Complementary Color Tuning by HCl via Phosphorescence-to-Fluorescence Conversion on Insulated Metallopolymer Film and Its Light-Induced Acceleration


Abstract
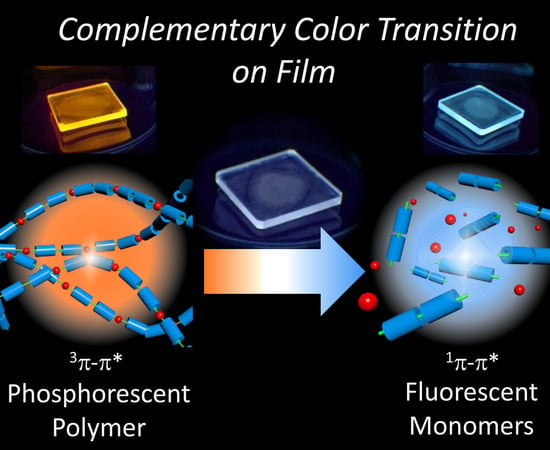
:
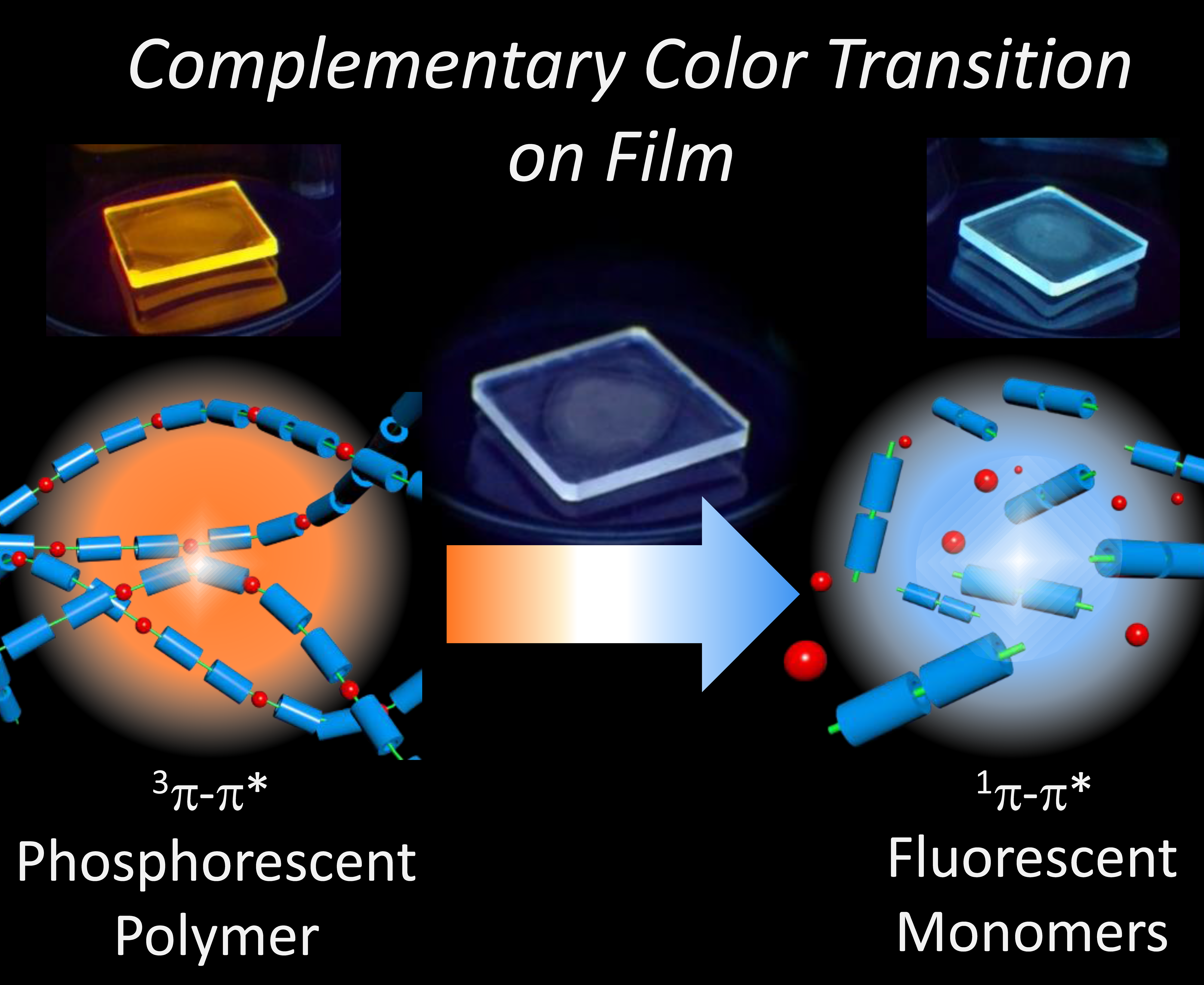{kind=link}
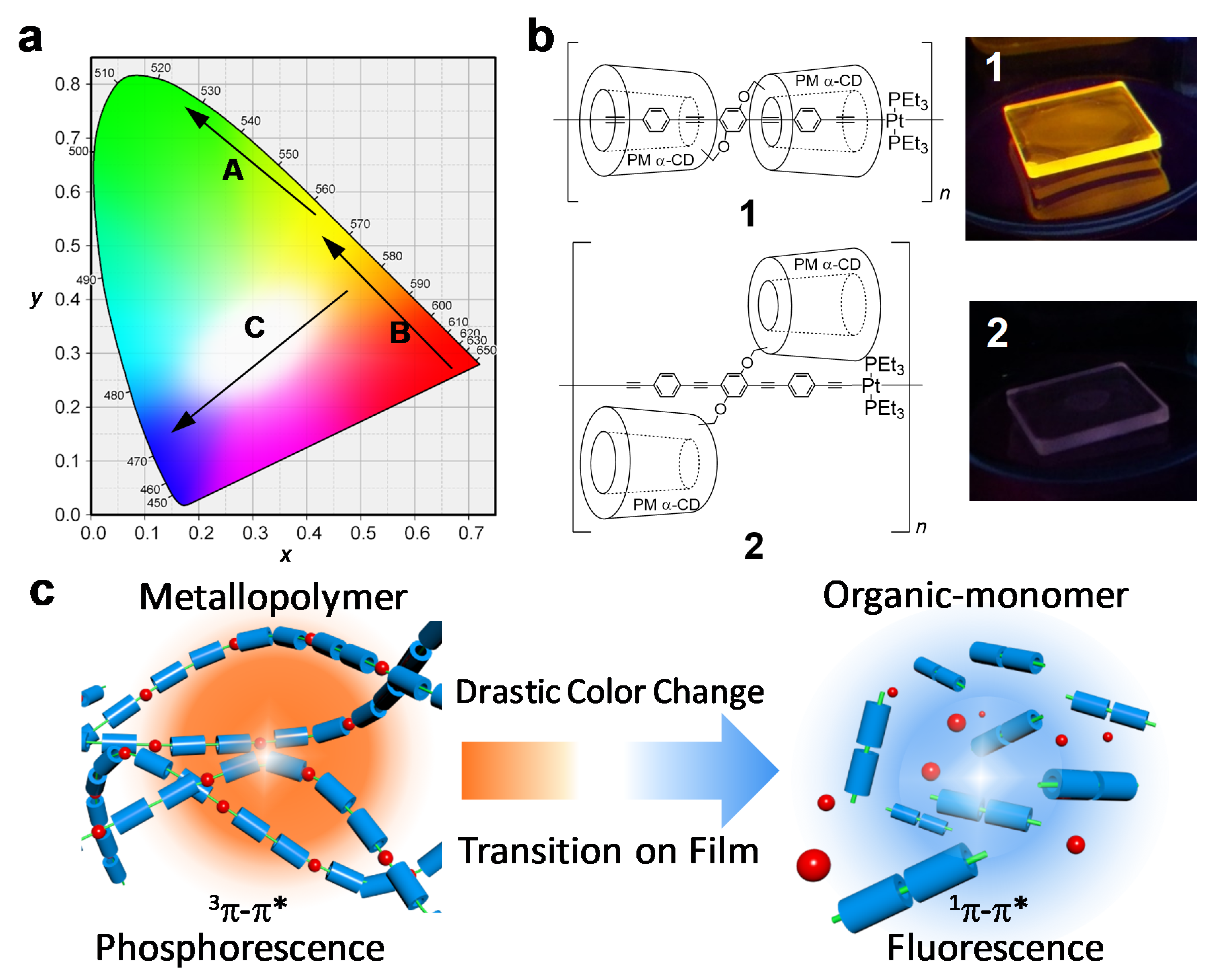{kind=link}
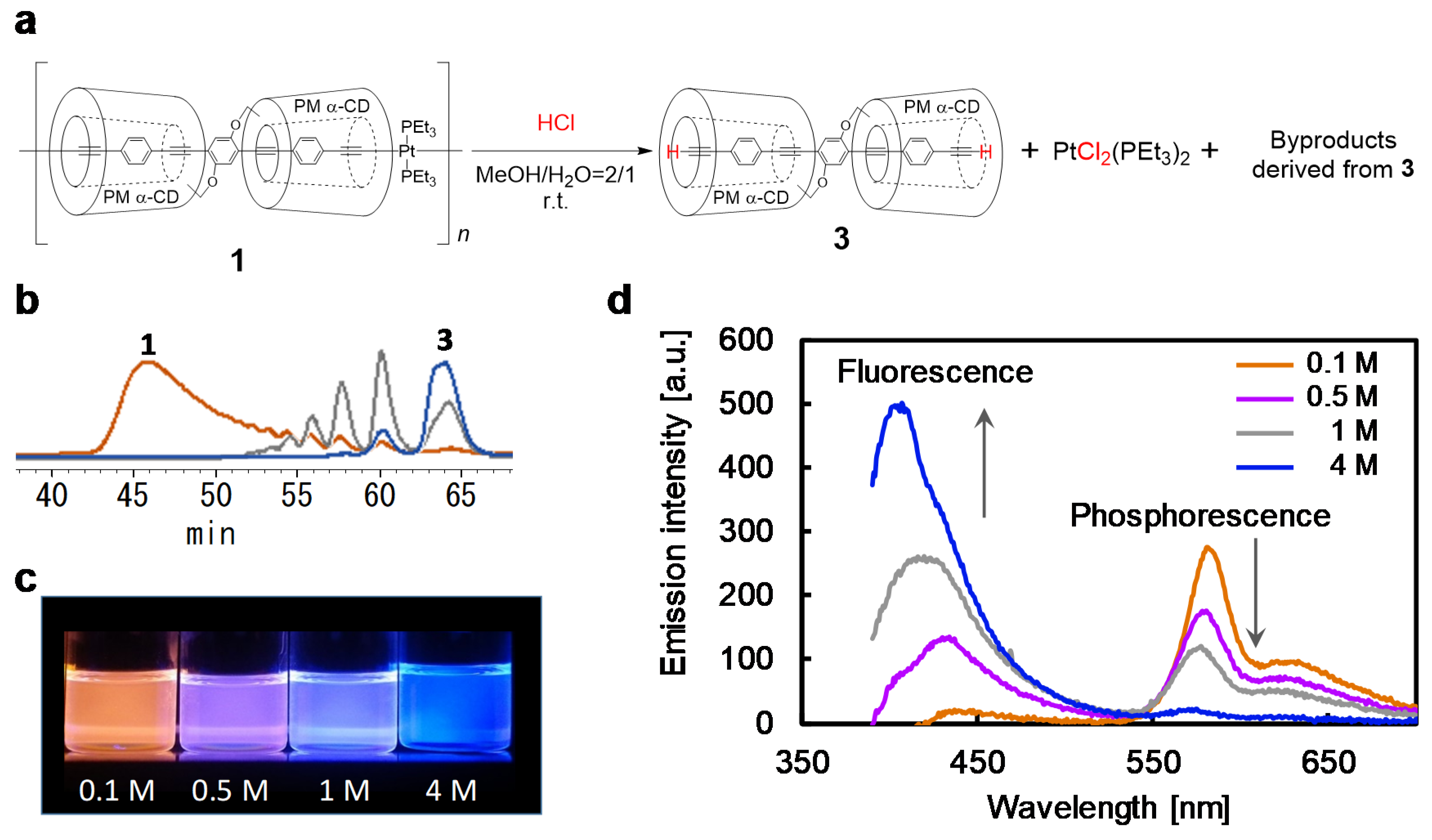{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis of 4

2.3. General Procedure of HCl Depolymerizing Experiment in the Solution State
2.4. General Procedure of Depolymerizing Experiment in the Solid State
3. Results and Discussion
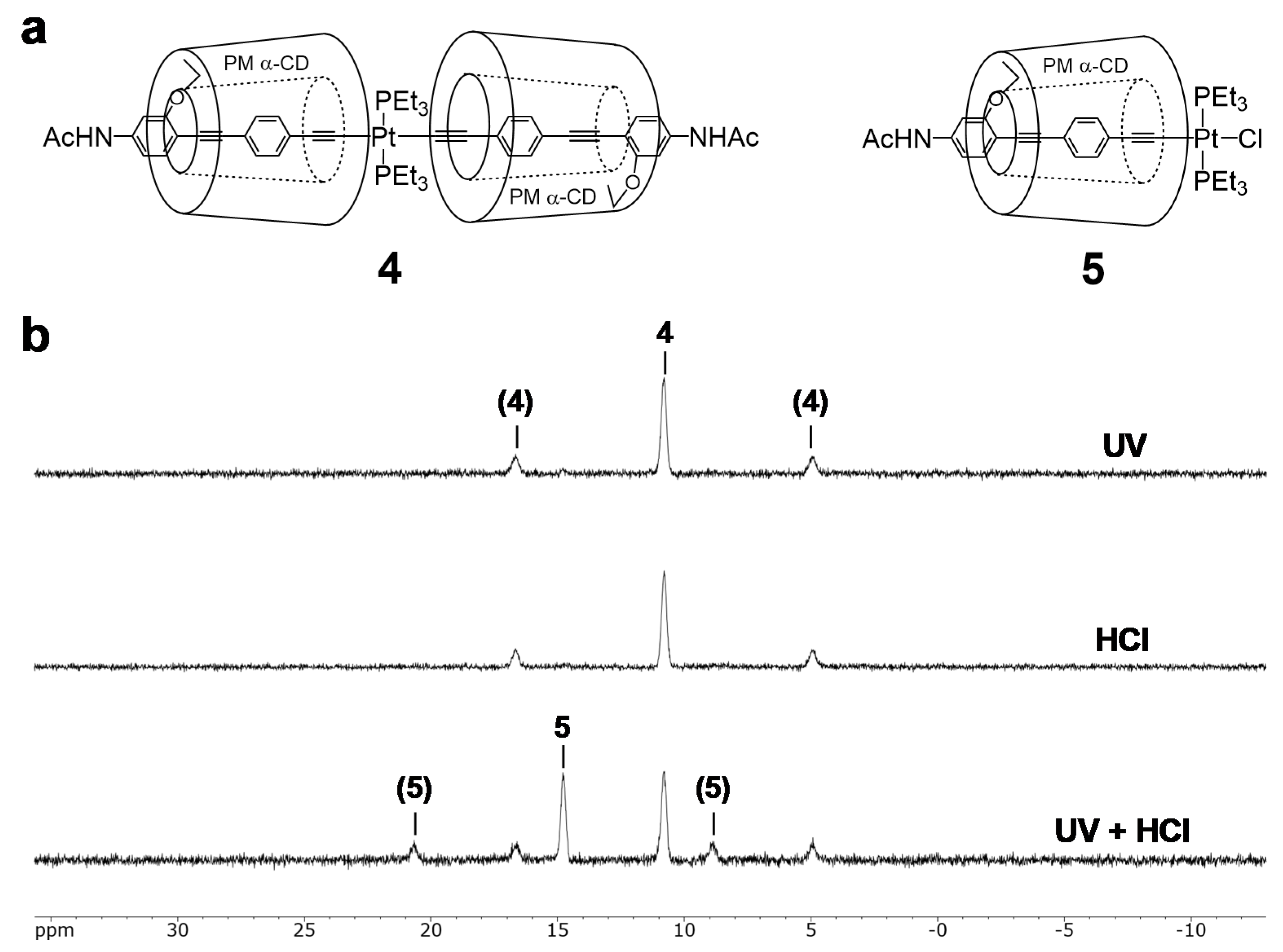
3.1. Depolymerizing Reaction of Pt-Acetylide Polymer 1 and 2
3.2. Color-Tunability in the Solution State
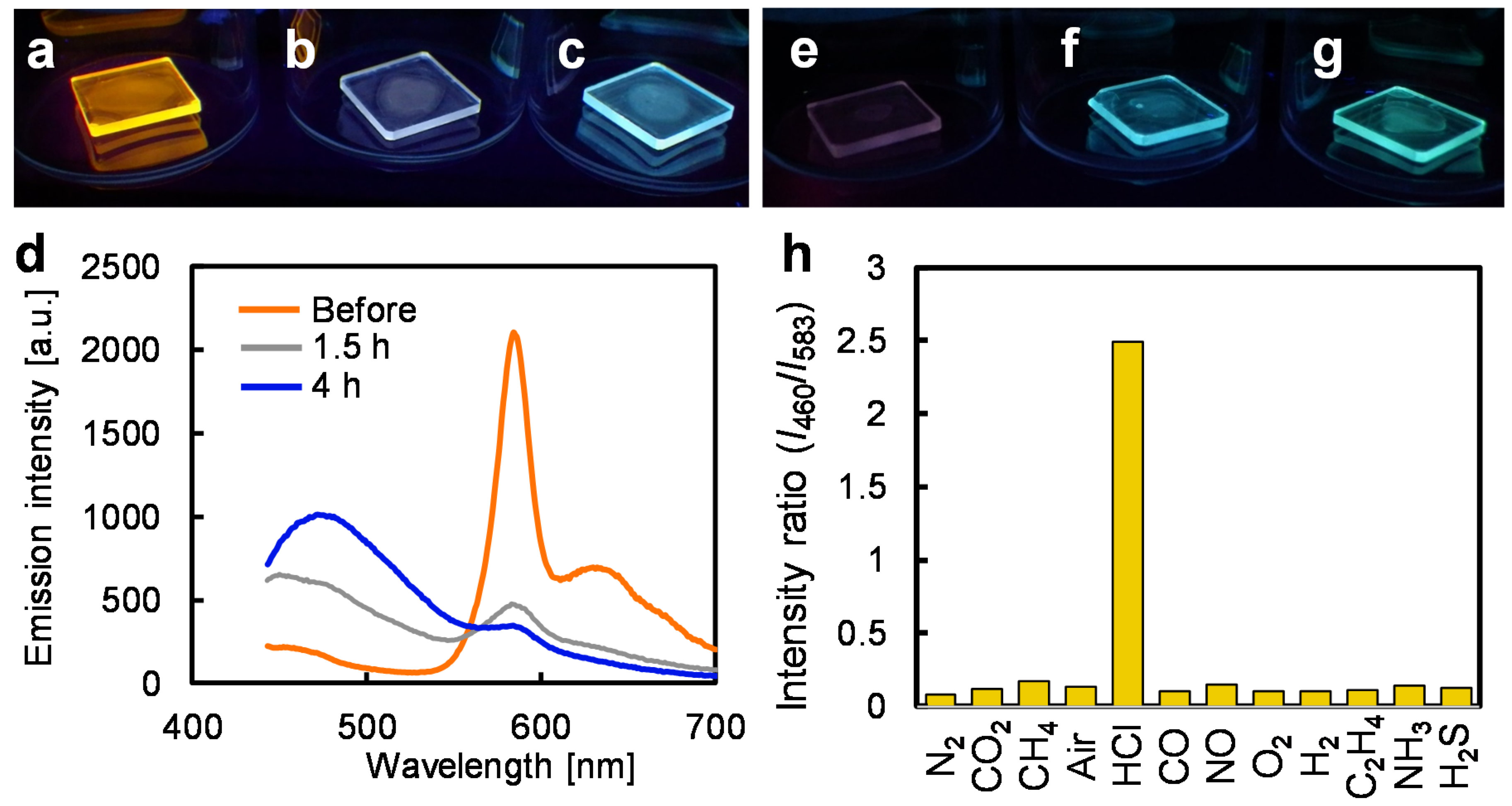
3.3. Depolymerizing Reaction in the Solid State
3.4. Chemospecific Reactivity for Pt–Acetylide Bonds with HCl Gas
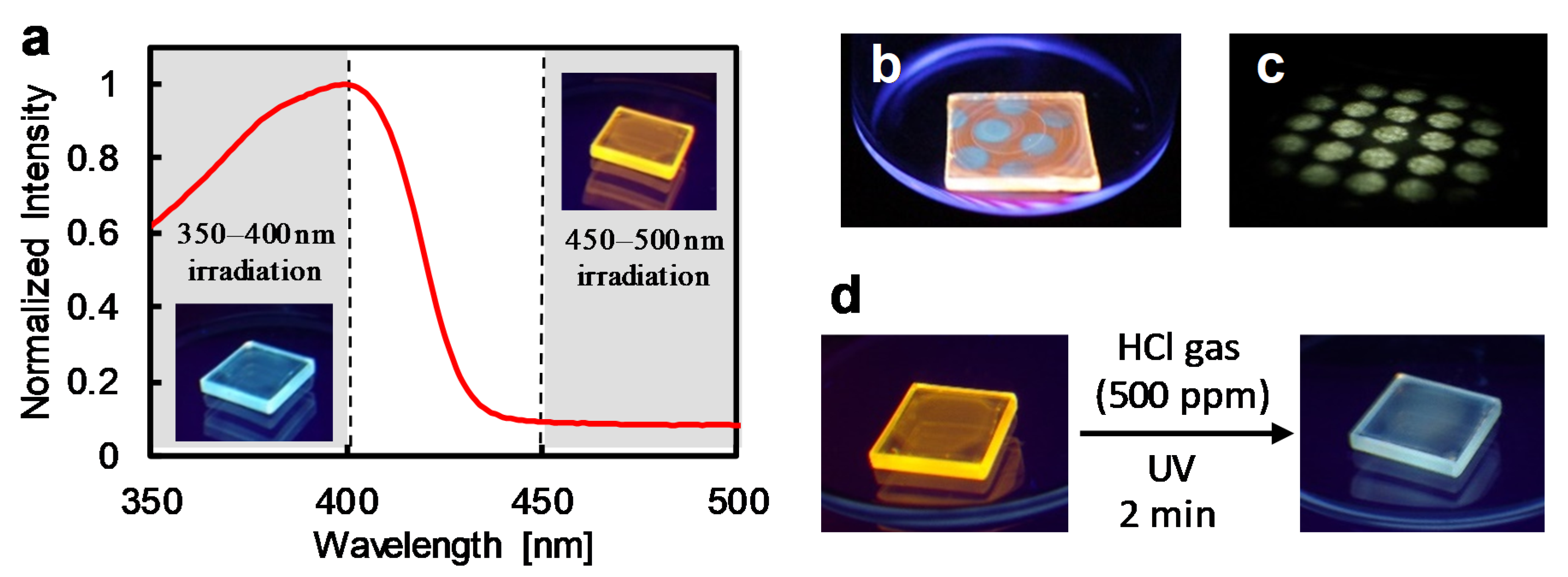
3.5. Light-Induced Acceleration for the Depolymerization
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lim, S.H.; Feng, L.; Kemling, J.W.; Musto, C.J.; Suslick, K.S. An optoelectronic nose for the detection of toxic gases. Nat. Chem. 2009, 1, 562–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wenger, O.S. Vapochromism in Organometallic and Coordination Complexes: Chemical Sensors for Volatile Organic Compounds. Chem. Rev. 2013, 113, 3686–3733. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.-L.; Ma, V.P.-Y.; Chan, D.S.-H.; Leung, K.-H.; He, H.-Z.; Leung, C.-H. Recent advances in luminescent heavy metal complexes for sensing. Coord. Chem. Rev. 2012, 256, 3087–3113. [Google Scholar] [CrossRef]
- Yoon, B.; Lee, J.; Park, I.S.; Jeon, S.; Lee, J.; Kim, J.-M. Recent functional material based approaches to prevent and detect counterfeiting. J. Mater. Chem. C 2013, 1, 2388. [Google Scholar] [CrossRef]
- Hou, X.; Ke, C.; Bruns, C.J.; McGonigal, P.R.; Pettman, R.B.; Stoddart, J.F. Tunable solid-state fluorescent materials for supramolecular encryption. Nat. Commun. 2015, 6, 6884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guan, W.; Zhou, W.; Lu, J.; Lu, C. Luminescent films for chemo- and biosensing. Chem. Soc. Rev. 2015, 44, 6981–7009. [Google Scholar] [CrossRef]
- Sun, X.; Chen, T.; Huang, S.; Li, L.; Peng, H. Chromatic polydiacetylene with novel sensitivity. Chem. Soc. Rev. 2010, 39, 4244–4257. [Google Scholar] [CrossRef]
- Pridmore, R.W. Complementary colors theory of color vision: Physiology, color mixture, color constancy and color perception. Color Res. Appl. 2011, 36, 394–412. [Google Scholar] [CrossRef]
- Heo, J.H.; Cho, H.H.; Leea, J.W.; Lee, J.H. Achromatic-chromatic colorimetric sensors for on-off type detection of analytes. Analyst 2014, 139, 6486–6493. [Google Scholar] [CrossRef]
- Kido, J.; Kimura, M.; Nagai, K. Multilayer white light-emitting organic electroluminescent device. Science 1995, 267, 1332–1334. [Google Scholar] [CrossRef] [Green Version]
- Fleetham, T.; Li, G.; Li, J. Phosphorescent Pt(II) and Pd(II) Complexes for Efficient, High-Color-Quality, and Stable OLEDs. Adv. Mater. 2017, 29, 1601861. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Kwon, J.E.; Kim, S.H.; Seo, J.; Chung, K.; Park, S.-Y.; Jang, D.-J.; Medina, B.M.; Gierschner, J.; Park, S.Y. White-Light-Emiting Molecule: Frustrated Energy Transfer between Constituent Emitting Centors. J. Am. Chem. Soc. 2009, 131, 14043–14049. [Google Scholar] [CrossRef] [PubMed]
- Fleetham, T.; Ecton, J.; Wang, Z.; Bakken, N.; Li, J. Single-doped white organic light-emitting device with an external quantum efficiency over 20%. Adv. Mater. 2013, 25, 2573–2576. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Li, Q.; Grindy, S.; Holten-Andersen, N. White-Light-Emitting Lanthanide Metallogels with Tunable Luminescence and Reversible Stimuli-Responsive Properties. J. Am. Chem. Soc. 2015, 137, 1159–11593. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, K.; Murai, T.; Guo, J.D.; Sasamori, T.; Tokitoh, N. Acid-Responsive Absorption and Emission of 5-N-Arylaminothiazoles: Emission of White Light from a Single Fluorescent Dye and a Lewis Acid. ChemistryOpen 2016, 5, 396. [Google Scholar] [CrossRef]
- Radotić, K.; Melø, T.B.; Leblanc, R.M.; Yousef, Y.A.; Naqvi, K.R. Fluorescence and phosphorescence of tryptophan in peptides of different length and sequence. J. Photochem. Photobiol. B Biol. 2016, 157, 120–128. [Google Scholar] [CrossRef]
- Xu, R.; Wang, Y.; Duan, X.; Lu, K.; Micheroni, D.; Hu, A.; Lin, W. Nanoscale Metal–Organic Frameworks for Ratiometric Oxygen Sensing in Live Cells. J. Am. Chem. Soc. 2016, 138, 2158–2161. [Google Scholar] [CrossRef] [Green Version]
- Zang, L.; Zhao, H.; Hua, J.; Qin, F.; Zheng, Y.; Zhang, Z.; Cao, W. Ratiometric dissolved oxygen sensitive indicator based on lutetium labeled hematoporphyrin monomethyl ether with balanced phosphorescence and fluorescence dual emission. Sens. Actuators B Chem. 2016, 231, 539–546. [Google Scholar] [CrossRef]
- Wu, H.; Hang, C.; Li, X.; Yin, L.; Zhu, M.; Zhang, J.; Zhou, Y.; Ågren, H.; Zhang, Q.; Zhu, L. Molecular stacking dependent phosphorescence–fluorescence dual emission in a single luminophore for self-recoverable mechanoconversion of multicolor luminescence. Chem. Commun. 2017, 53, 2661–2664. [Google Scholar] [CrossRef]
- Mao, Z.; Yang, Z.; Mu, Y.; Zhang, Y.; Wang, Y.F.; Chi, Z.; Lo, C.C.; Liu, S.; Lien, A.; Xu, J. Linearly Tunable Emission Colors Obtained from a Fluorescent-Phosphorescent Dual-Emission Compound by Mechanical Stimuli. Angew. Chem. Int. Ed. Engl. 2015, 54, 6270–6273. [Google Scholar] [CrossRef]
- Gong, S.; Yang, C.; Qin, J. Efficient phosphorescent polymer light-emitting diodes by suppressing triplet energy back transfer. Chem. Soc. Rev. 2012, 41, 4797–4807. [Google Scholar] [CrossRef] [PubMed]
- Terao, J.; Wadahama, A.; Matono, A.; Tada, T.; Watanabe, S.; Seki, S.; Fujihara, T.; Tsuji, Y. Design principle for increasing charge mobility of π-conjugated polymers using regularly localized molecular orbitals. Nat. Commun. 2013, 4, 1691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masai, H.; Terao, J.; Seki, S.; Nakashima, S.; Kiguchi, M.; Okoshi, K.; Fujihara, T.; Tsuji, Y. Synthesis of One-Dimensional Metal-Containing Insulated Molecular Wire with Versatile Properties Directed toward Molecular Electronics Materials. J. Am. Chem. Soc. 2014, 136, 1742–1745. [Google Scholar] [CrossRef] [PubMed]
- Masai, H.; Terao, J.; Makuta, S.; Tachibana, Y.; Fujihara, T.; Tsuji, Y. Enhancement of Phosphorescence and Unimolecular Behavior in the Solid State by Perfect Insulation of Platinum—Acetylide Polymers. J. Am. Chem. Soc. 2014, 136, 14714–14717. [Google Scholar] [CrossRef] [PubMed]
- Masai, H.; Terao, J. Stimuli-responsive functionalized insulated conjugated polymers. Polym. J. 2017, 49, 805–814. [Google Scholar] [CrossRef]
- Hosomi, T.; Masai, H.; Fujihara, T.; Tsuji, Y.; Terao, J. A Typical Metal-Ion-Responsive Color-Tunable Emitting Insulated π-Conjugated Polymer Film. Angew. Chem. Int. Ed. 2016, 55, 13427–13431. [Google Scholar] [CrossRef]
- Miyagishi, H.V.; Tamaki, T.; Masai, H.; Terao, J. Synthesis and Acid-Responsiveness of an Insulatedπ-Conjugated Polymer Containing Spiropyrans in Its Backbone. Molecules 2019, 24, 1301. [Google Scholar] [CrossRef] [Green Version]
- Russell, G.M.; Inamori, D.; Masai, H.; Tamaki, T.; Terao, J. Luminescent and mechanical enhancement of phosphorescent hydrogel through cyclic insulation of platinum-acetylide crosslinker. Polym. Chem. 2019, 10, 5280. [Google Scholar] [CrossRef]
- Perera, S.D.; Shaw, B.L. Complexes including acetylides formed from 3-diphenylphosphinocamphor and platinum or palladium. J. Organomet. Chem. 1991, 402, 133–138. [Google Scholar] [CrossRef]
- Liu, Y.; Jiang, S.; Glusac, K.; Powell, D.H.; Anderson, D.F.; Schanze, K.S. Photophysics of Monodisperse Platinum-Acetylide Oligomers: Delocalizaton in the Singlet and Triplet Excited States. J. Am. Chem. Soc. 2002, 124, 12412–12413. [Google Scholar] [CrossRef]
- Terao, J.; Masai, H.; Fujihara, T.; Tsuji, Y. Synthesis of Insulated Pt—Alkynyl Complex Polymer. Chem. Lett. 2012, 41, 652–653. [Google Scholar] [CrossRef]
- Terao, J.; Tsuda, S.; Tanaka, Y.; Okoshi, K.; Fujihara, T.; Tsuji, Y.; Kambe, N. Synthesis of Organic-Soluble Conjugated Polyrotaxanes by Polymerization of Linked Rotaxanes. J. Am. Chem. Soc. 2009, 131, 16004–16005. [Google Scholar] [CrossRef] [PubMed]
- Masai, H.; Terao, J.; Fujihara, T.; Tsuji, Y. Rational design for rotaxane synthesis through intramolecular slippage: Control of activation energy by rigid axle length. Chem. A Eur. J. 2016, 22, 6624–6630. [Google Scholar] [CrossRef] [PubMed]
- Rahn, J.A.; Nelson, J.H.; Baltusis, L. Solid-state structures of platinum phosphine, (R3P)2PtX2 complexes as determined by a combination of 13C{1H} and 31P{1H} NMR spectroscopy. Inorg. Chem. 1999, 29, 750–755. [Google Scholar] [CrossRef]
- Sebald, A.; Stader, C.; Wrackmeyer, B.; Bensch, W. Alkynyl/chloride exchange between trans-platinum(II) and -palladium(II) chlorides and alkynylstannanes. Crystalstructure of trans-[bis(1-propynyl)-bis(triethylphosphine)platinum(II)]. J. Organomet. Chem. 1986, 311, 233–241. [Google Scholar] [CrossRef]
- Zhou, X.; Lee, S.; Xu, Z.; Yoon, J. Recent Progress on the Development of Chemosensors for Gases. Chem. Rev. 2015, 115, 7944. [Google Scholar]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaneko, S.; Masai, H.; Yokoyama, T.; Liu, M.; Tachibana, Y.; Fujihara, T.; Tsuji, Y.; Terao, J. Complementary Color Tuning by HCl via Phosphorescence-to-Fluorescence Conversion on Insulated Metallopolymer Film and Its Light-Induced Acceleration. Polymers 2020, 12, 244. https://doi.org/10.3390/polym12010244
Kaneko S, Masai H, Yokoyama T, Liu M, Tachibana Y, Fujihara T, Tsuji Y, Terao J. Complementary Color Tuning by HCl via Phosphorescence-to-Fluorescence Conversion on Insulated Metallopolymer Film and Its Light-Induced Acceleration. Polymers. 2020; 12(1):244. https://doi.org/10.3390/polym12010244
Chicago/Turabian StyleKaneko, Shunichi, Hiroshi Masai, Takuya Yokoyama, Maning Liu, Yasuhiro Tachibana, Tetsuaki Fujihara, Yasushi Tsuji, and Jun Terao. 2020. "Complementary Color Tuning by HCl via Phosphorescence-to-Fluorescence Conversion on Insulated Metallopolymer Film and Its Light-Induced Acceleration" Polymers 12, no. 1: 244. https://doi.org/10.3390/polym12010244