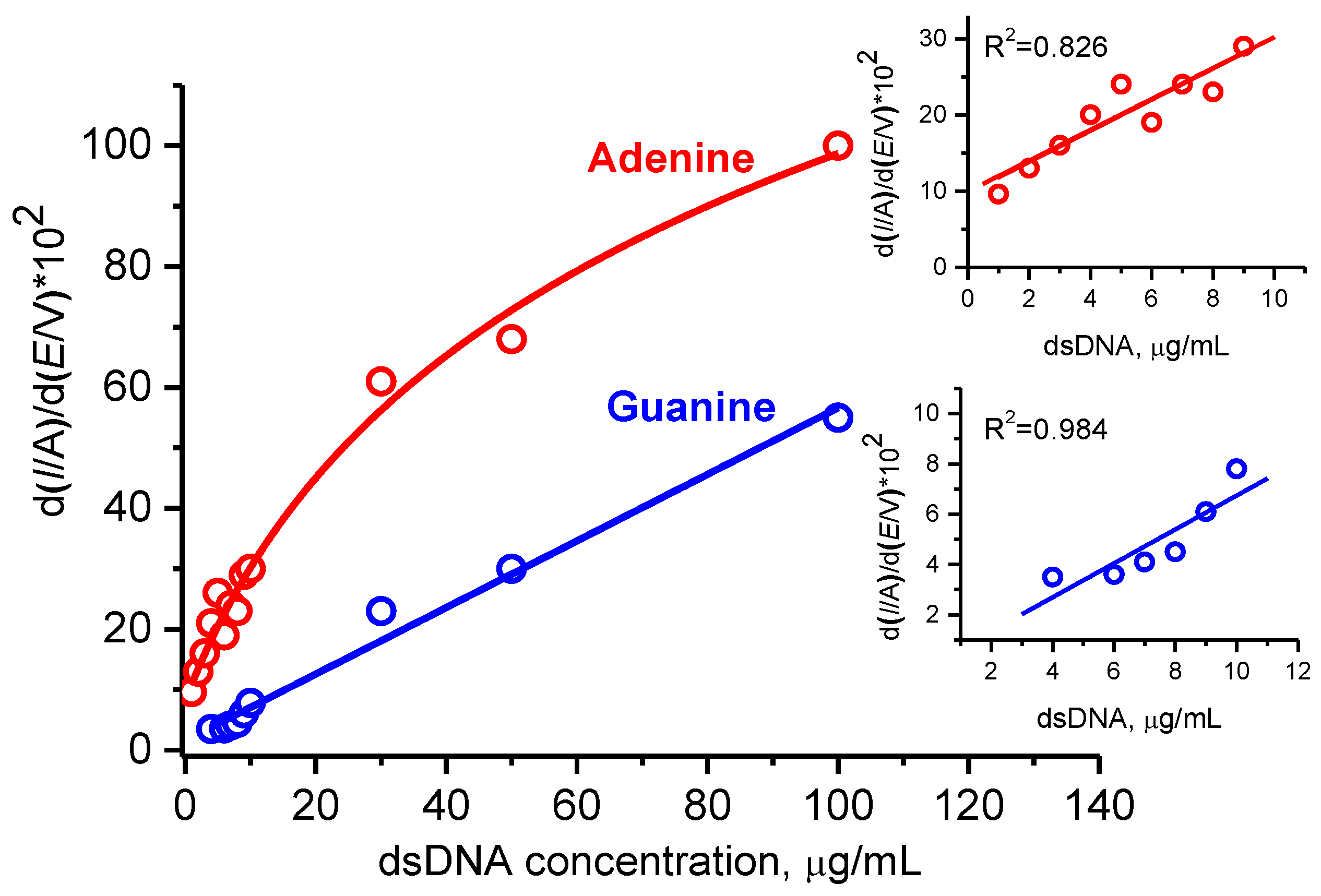
3.3. Characterization and Quantification of Electrochemical dsDNA Assay
3.3.1. Optimization of Electrochemistry of dsDNA for Different Electrode Modifications
3.3.2. Electroanalysis of dsDNA in the Presence of Human Serum
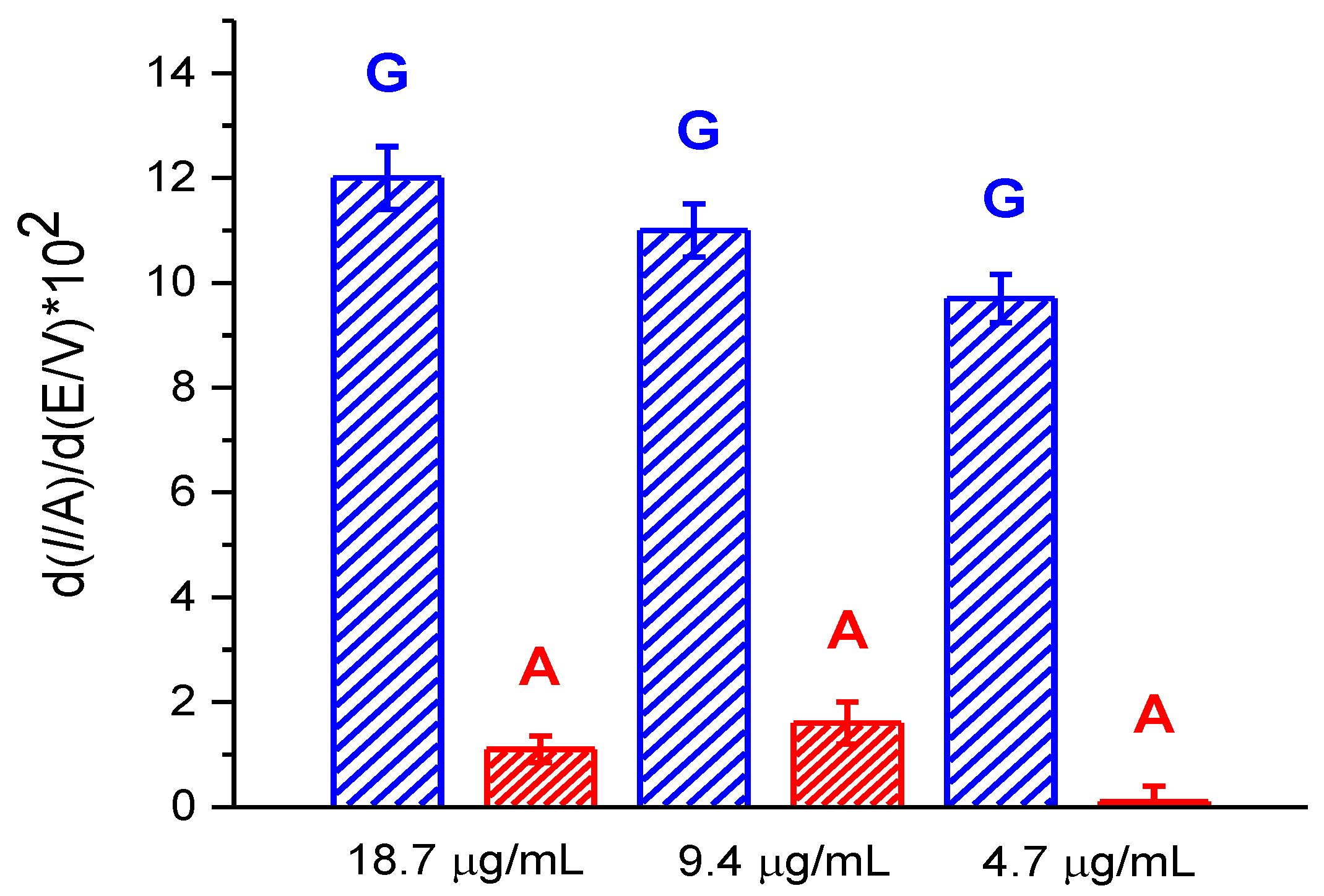
3.3.3. Electroanalysis of dsDNA at Low Concentrations
3.3.4. Electroanalysis of Leukocyte DNA
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Campos-Carrillo, A.; Weitzel, J.N.; Sahoo, P.; Rockne, R.; Mokhnatkin, J.V.; Murtaza, M.; Gray, S.W.; Goetz, L.; Goel, A.; Schork, N.; et al. Circulating tumor DNA as an early cancer detection tool. Pharmacol. Therap. 2020, 207, 107458. [Google Scholar] [CrossRef]
- Hasanzadeh, M.; Shadjou, N.; de la Guardia, M. Early stage diagnosis of programmed cell death (apoptosis) using electroanalysis: Nanomaterial and methods overview. TrAC Trends in Anal. Chem. 2017, 93, 199–211. [Google Scholar] [CrossRef]
- Yin, J.; Miao, P. Apoptosis Evaluation by Electrochemical Techniques. Chem. Asian, J. 2016, 11, 632–641. [Google Scholar] [CrossRef] [PubMed]
- Kogikoski, S., Jr.; Paschoalino, W.J.; Cantelli, L.; Silva, W.; Kubota, L.T. Electrochemical sensing based on DNA nanotechnology. TrAC Trends in Anal. Chem. 2019, 118, 597–605. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, W.B.; Liu, C.; Zhang, P.; Balaeff, A.; Beratan, D.N. DNA charge transport: Moving beyond 1D. Surf. Sci. 2016, 652, 33–38. [Google Scholar] [CrossRef] [Green Version]
- Palecek, E.; Bartosik, M. Electrochemistry of nucleic acids. Chem. Rev. 2012, 112, 3427–3481. [Google Scholar] [CrossRef]
- Machera, H.C.; García-Fernándeza, N.; Adsuar-Gómez, A.; Porras-Lópezc, M.; González-Calleb, A.; Noval-Padilloa, J.; Guerrero, J.M.; Molinerod, P.; Borrego-Domínguezb, J.M.; Herruzo-Avilésc, Á.; et al. Donor-specific circulating cell free DNA as a noninvasive biomarker of graft injury in heart transplantation. Clin. Chim. Acta 2019, 495, 590–597. [Google Scholar] [CrossRef]
- Udomsinprasert, W.; Poovorawan, Y.; Chongsrisawat, V.; Vejchapipat, P.; Jittikoon, J.; Honsawek, S. Leukocyte mitochondrial DNA copy number as a potential biomarker indicating poor outcome in biliary atresia and its association with oxidative DNA damage and telomere length. Mitochondrion 2019, 47, 1–9. [Google Scholar] [CrossRef]
- Arvand, M.; Niazi, A.; Mazhabi, R.M.; Biparva, P. Direct electrochemistry of adenine on multiwalled carbon nanotube-ionic liquid composite film modified carbon paste electrode and its determination in DNA. J. Mol. Liquids 2012, 173, 1–7. [Google Scholar] [CrossRef]
- Zhu, Z.; Li, X.; Zeng, Y.; Sun, W. Ordered mesoporous carbon modified carbon ionic liquid electrode for the electrochemical detection of double-stranded DNA, Biosens. Bioelectron 2010, 25, 2313–2317. [Google Scholar] [CrossRef]
- Hasoň, S.; Fojta, M.; Ostatná, V. Label-free electrochemical analysis of purine nucleotides and nucleobaes at disposable carbon electrodes in microliter volumes. J. Electroanal. Chem. 2019, 847, 113252. [Google Scholar] [CrossRef]
- Sanjuán, I.; Martín-Gómez, A.N.; Graham, J.; Hernández-Ibánez, N.; Banks, C.; Thiemann, T.; Iniesta, J. The electrochemistry of 5-halocytosines at carbon based electrodes towards epigenetic sensing. Electrochim Acta 2018, 282, 459–468. [Google Scholar] [CrossRef]
- Abi, A.; Mohammadpour, Z.; Zuo, X.; Safavi, A. Nucleic acid-based electrochemical nanobiosensors. Biosens. Bioelectron. 2018, 102, 479–489. [Google Scholar] [CrossRef] [PubMed]
- Bartosik, M.; Jirakova, L. Electrochemical analysis of nucleic acids as potential cancer biomarkers. Curr. Opin. Electrochem. 2019, 14, 96–103. [Google Scholar] [CrossRef]
- Carinelli, S.; Kühnemund, M.; Nilsson, M.; Pividori, M.I. Yoctomole electrochemical genosensing of Ebola virus cDNA by rolling circle and circle to circle amplification. Biosens. Bioelectron. 2017, 93, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Lee, E.-C. Carbon nanotube/polymer composite electrodes for flexible, attachable electrochemical DNA sensors. Biosens. Bioelectron. 2015, 71, 414–419. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Murthy, C.N.; Ratna Prabha, C. Recent advances in carbon nanotube based electrochemical biosensors. Int. J. Biol. Macromol. 2018, 108, 687–703. [Google Scholar] [CrossRef]
- Sharma, V.K.; Jelen, F.; Trnkova, L. Functionalized solid electrodes for electrochemical biosensing of purine nucleobases and their analogues: A Review. Sensors 2015, 15, 1564–1600. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Yang, X.-L.; Xiao, B.-L.; Geng, F.-Y.; Hong, J.; Sheibani, N.; Moosavi-Movahedi, A.A. Detection of guanine and adenine using an aminated reduced graphene oxide functional membrane-modified glassy carbon electrode. Sensors 2017, 17, 1652. [Google Scholar] [CrossRef] [Green Version]
- Barman, K.; Jasimuddin, S. Electrochemical detection of adenine and guanine using a self-assembled copper(ii)–thiophenyl-azo-imidazole complex monolayer modified gold electrode. RSC Adv. 2014, 4, 49819–49826. [Google Scholar] [CrossRef]
- Oliveira-Brett, A.M.; Piedade, J.A.P.; Silva, L.A.; Diculescu, V.C. Voltammetric determination of all DNA nucleotides. Anal. Bichem. 2004, 332, 321–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, L.; Yu, S.; Zhou, X.; Bao, Y.; Yang, F.; Kang, W.; Zhang, X. Modification of electron structure on the semiconducting singlewalled carbon nanotubes for effectively electrosensing guanine and adenine, Anal. Chim. Acta 2019, 1079, 86–93. [Google Scholar] [CrossRef]
- Sigolaeva, L.V.; Bulko, T.V.; Kozin, M.S.; Zhang, W.; Köhler, M.; Romanenko, I.; Yuan, J.; Schacher, F.H.; Pergushov, D.V.; Shumyantseva, V.V. Long-term stable poly(ionic liquid)/MWCNTs inks enable enhanced surface modification for electrooxidative detection and quantification of dsDNA. Polymer 2019, 168, 95–103. [Google Scholar] [CrossRef] [Green Version]
- Bartlett, P.N. Biolectrochemistry: Fundamentals, Experimental Techniques and Applications; Wiley: Chichester, UK, 2008; p. 494. [Google Scholar]
- Luque, G.L.; Ferreyra, N.F.; Granero, A.; Bollo, S.; Rivas, G.A. Electrooxidation of DNA at glassy carbon electrodes modified with multiwall carbon nanotubes dispersed in polyethylenimine. Electrochim. Acta 2011, 56, 9121–9126. [Google Scholar] [CrossRef]
- Campuzano, S.; Yáñez-Sedeño, P.; Pingarrón, J.M. Nanoparticles for nucleic-acid-based biosensing: Opportunities, challenges, and prospects. Anal. Bioanal. Chem. 2019, 411, 1791–1806. [Google Scholar] [CrossRef]
- Li, J.; Liu, Q.; Liu, Y.; Liu, S.; Yao, S. DNA biosensor based on chitosan film doped with carbon nanotubes. Anal. Biochem. 2005, 346, 107–114. [Google Scholar] [CrossRef]
- Kuzmicz, D.; Coupillaud, P.; Men, Y.; Vignolle, J.; Vendraminetto, G.; Ambrogi, M.; Taton, D.; Yuan, J. Functional mesoporous poly(ionic liquid)-based copolymer monoliths: From synthesis to catalysis and microporous carbon production. Polymer 2014, 55, 3423–3430. [Google Scholar] [CrossRef]
- Kim, T.; Tung, T.T.; Lee, T.; Kim, J.; Suh, K.S. Poly(ionic liquid)-Mediated Hybridization of Single-Walled Carbon Nanotubes and Conducting Polymers. Chem. Asian, J. 2010, 5, 256–260. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.C.; Chang, C.C.; Yang, K.H.; Mai, F.D.; Tseng, C.L.; Chen, L.Y.; Hwang, B.J.; Liu, Y.C. Polypyrrole electrode with a greater electroactive surface electrochemically polymerized in plasmon-activated water. J. Taiwan Inst. Chem. Eng. 2018, 82, 252–260. [Google Scholar] [CrossRef]
- Arduini, F.; Micheli, L.; Moscone, D.; Palleschi, G.; Piermarini, S.; Ricci, F.; Volpe, G. Electrochemical biosensors based on nanomodified screen-printed electrodes: Recent applications in clinical analysis. TrAC Trends in Anal. Chem. 2016, 79, 114–126. [Google Scholar] [CrossRef] [Green Version]
- Gutiérrez, A.; Gutierrez, F.; Eguílaz, M.; González-Domíngues, J.M.; Hernández-Ferrer, J.; Ansón-Casaos, A.; Martínez, M.T.; Rivas, G.A. Electrochemical sensing of guanine, adenine and 8-hydroxy-2’-deoxyguanosine at glassy carbon modified with single-walled carbon nanotubes covalently functionalized with lysine. RSC Adv. 2016, 6, 13469–13477. [Google Scholar] [CrossRef] [Green Version]
- Palecek, E.; Flojta, M.; Jelen, F.; Vetteri, V. Electrochemical Analysis of nucleic acids. In The Encyclopedia of Electrochemistry, Bard, A.J., Stratmann, M., Eds.; Wiley-VCH: Weinheim, Germany, 2002; Volume 9, pp. 365–429. [Google Scholar]
- Shumyantseva, V.V.; Bulko, T.V.; Kuzikov, A.V.; Masamrekh, R.A.; Konyakhina, A.Y.; Romanenko, I.; Max, J.B.; Köhler, M.; Gilep, A.A.; Usanov, S.A.; et al. All-electrochemical nanocomposite two-electrode setup for quantification of drugs and study their electrocatalytical conversion by cytochromes P450. Electrochim. Acta 2020, 336, 135579. [Google Scholar] [CrossRef]
- Shumyantseva, V.V.; Sigolaeva, L.V.; Agafonova, L.E.; Bulko, T.V.; Pergushov, D.V.; Schacher, F.H.; Archakov, A.I. Facilitated biosensing via direct electron transfer of myoglobin integrated into diblock copolymer/multi-walled carbon nanotube nanocomposites. J. Mater. Chem. B 2015, 3, 5467–5477. [Google Scholar] [CrossRef] [PubMed]
- Shumyantseva, V.V.; Bulko, T.V.; Kuzikov, A.V.; Masamrekh, R.A.; Pergushov, D.V.; Schacher, F.H.; Sigolaeva, L.V. Electrochemical fingerprint of cytochrome c on a MWCNT/polymer nanocomposite electrode. Mend. Commun. 2020, 30, 299–301. [Google Scholar] [CrossRef]
- Narrainen, A.P.; Pascual, S.; Haddleton, D.M. Amphiphilic diblock, triblock, and star block copolymers by living radical polymerization: Synthesis and aggregation behavior. J. Polym. Sci., Part. A: Polym. Chem. 2002, 40, 439–450. [Google Scholar] [CrossRef]
- Max, J.B.; Pergushov, D.V.; Sigolaeva, L.V.; Schacher, F.H. Polyampholytic graft copolymers based on polydehydroalanines (PDha) - synthesis, solution behavior and application as dispersants for carbon nanotubes. Polymer Chem. 2019, 10, 3006–3019. [Google Scholar] [CrossRef]
- Ahn, H.; Lee, Y.; Lee, H.; Han, Y.S.; Seong, B.S.; Ryu, D.Y. Various phase behaviors of weakly interacting binary block copolymer blends. Macromolecules 2013, 46, 4454–4461. [Google Scholar] [CrossRef]
- Sigolaeva, L.V.; Günther, U.; Pergushov, D.V.; Gladyr, S.Y.; Kurochkin, I.N.; Schacher, F.H. Sequential pH-dependent adsorption of ionic amphiphilic diblock copolymer micelles and choline oxidase onto conductive substrates: Toward the design of biosensors. Macromol. Biosci. 2014, 14, 1039–1051. [Google Scholar] [CrossRef]
- Wang, J. Analytical Electrochemistry, 3rd ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2006; p. 32. [Google Scholar]
- Sigolaeva, L.V.; Pergushov, D.V.; Synatschke, C.V.; Wolf, A.; Dewald, I.; Kurochkin, I.N.; Fery, A.; Müller, A.H.E. Co-assemblies of micelle-forming diblock copolymers and enzymes on graphite substrate for an improved design of biosensor systems. Soft Matter 2013, 9, 2858–2868. [Google Scholar] [CrossRef]
- Muti, M.; Muti, M. Electrochemical monitoring of the interaction between anticancer drug and DNA in the presence of antioxidant. Talanta 2018, 178, 1033–1039. [Google Scholar] [CrossRef]
- Bagni, G.; Osella, D.; Sturchio, E.; Mascini, M. Deoxyribonucleic acid (DNA) biosensors for environmental risk assessment and drug studies. Anal. Chim. Acta 2006, 573-574, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Švorc, L.; Kalcher, K. Modification-free electrochemical approach for sensitive monitoring of purine DNA bases: Simultaneous determination of guanine and adenine in biological samples using boron-doped diamond electrode. Sens. Actuat. B-Chemical 2014, 194, 332–342. [Google Scholar] [CrossRef]
- Ren, S.; Wang, H.; Zhang, H.Y.; Yu, L.Q.; Li, M.J.; Li, M. Direct electrocatalytic and simultaneous determination of purine and pyrimidine DNA bases using novel mesoporous carbon fibers as electrocatalyst. J. Electroanal. Chem. 2015, 750, 65–73. [Google Scholar] [CrossRef]
- Li, Q.; Batchelor-McAuley, C.; Compon, R.G. Electrochemical oxidation of guanine: Electrode reaction mechanism and tailoring carbon electrode surface to switch between adsorptive and diffusional responses. J. Phys. Chem. B 2010, 114, 7423–7428. [Google Scholar] [CrossRef]
- Gonçalves, L.M.; Bachelor-McAuley, C.; Barros, A.; Comton, R.G. Electrochemical oxidation of adenine: A mixed adsorption and diffusion response on an edge-plane pyrolytic graphite electrode. J. Phys. Chem. 2010, 114, 14213–14219. [Google Scholar] [CrossRef]
- Dryhurst, G.; Elving, P. Electrochemical oxidation of adenine: Reaction products and mechanisms. J. Electrochem. Soc. 1968, 5, 1014–1022. [Google Scholar] [CrossRef]
- Ni, G.; Qin, J.; Chen, Z.; Li, H.; Zhou, J.; Huang, M.; Zhou, L. Associations between genetic variation in one-carbon metabolism and leukocyte DNA methylation in valproate-treated patients with epilepsy. Clin. Nutrition 2018, 37, 308–312. [Google Scholar] [CrossRef]
- Babazadeh, Z.; Razavi, S.; Tavalaee, M.; Deemeh, M.R.; Shahidi, M.; Nasr-Esfahani, M.H. Sperm DNA damage and its relation with leukocyte DNA damage. Reproductive Toxicol. 2010, 29, 120–124. [Google Scholar] [CrossRef]
- Trotter, M.; Borst, N.; Thewes, R.; von Stetten, F. Review: Electrochemical DNA sensing – Principles, commercial systems, and applications. Biosens. Bioelectron. 2020, 154, 112069. [Google Scholar] [CrossRef]
- Ferapontova, E.E. Hybridization biosensors relying on electrical properties of nucleic acids. Electroanalysis 2017, 29, 6–13. [Google Scholar] [CrossRef]
- Ferapontova, E.E. DNA electrochemistry and electrochemical sensor for nucleic acids. Ann. Rev. Anal. Chem. 2018, 11, 197–218. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Modification Code | MWCNT Content, g/L | Ered, mV | Eox, mV | ΔE, mV | E1/2, mV | Ired, μμ | Iox, μμ | Electroactive Surface Area, A, cm2 | Electroactive Surface Area Relative to the Reference Electrode (naked SPE) |
|---|---|---|---|---|---|---|---|---|---|
| Naked SPE | - | −87 | 421 | 499 | 167 | 6 | 5 | 0.0024 (8% 2) | 1 |
| SPE/(PnBMA40-b-PDMAEMA40+MWCNT_1) | 1.0 | 130 | 235 | 105 | 183 | 44 | 36 | 0.012 (37%) | 5 |
| SPE/(PnBMA40-b-PDMAEMA120+MWCNT_1) | 1.0 | 141 | 240 | 100 | 190 | 38 | 30 | 0.014 (45%) | 6 |
| SPE/(PnBMA70-b-PDMAEMA120+MWCNT_1) | 1.0 | 125 | 220 | 95 | 173 | 104 | 69 | 0.027 (85%) | 11 |
| SPE/(PnBMA40-b-PDMAEMA120+MWCNT_1) | 1.0 | 141 | 240 | 100 | 190 | 38 | 30 | 0.014 (45%) | 6 |
| SPE/(PnBMA40-b-PDMAEMA120+MWCNT_1.5) | 1.5 | 125 | 160 | 35 | 143 | 197 | 96 | 0.069 (220%) | 29 |
| SPE/(PnBMA40-b-PDMAEMA120+MWCNT_2) | 2.0 | 125 | 160 | 35 | 143 | 247 | 103 | 0.081 (259%) | 34 |
| SPE/(PnBMA40-b-PDMAEMA120+MWCNT_2.5) | 2.5 | 125 | 150 | 25 | 138 | 227 | 87 | 0.054 (173%) | 23 |
| SPE/(PnBMA40-b-PDMAEMA120+MWCNT_3) | 3.0 | 120 | 150 | 30 | 135 | 254 | 106 | 0.076 (241%) | 32 |
| Modification | Guanine (G) Residue | Adenine (A) Residue | ||
|---|---|---|---|---|
| V, mV | I, µA | V, mV | I, µA | |
| Naked SPE | - | - | 935 ± 17 | 0.005 ± 0.0003 |
| SPE/(PnBMA40-b-PDMAEMA40+MWCNT_1)/dsDNA | 644 ± 20 | 0.6 ± 0.1 | 947 ± 40 | 0.6 ± 0.2 |
| SPE/(PnBMA40-b-PDMAEMA120+MWCNT_1)/dsDNA | 610 ± 10 | 1.5 ± 0.1 | 898 ± 40 | 3.0 ± 0.3 |
| SPE/(PnBMA70-b-PDMAEMA120+MWCNT_1)/dsDNA | 610 ± 10 | 1.4 ± 0.2 | 898 ± 40 | 2.4 ± 0.3 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sigolaeva, L.V.; Bulko, T.V.; Konyakhina, A.Y.; Kuzikov, A.V.; Masamrekh, R.A.; Max, J.B.; Köhler, M.; Schacher, F.H.; Pergushov, D.V.; Shumyantseva, V.V. Rational Design of Amphiphilic Diblock Copolymer/MWCNT Surface Modifiers and Their Application for Direct Electrochemical Sensing of DNA. Polymers 2020, 12, 1514. https://doi.org/10.3390/polym12071514
Sigolaeva LV, Bulko TV, Konyakhina AY, Kuzikov AV, Masamrekh RA, Max JB, Köhler M, Schacher FH, Pergushov DV, Shumyantseva VV. Rational Design of Amphiphilic Diblock Copolymer/MWCNT Surface Modifiers and Their Application for Direct Electrochemical Sensing of DNA. Polymers. 2020; 12(7):1514. https://doi.org/10.3390/polym12071514
Chicago/Turabian StyleSigolaeva, Larisa V., Tatiana V. Bulko, Apollinariya Yu. Konyakhina, Alexey V. Kuzikov, Rami A. Masamrekh, Johannes B. Max, Moritz Köhler, Felix H. Schacher, Dmitry V. Pergushov, and Victoria V. Shumyantseva. 2020. "Rational Design of Amphiphilic Diblock Copolymer/MWCNT Surface Modifiers and Their Application for Direct Electrochemical Sensing of DNA" Polymers 12, no. 7: 1514. https://doi.org/10.3390/polym12071514