A Mechanistic Physiologically-Based Biopharmaceutics Modeling (PBBM) Approach to Assess the In Vivo Performance of an Orally Administered Drug Product: From IVIVC to IVIVP

 , , ,
, , ,
Abstract
:
1. Introduction
2. Materials and Methods
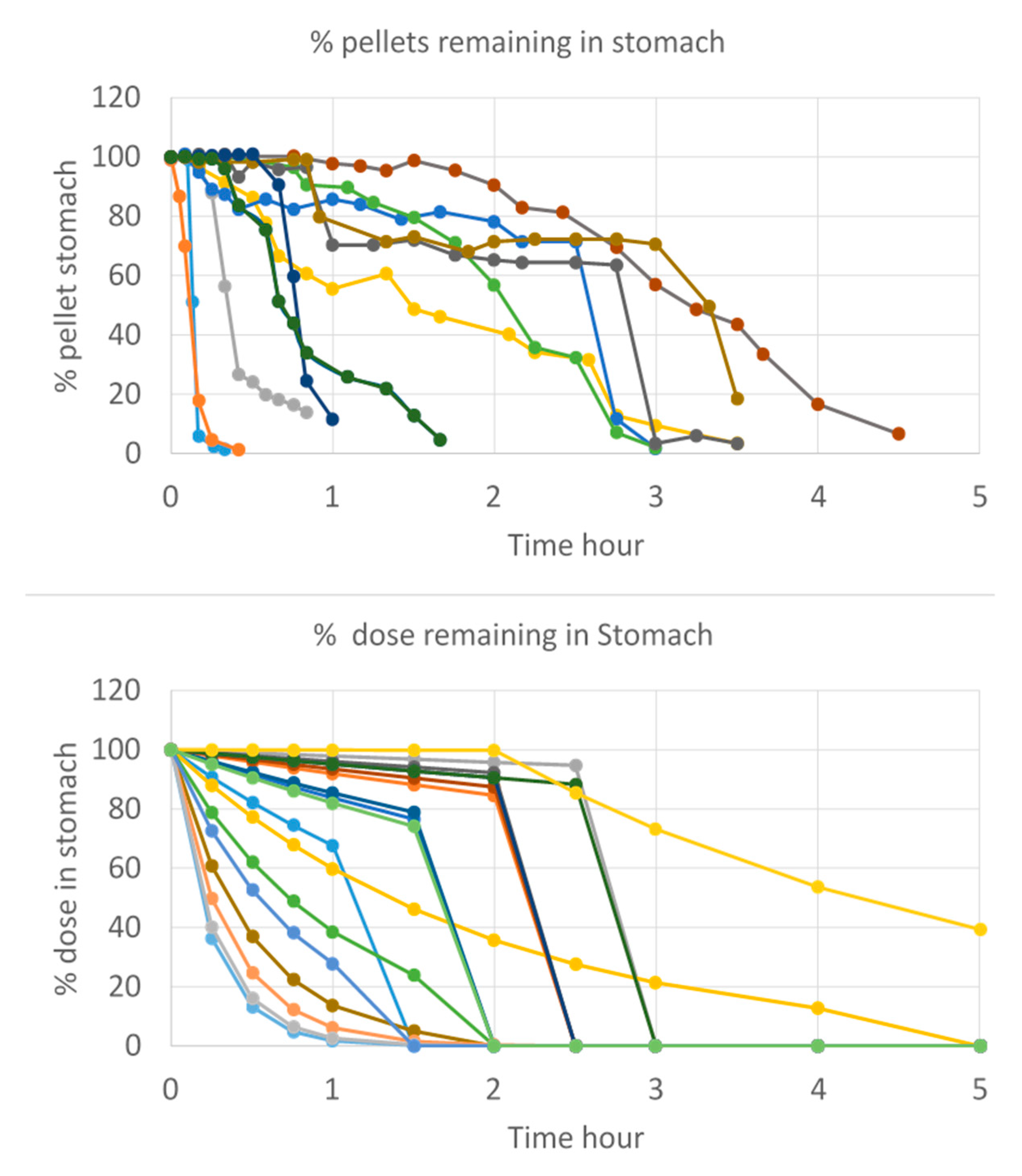
2.1. Reference Intraluminal and Systemic Data of Ibuprofen
2.1.1. Intraluminal and Systemic Profiling of Ibuprofen in Healthy Volunteers
2.1.2. Recording of Post-Dose Phase III Contractions
2.1.3. Thermodynamic Equilibrium Solubility of Ibuprofen in Fasted-State Human Gastric and Intestinal Fluids (Fahgf/Fahif)
2.1.4. Bioanalysis of Ibuprofen by HPLC
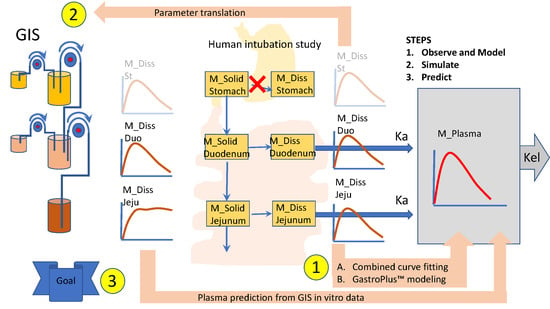
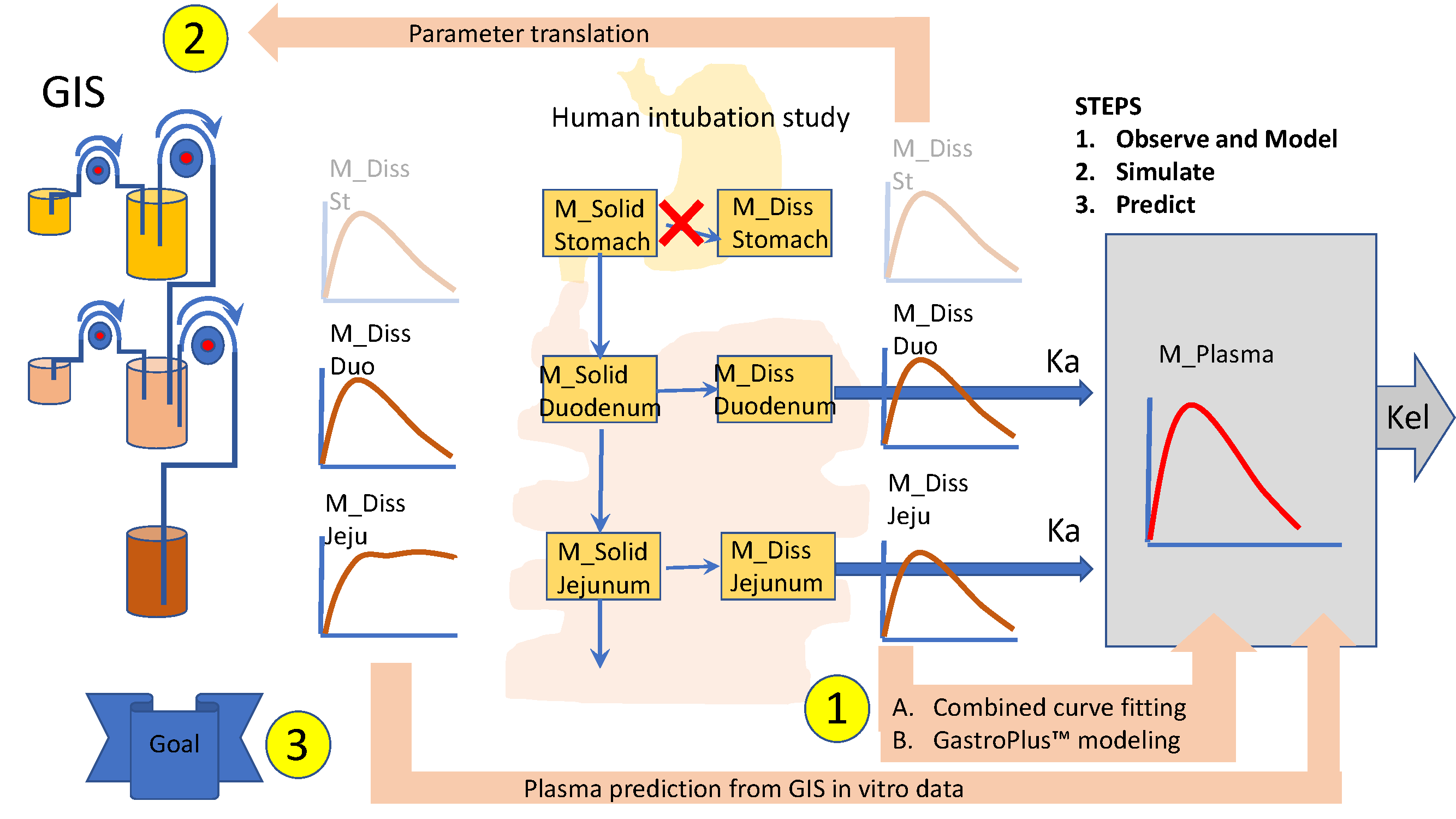
2.2. Mechanistic Oral Absorption Modeling in Phoenix WinNonlin®
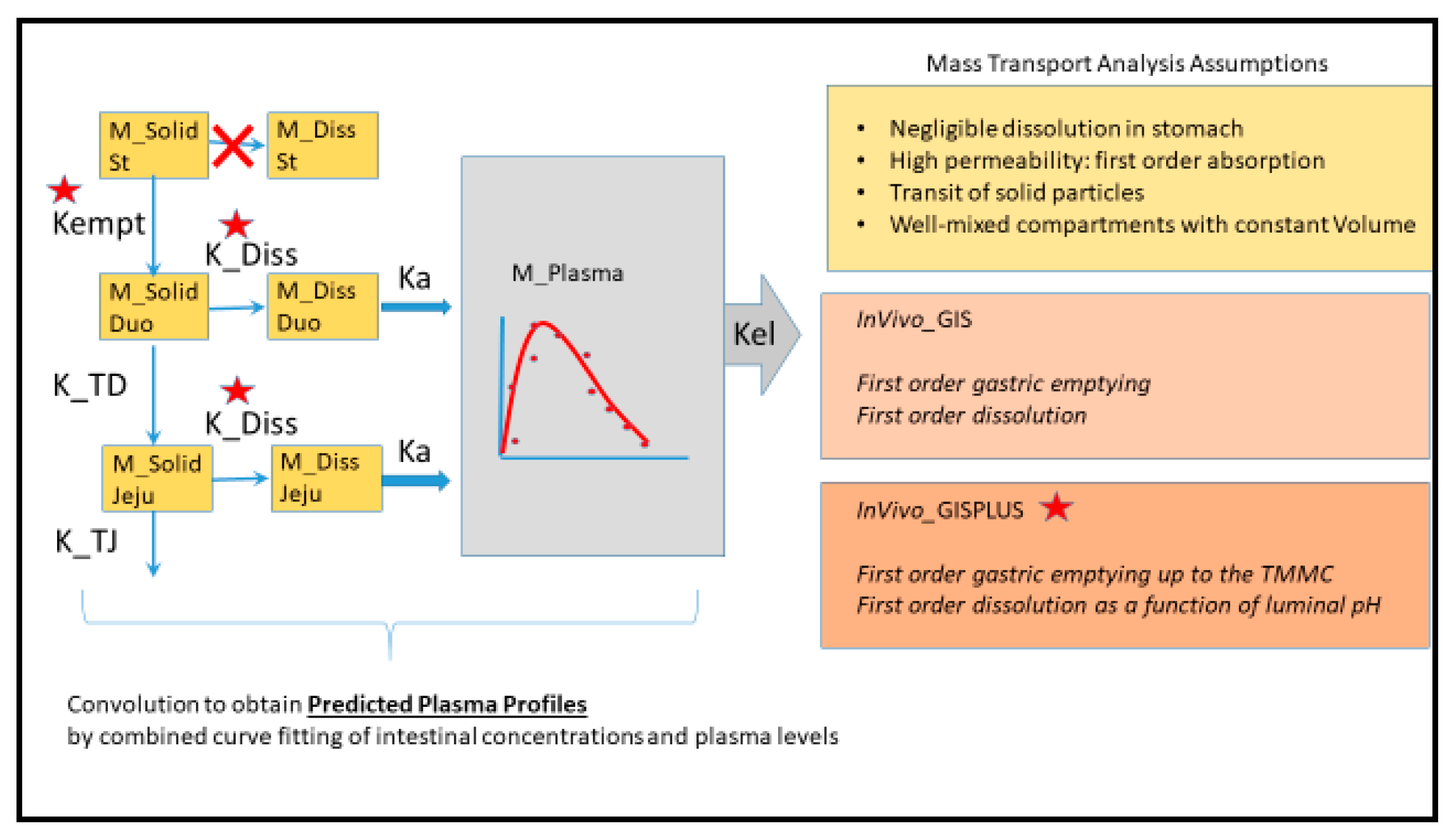
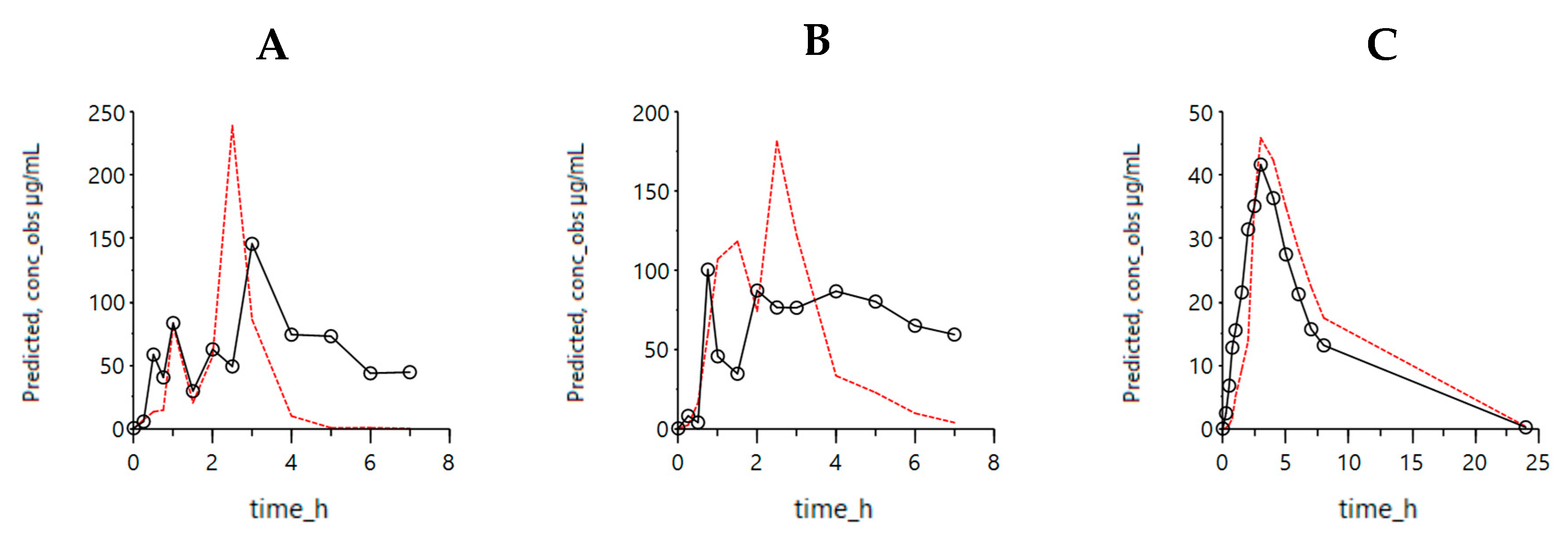
WinNonlin User-Customized Mechanistic Model to Stress the Pivotal Underlying GI Variables: InVivo_GIS versus InVivo_GISPlus
- Ibuprofen dissolution was considered negligible in the stomach chamber due to the acidic pH (pH < pKa). The administered oral tablet disintegrates in the stomach and particles will not be dissolved but emptied in the next segment, i.e., the duodenum. In addition, no significant absorption can occur from the stomach.
- Gastric emptying follows a first-order kinetic process.
- Dissolution follows a first-order process in the duodenal and jejunal segment. The dissolution rates are proportional to the remaining amount of solid ibuprofen.
- Duodenal and jejunal compartments are well-mixed, resulting in homogenous drug concentrations.
- The permeability of the intestinal membrane is high for ibuprofen, indicating that dissolved ibuprofen will be immediately absorbed. Only solid particles transit from the duodenum to the jejunum.
- Transit from the duodenum to the jejunum is faster than the transit from the jejunum to the more distal parts.
- Drug degradation does not occur in the GI lumen.
- Gastric emptying follows a first-order kinetic up to the next post-dose phase III contractions when all the remaining stomach content is suddenly emptied in the duodenum.
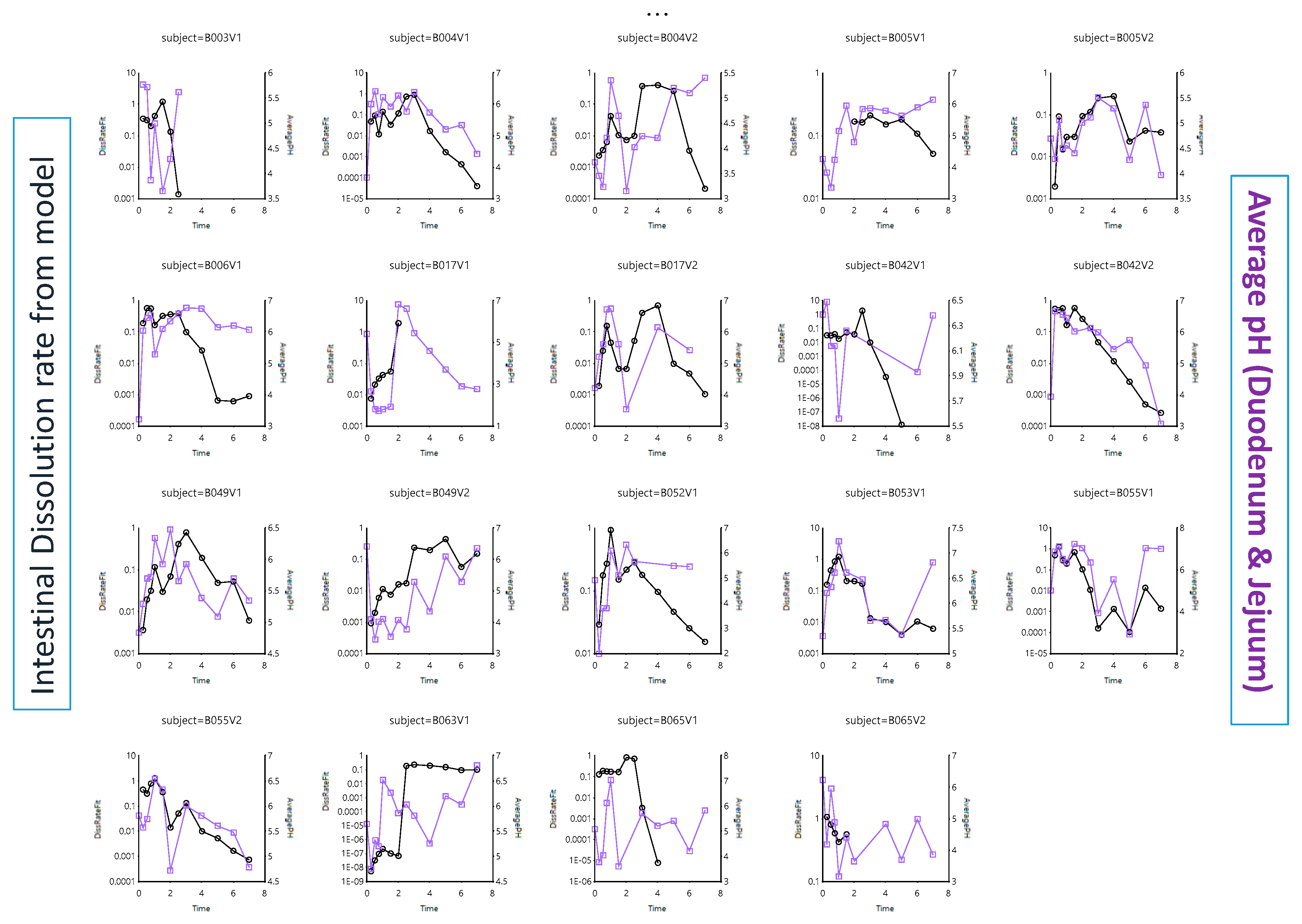
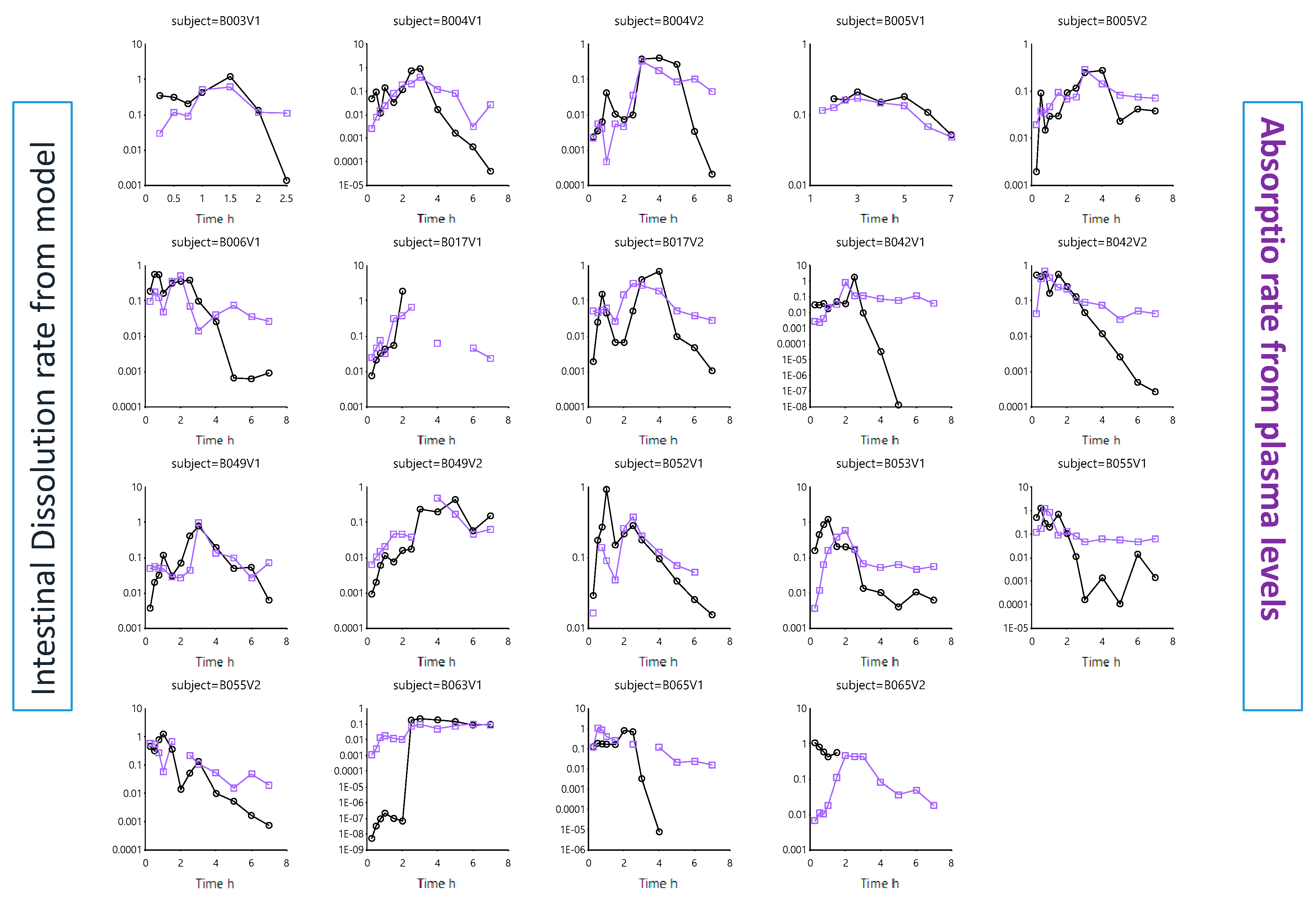
- Dissolution follows a first-order process in the duodenal and jejunal segment. The dissolution rates are proportional to the remaining amount of solid ibuprofen. The dissolution rate is modeled as a function of luminal pH values. Ibuprofen solubility is re-calculated at each time point with the duodenum or jejunal pH at that specific moment.
- Duodenal and jejunum average fluid volume values during the sampling time, V1 and V2, respectively;
- Transit rate coefficients from the duodenal to the jejunal segment, K_TD; and from the jejunum to the more distal segment, K_TJ;
- First-order rate coefficient of gastric emptying, Kempt;
- Dissolution rate coefficient, K_Diss;
- First-order absorption rate constants, Ka.
2.3. Mechanistic Oral Absorption Modeling in GastroPlus™
2.3.1. GastroPlus™ Advanced Compartmental Absorption Transit (ACAT™) Mechanistic Absorption Model
2.3.2. Data Presentation
3. Results and Discussion
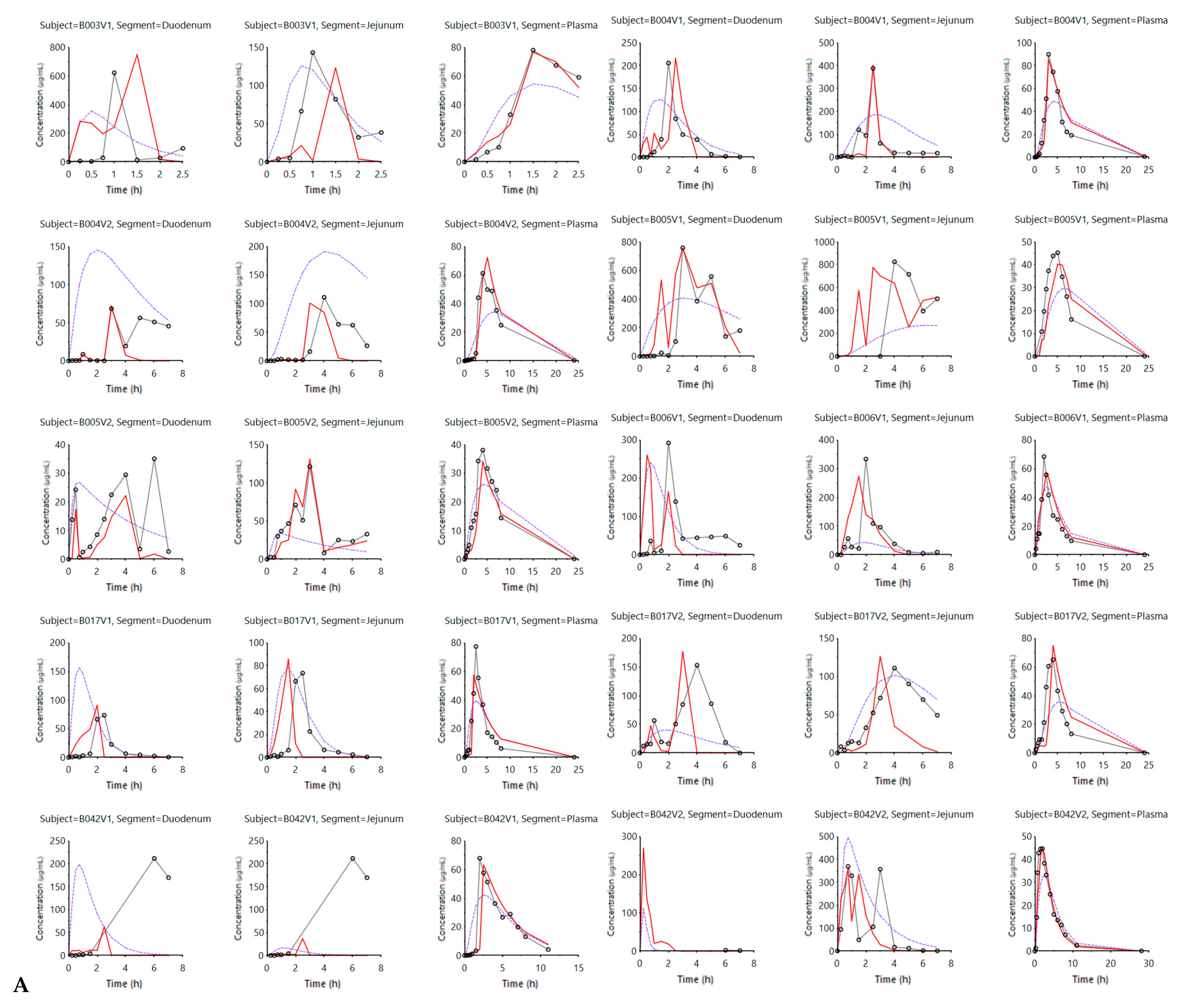
3.1. Mechanistic Oral Absorption Modeling in WinNonlin
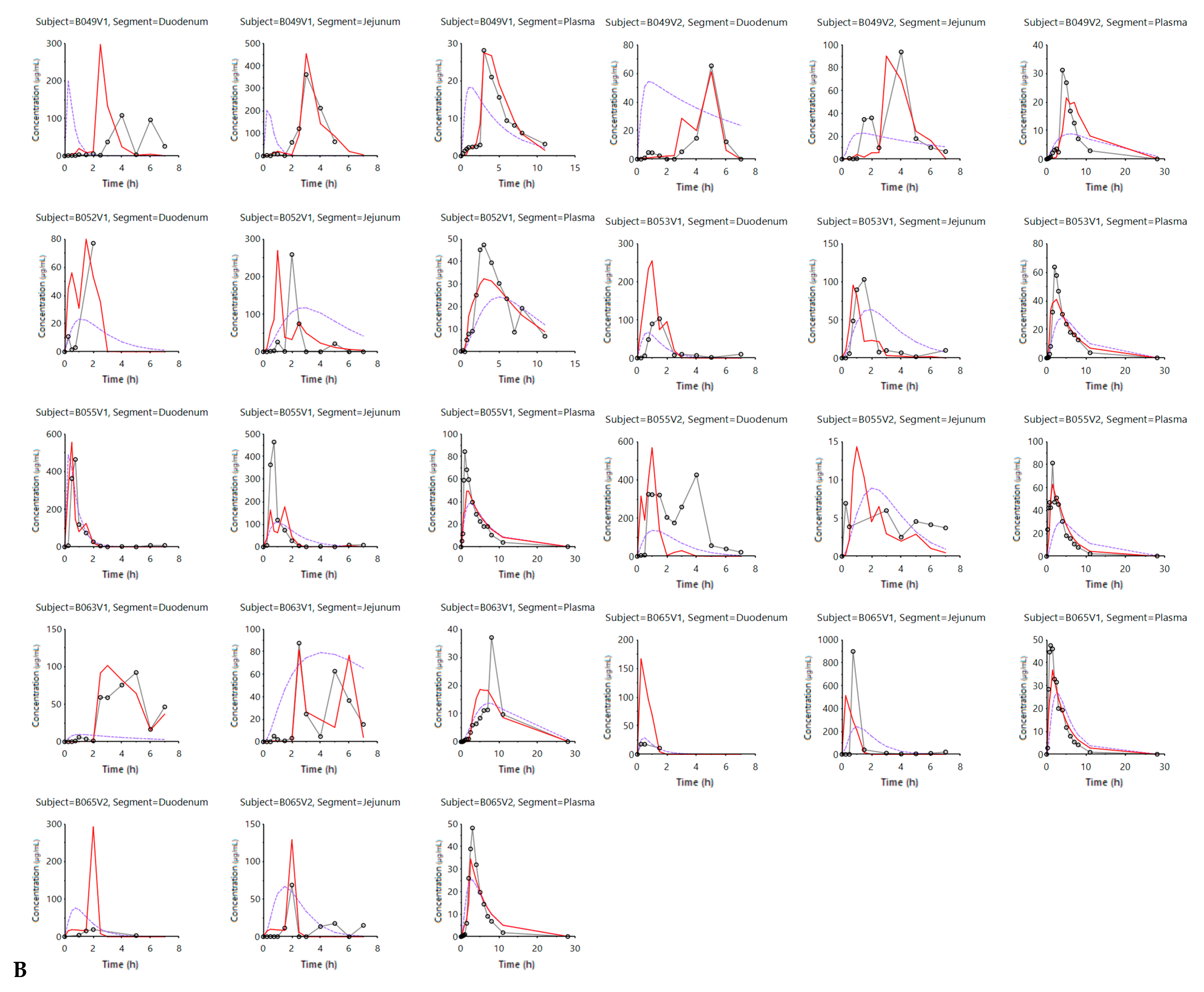
3.2. Mechanistic Oral Absorption Modeling in GastroPlus™
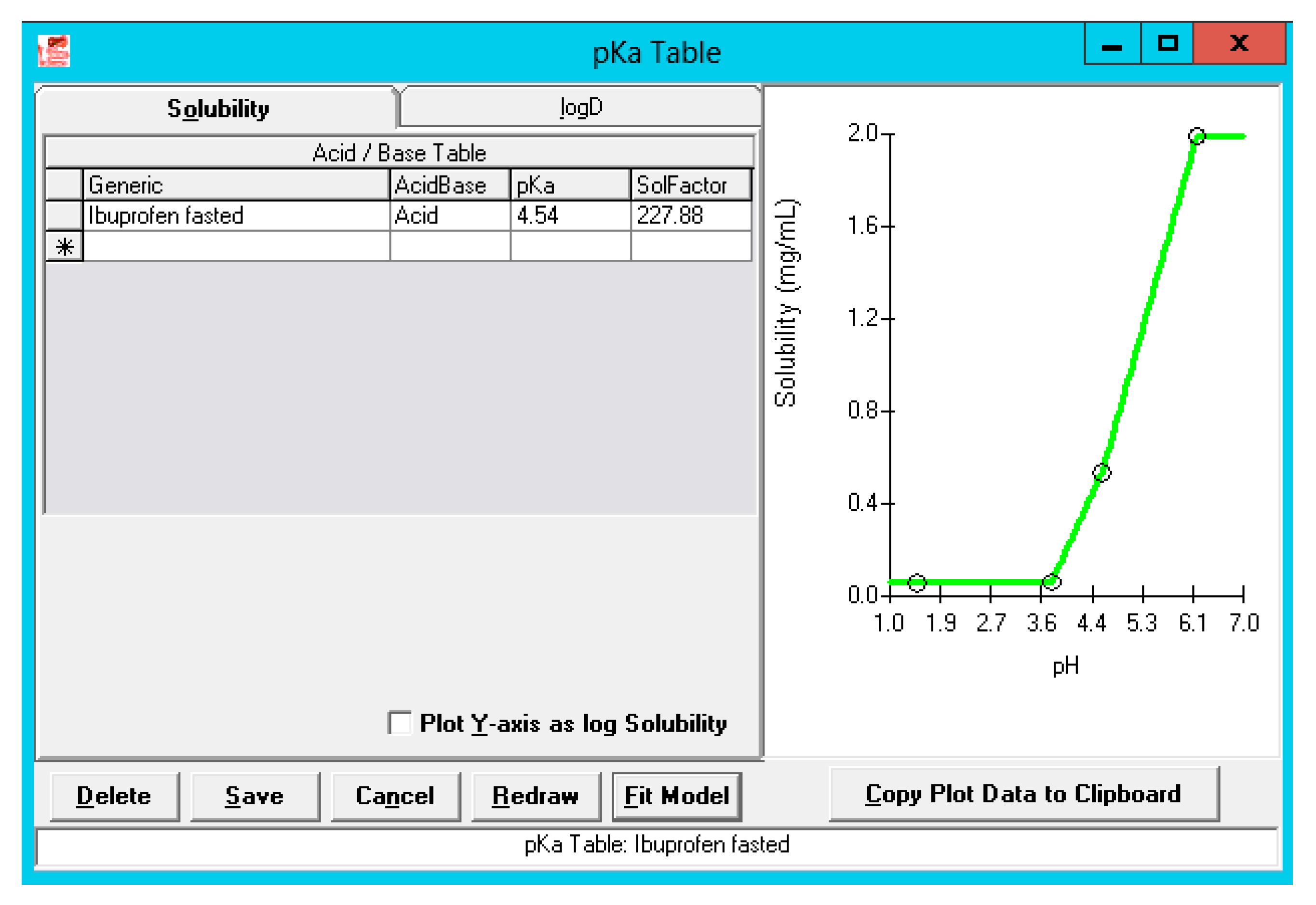
3.2.1. Solubility versus pH: pH-Driven Dissolution
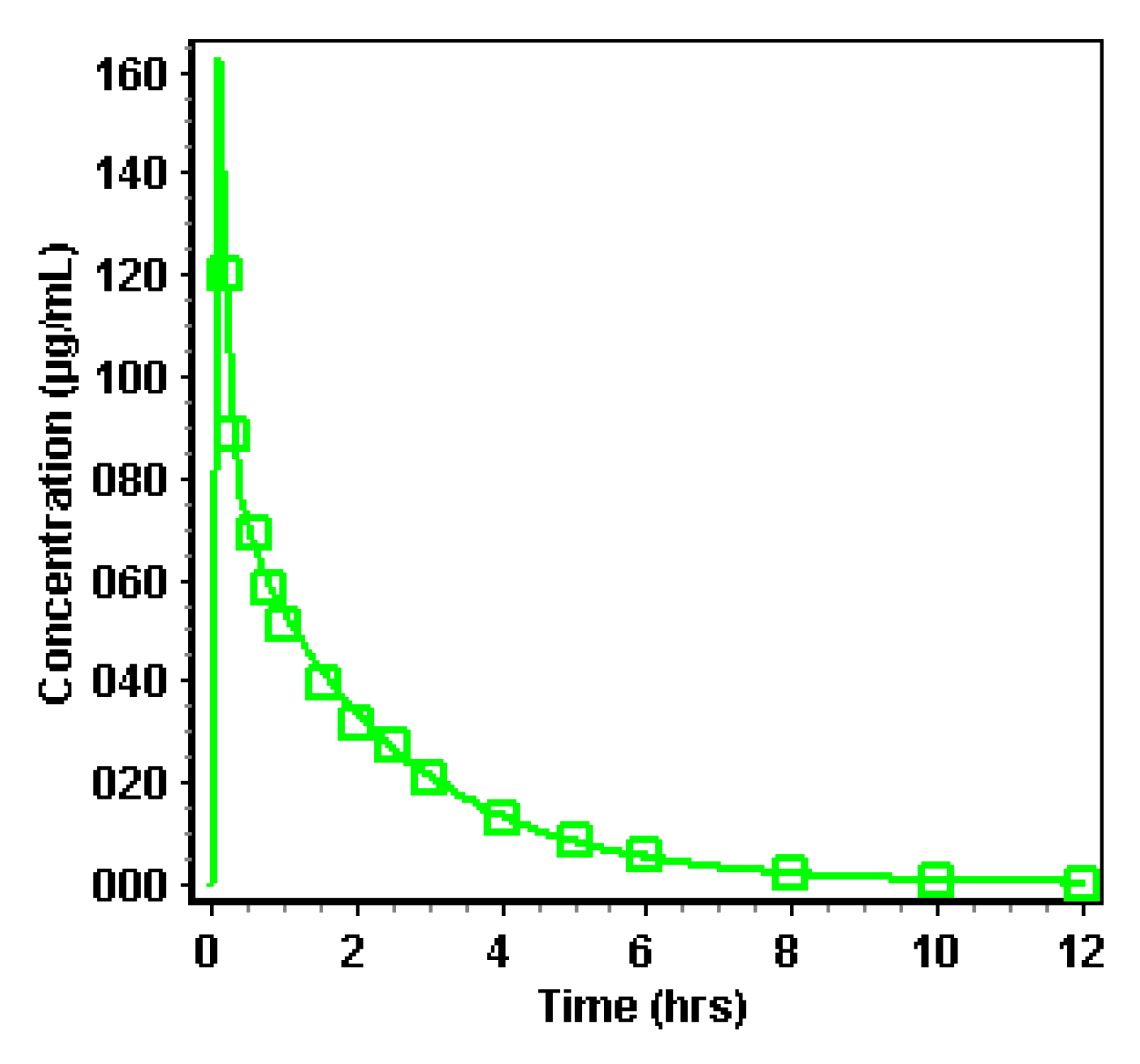
3.2.2. Simulation of Distribution and Clearance of Ibuprofen: A Two-Compartmental Pharmacokinetic (PK) Approach
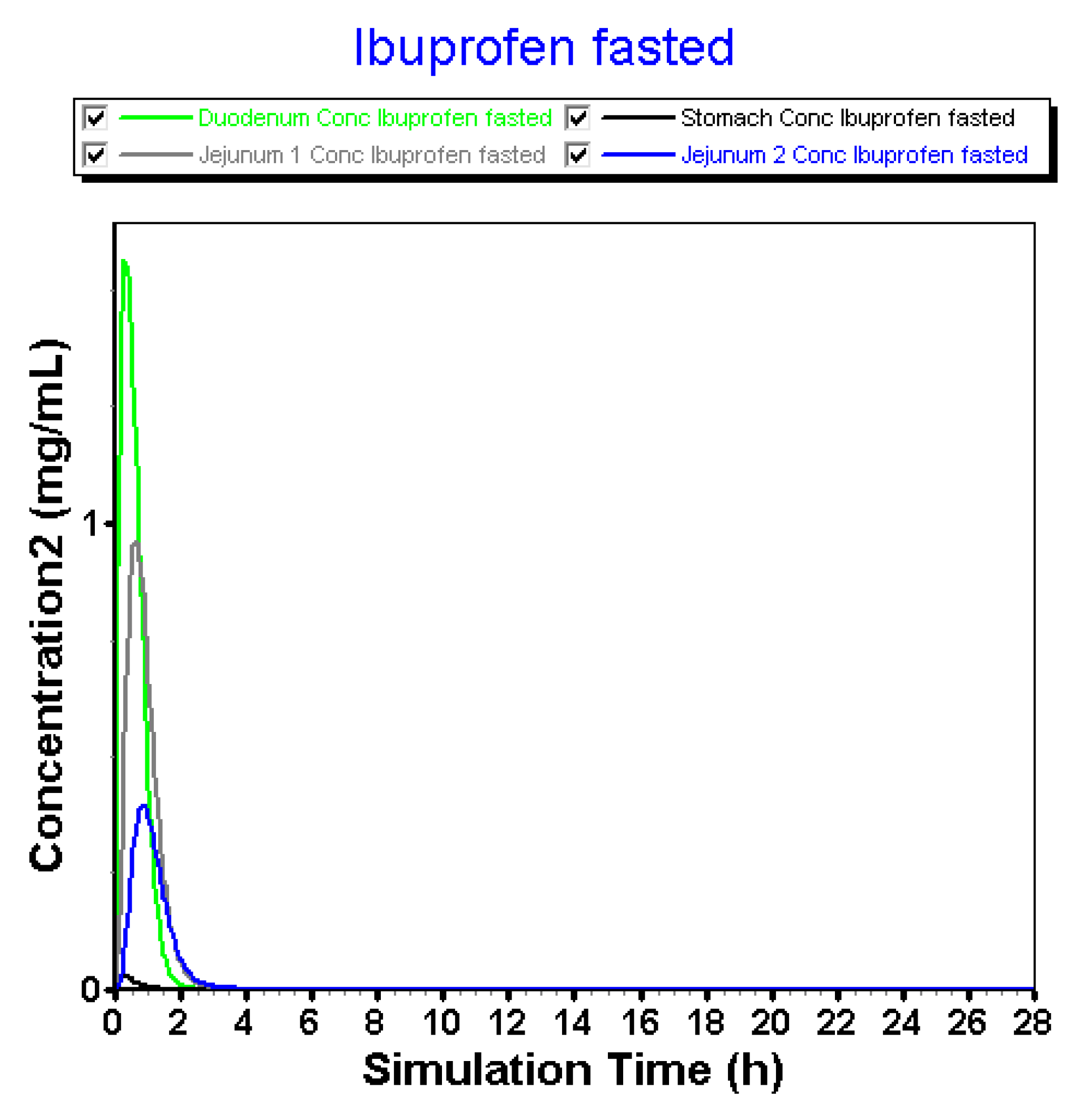
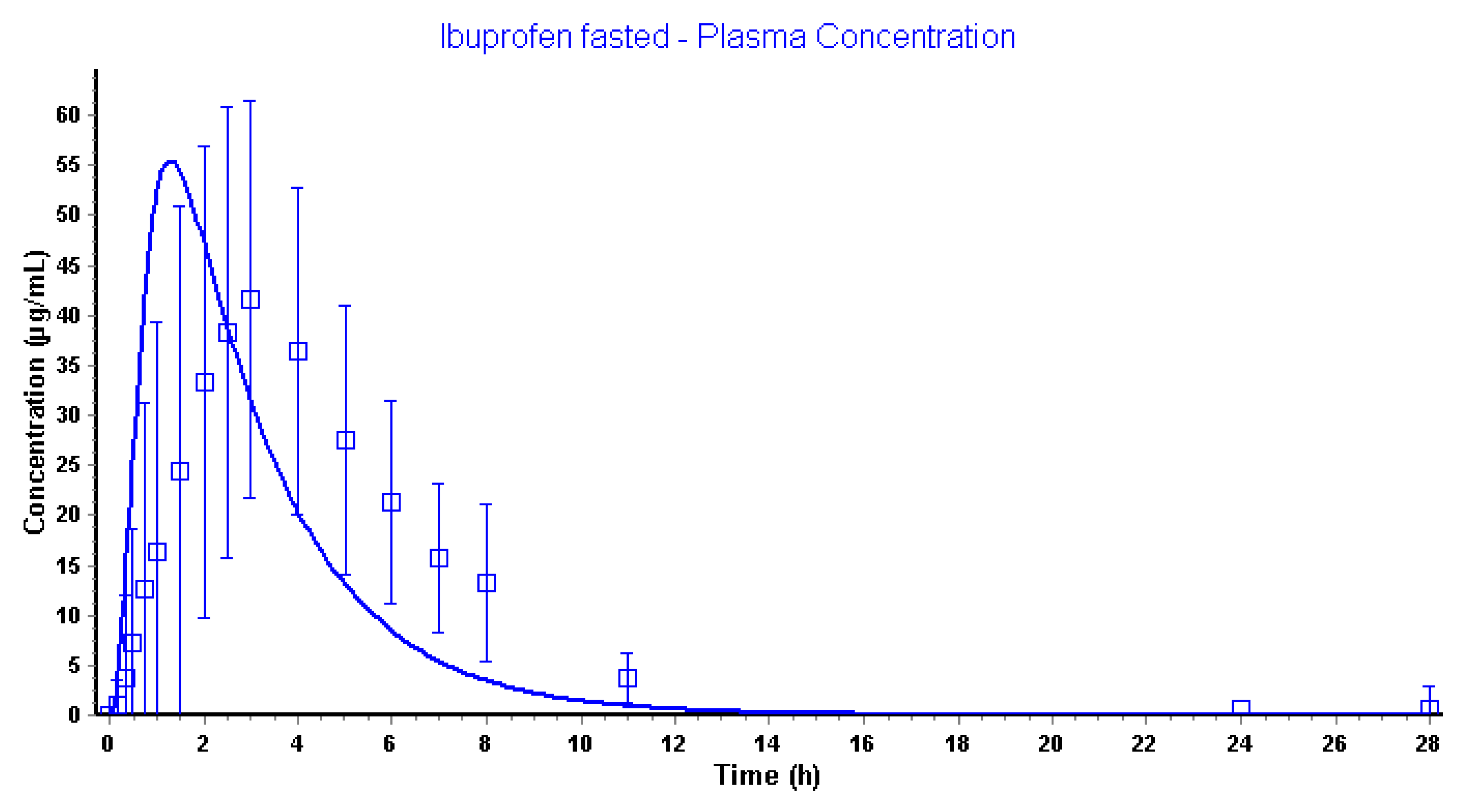
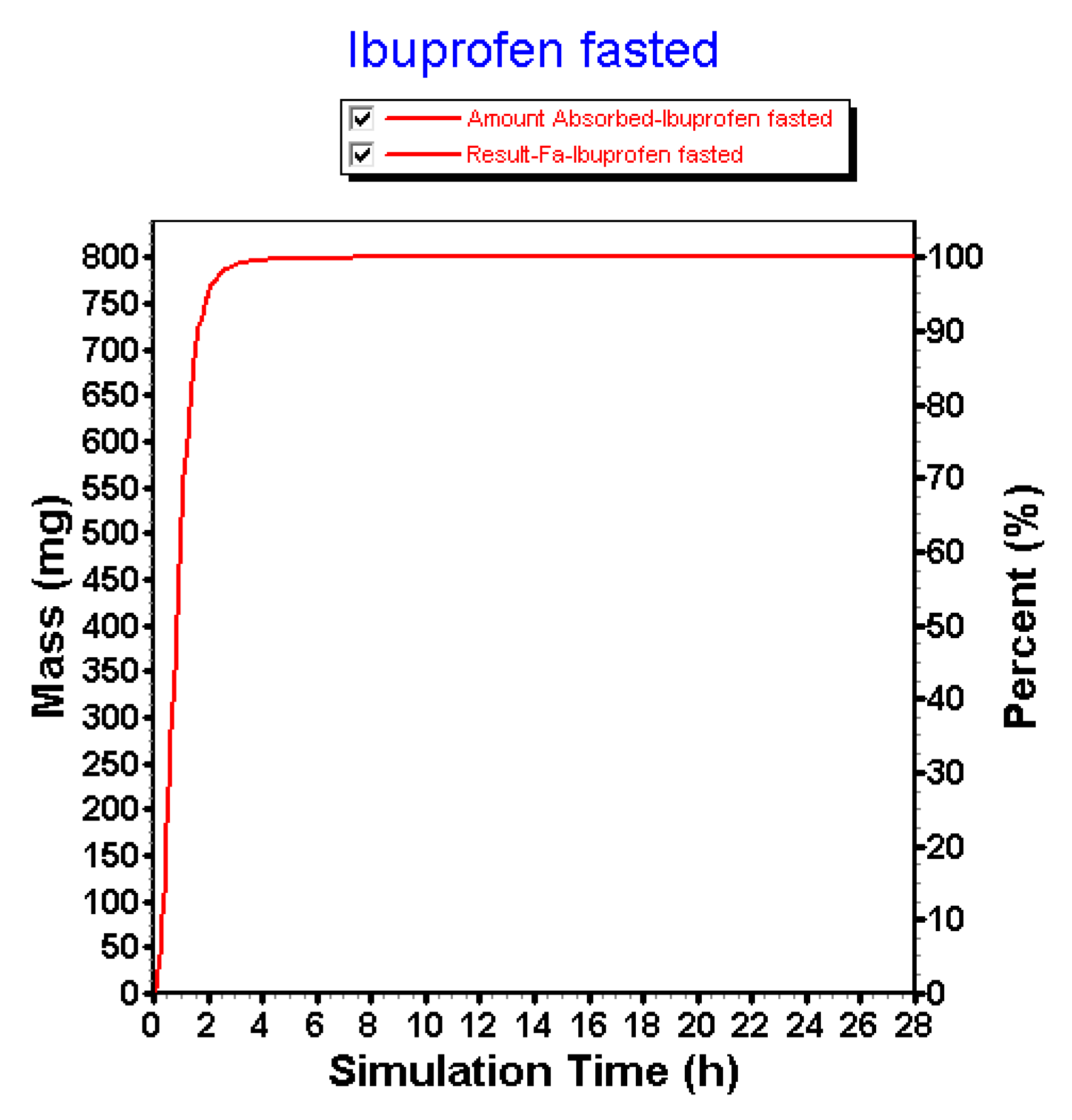
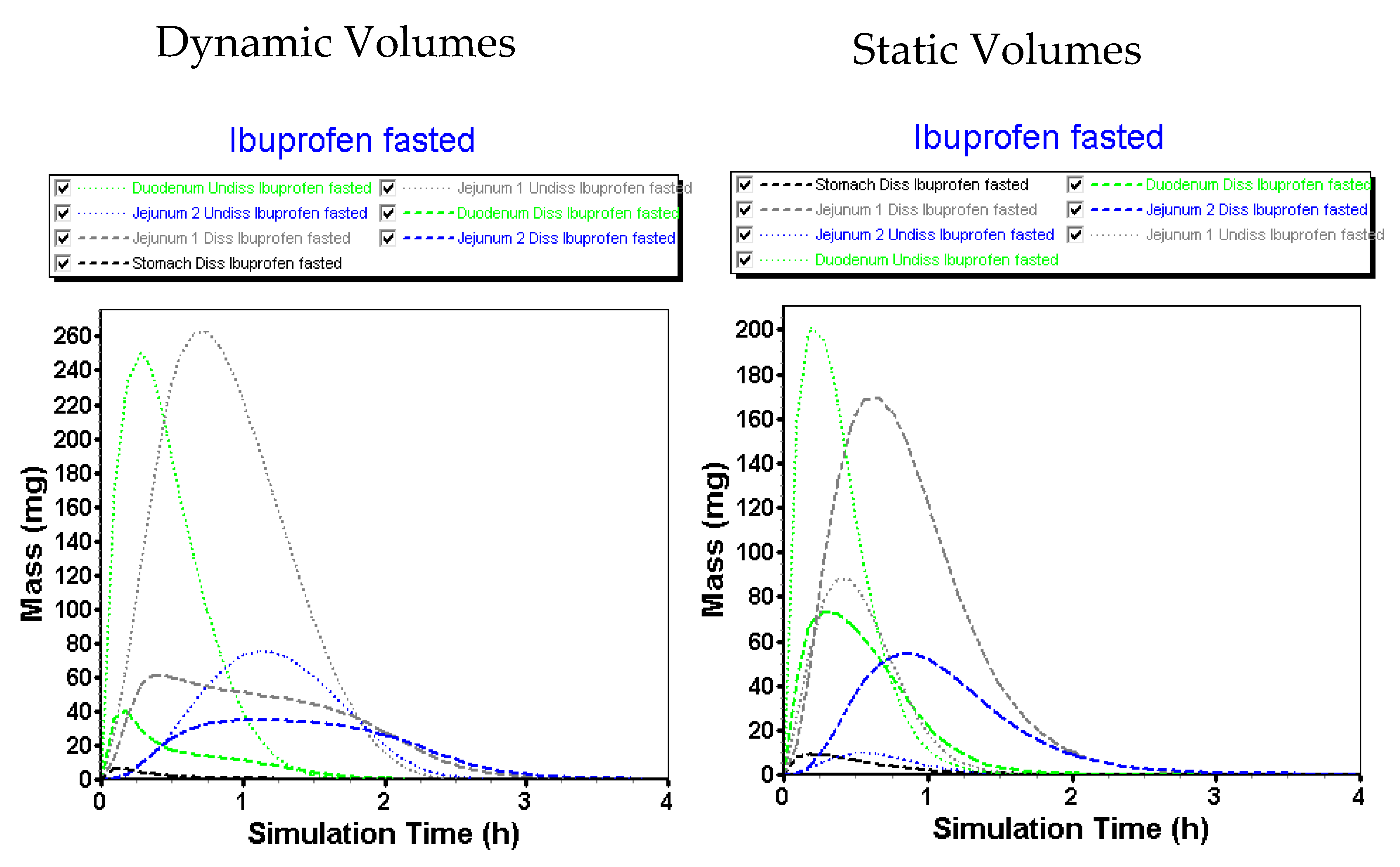
3.2.3. Advanced Compartmental Absorption and Transit Simulations: Static Simulations with Default Settings
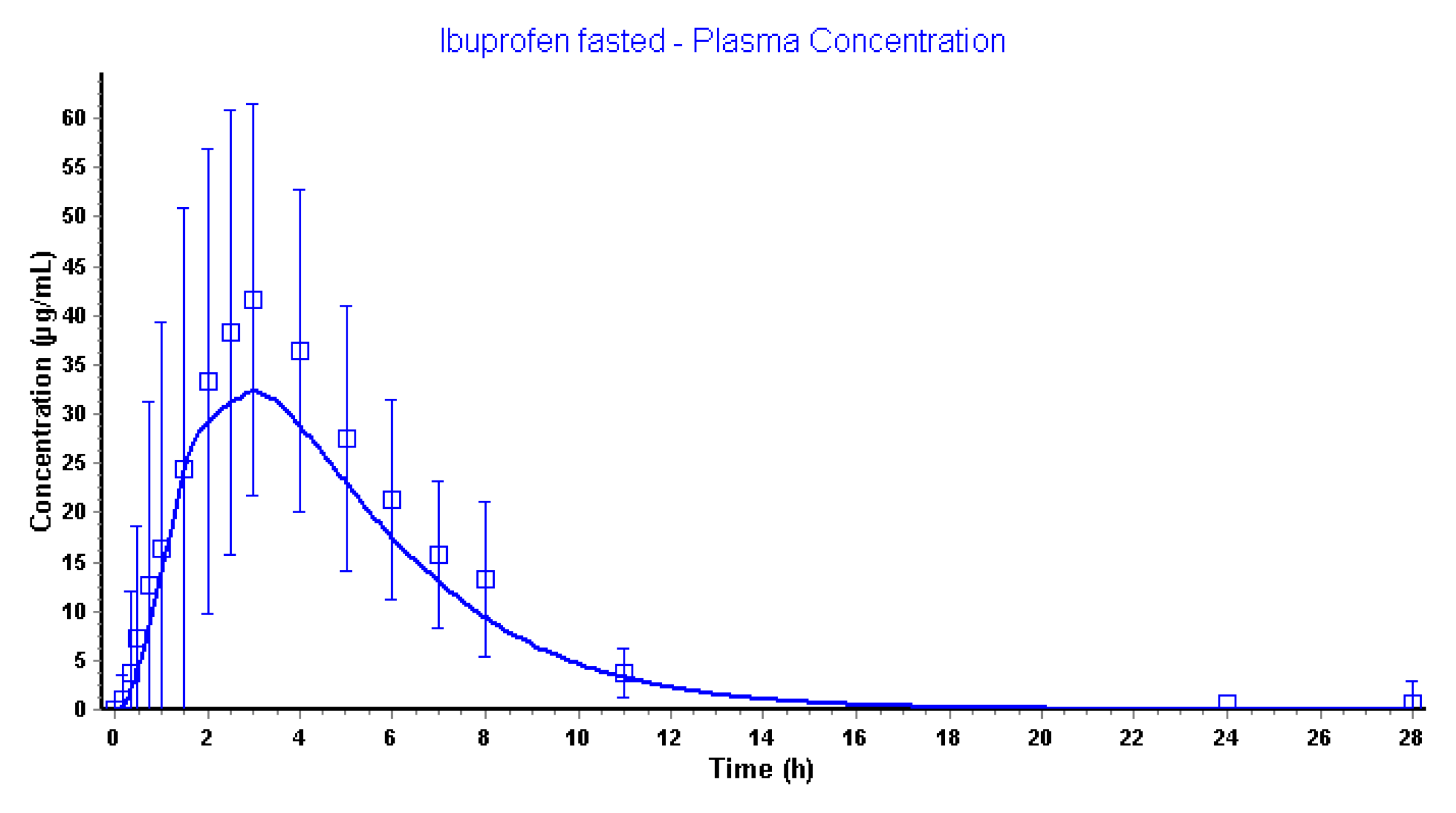
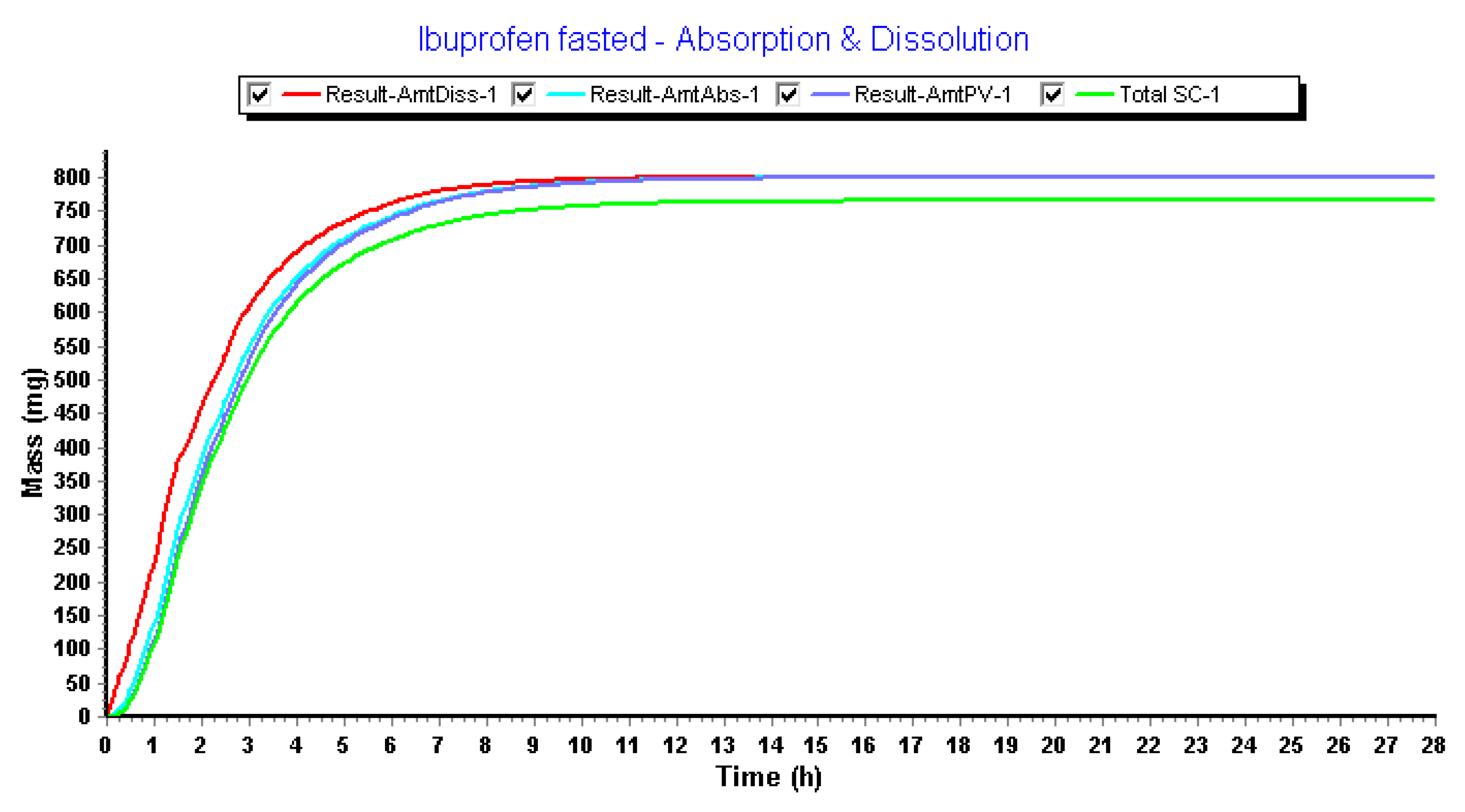
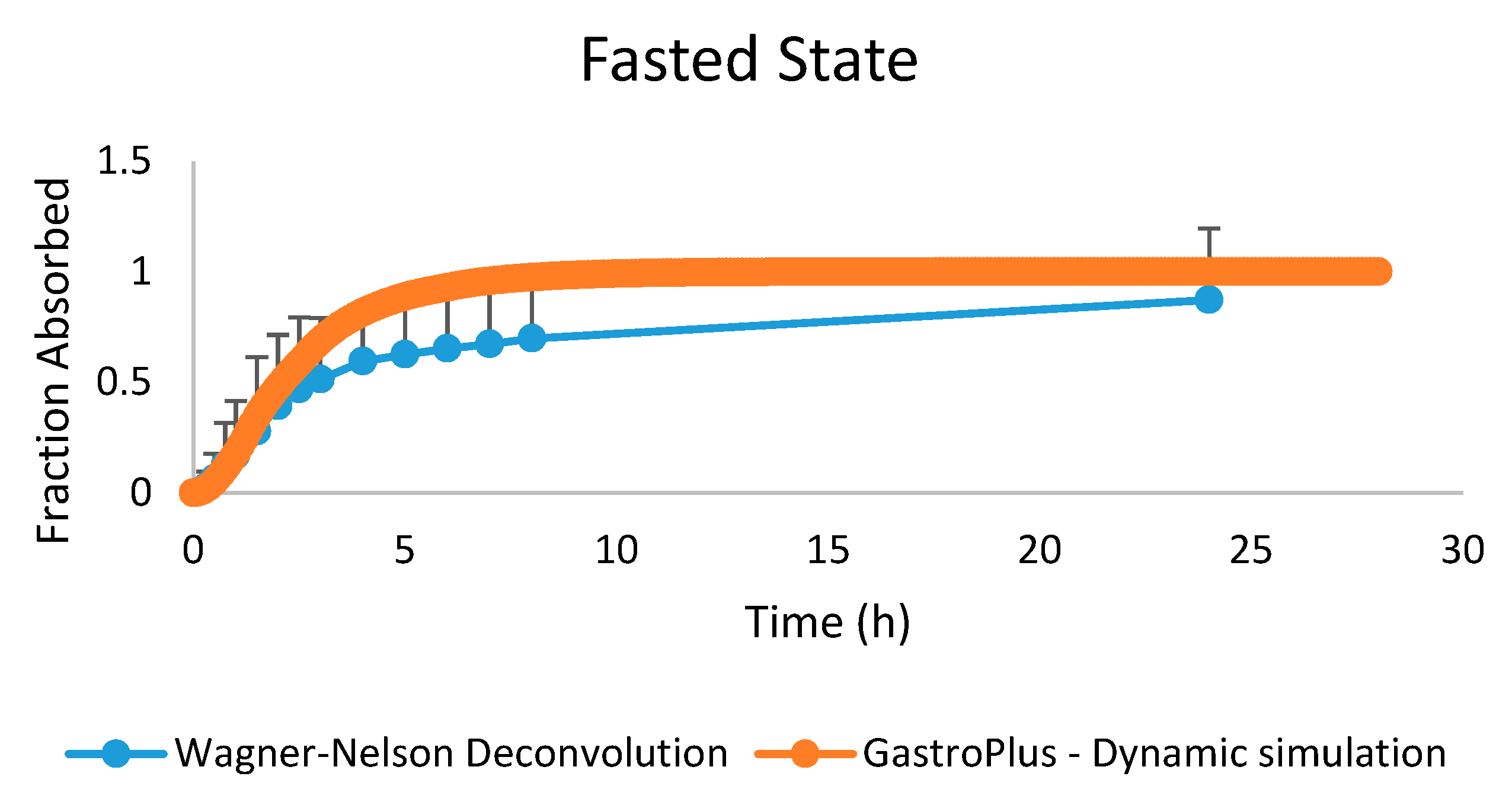
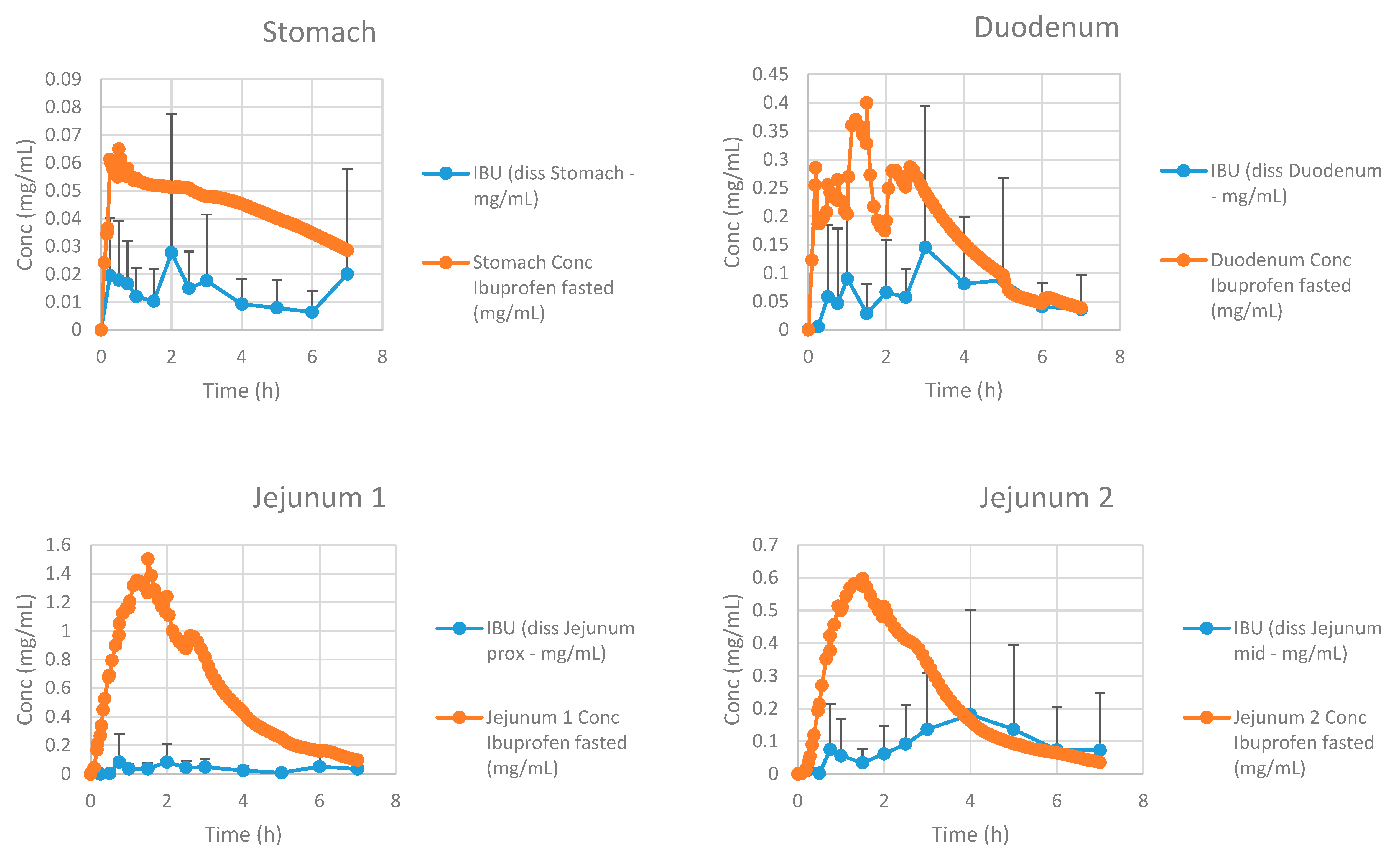
3.2.4. Advanced Compartmental Absorption and Transit Simulations: Dynamic Simulations with Adjusted Settings
4. Conclusions and Future Directions: Requesting Biowaivers?
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hens, B.; Corsetti, M.; Spiller, R.; Marciani, L.; Vanuytsel, T.; Tack, J.; Talattof, A.; Amidon, G.L.; Koziolek, M.; Weitschies, W.; et al. Exploring Gastrointestinal Variables Affecting Drug and Formulation Behavior: Methodologies, Challenges and Opportunities. Int. J. Pharm. 2016, 59, 79–97. [Google Scholar] [CrossRef] [PubMed]
- DeSesso, J.M.; son, C.F. Anatomical and physiological parameters affecting gastrointestinal absorption in humans and rats. Food Chem. Toxicol. 2001, 39, 209–228. [Google Scholar] [CrossRef]
- Hens, B.; Brouwers, J.; JacobCorsetti, M.; Augustijns, P. Supersaturation and Precipitation of Posaconazole Upon Entry in the Upper Small Intestine in Humans. J. Pharm. Sci. 2016, 105, 2677–2684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hens, B.; Corsetti, M.; Brouwers, J.; Augustijns, P. Gastrointestinal and Systemic Monitoring of Posaconazole in Humans After Fasted and Fed State Administration of a Solid Dispersion. J. Pharm. Sci. 2016, 105, 2904–2912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hens, B.; Brouwers, J.; Corsetti, M.; Augustijns, P. Gastrointestinal behavior of nano- and microsized fenofibrate: In vivo evaluation in man and in vitro simulation by assessment of the permeation potential. Eur. J. Pharm. Sci. 2015, 77, 40–47. [Google Scholar] [CrossRef]
- Hens, B.; Sinko, P.D.; Job, N.; Dean, M.; Al-Gousous, J.; Salehi, N.; Ziff, R.M.; Tsume, Y.; Bermejo, M.; Paixão, P.; et al. Formulation predictive dissolution (fPD) testing to advance oral drug product development: An introduction to the US FDA funded “21st Century BA/BE” project. Int. J. Pharm. 2018, 548, 120–127. [Google Scholar] [CrossRef]
- Psachoulias, D.; Vertzoni, M.; Goumas, K.; Kalioras, V.; Beato, S.; Butler, J.; Reppas, C. Precipitation in and supersaturation of contents of the upper small intestine after administration of two weak bases to fasted adults. Pharm. Res. 2011, 28, 3145–3158. [Google Scholar] [CrossRef]
- Kostewicz, E.S.; Abrahamsson, B.; Brewster, M.; Brouwers, J.; Butler, J.; Carlert, S.; Dickinson, P.A.; Dressman, J.; Holm, R.; Klein, S.; et al. In vitro models for the prediction of in vivo performance of oral dosage forms. Eur. J. Pharm. Sci. 2014, 57, 342–366. [Google Scholar] [CrossRef]
- Ehrhardt, C.; Kim, K. (Eds.) Drug Absorption Studies: In Situ, In Vitro and In Silico Models; Biotechnology: Pharmaceutical Aspects; Springer: New York, NY, USA, 2008; ISBN 978-0-387-74900-6. [Google Scholar]
- Hens, B.; Bermejo, M.; Tsume, Y.; Gonzalez-Alvarez, I.; Ruan, H.; Matsui, K.; Amidon, G.E.; Cavanagh, K.; Kuminek, G.; Benninghoff, G.; et al. Evaluation and optimized selection of supersaturating drug delivery systems of posaconazole (BCS class 2b) in the gastrointestinal simulator (GIS): An in vitro-in silico-in vivo approach. Eur. J. Pharm. Sci. 2018, 115, 258–269. [Google Scholar] [CrossRef]
- Kostewicz, E.S.; Aarons, L.; Bergstrand, M.; Bolger, M.B.; Galetin, A.; Hatley, O.; Jamei, M.; Lloyd, R.; Pepin, X.; Rostami-Hodjegan, A.; et al. PBPK models for the prediction of in vivo performance of oral dosage forms. Eur. J. Pharm. Sci. 2014, 57, 300–321. [Google Scholar] [CrossRef]
- Deloose, E.; Janssen, P.; Depoortere, I.; Tack, J. The migrating motor complex: Control mechanisms and its role in health and disease. Nat. Rev. Gastroenterol. Hepatol. 2012, 9, 271–285. [Google Scholar] [CrossRef] [PubMed]
- Hens, B.; Tsume, Y.; Bermejo, M.; Paixao, P.; Koenigsknecht, M.J.; Baker, J.R.; Hasler, W.L.; Lionberger, R.; Fan, J.; Dickens, J.; et al. Low Buffer Capacity and Alternating Motility along the Human Gastrointestinal Tract: Implications for in vivo Dissolution and Absorption of Ionizable Drugs. Mol. Pharm. 2017, 14, 4281–4294. [Google Scholar] [CrossRef]
- Koenigsknecht, M.J.; Baker, J.R.; Wen, B.; Frances, A.; Zhang, H.; Yu, A.; Zhao, T.; Tsume, Y.; Pai, M.P.; Bleske, B.E.; et al. In vivo Dissolution and Systemic Absorption of Immediate Release Ibuprofen in Human Gastrointestinal Tract under Fed and Fasted Conditions. Mol. Pharm. 2017, 14, 4295–4304. [Google Scholar] [CrossRef] [PubMed]
- Andreas, C.J.; Rosenberger, J.; Butler, J.; Augustijns, P.; McAllister, M.; Abrahamsson, B.; Dressman, J. Introduction to the OrBiTo decision tree to select the most appropriate in vitro methodology for release testing of solid oral dosage forms during development. Eur. J. Pharm. Biopharm. 2018, 130, 207–213. [Google Scholar] [CrossRef] [PubMed]
- Berlin, M.; Ruff, A.; Kesisoglou, F.; Xu, W.; Wang, M.H.; Dressman, J.B. Advances and challenges in PBPK modeling—Analysis of factors contributing to the oral absorption of atazanavir, a poorly soluble weak base. Eur. J. Pharm. Biopharm. 2015, 93, 267–280. [Google Scholar] [CrossRef] [PubMed]
- Chow, E.C.Y.; Talattof, A.; Tsakalozou, E.; Fan, J.; Zhao, L.; Zhang, X. Using Physiologically Based Pharmacokinetic (PBPK) Modeling to Evaluate the Impact of Pharmaceutical Excipients on Oral Drug Absorption: Sensitivity Analyses. AAPS J. 2016, 18, 1500–1511. [Google Scholar] [CrossRef] [PubMed]
- Hens, B.; Talattof, A.; Paixão, P.; Bermejo, M.; Tsume, Y.; Löbenberg, R.; Amidon, G.L. Measuring the Impact of Gastrointestinal Variables on the Systemic Outcome of Two Suspensions of Posaconazole by a PBPK Model. AAPS J. 2018, 20, 57. [Google Scholar] [CrossRef] [PubMed]
- Margolskee, A.; Darwich, A.S.; Pepin, X.; Aarons, L.; Galetin, A.; Rostami-Hodjegan, A.; Carlert, S.; Hammarberg, M.; Hilgendorf, C.; Johansson, P.; et al. IMI—Oral biopharmaceutics tools project—Evaluation of bottom-up PBPK prediction success part 2: An introduction to the simulation exercise and overview of results. Eur. J. Pharm. Sci. 2017, 96, 610–625. [Google Scholar] [CrossRef] [Green Version]
- Paixão, P.; Bermejo, M.; Hens, B.; Tsume, Y.; Dickens, J.; Shedden, K.; Salehi, N.; Koenigsknecht, M.J.; Baker, J.R.; Hasler, W.L.; et al. Gastric emptying and intestinal appearance of nonabsorbable drugs phenol red and paromomycin in human subjects: A multi-compartment stomach approach. Eur. J. Pharm. Biopharm. 2018, 129, 162–174. [Google Scholar] [CrossRef]
- Food & Drug Administration Guidance for Industry: Bioanalytical Method Validation; U.S. Food and Drug Administration (U.S. FDA): Silver Spring, MD, USA, 2001.
- Bermejo, M.; Paixão, P.; Hens, B.; Tsume, Y.; Koenigsknecht, M.; Baker, J.; Hasler, W.; Lionberger, R.; Jianghong, F.; Dickens, J.; et al. The Impact of Gastrointestinal Motility and Luminal pH On the Oral Absorption of Ibuprofen in Humans—Part 1: Fasted State Conditions. Mol. Pharm. 2018, 5, 5454–5467. [Google Scholar] [CrossRef]
- Lozoya-Agullo, I.; Zur, M.; Beig, A.; Fine, N.; Cohen, Y.; González-Álvarez, M.; Merino-Sanjuán, M.; González-Álvarez, I.; Bermejo, M.; Dahan, A. Segmental-dependent permeability throughout the small intestine following oral drug administration: Single-pass vs. Doluisio approach to in-situ rat perfusion. Int. J. Pharm. 2016, 515, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Zakeri-Milani, P.; Valizadeh, H.; Tajerzadeh, H.; Azarmi, Y.; Islambolchilar, Z.; Barzegar, S.; Barzegar-Jalali, M. Predicting human intestinal permeability using single-pass intestinal perfusion in rat. J. Pharm. Pharm. Sci. 2007, 10, 368–379. [Google Scholar] [PubMed]
- Hens, B.; Bolger, M.B. Application of a Dynamic Fluid and pH Model to Simulate Intraluminal and Systemic Concentrations of a Weak Base in GastroPlusTM. J. Pharm. Sci. 2019, 108, 305–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mudie, D.M.; Murray, K.; Hoad, C.L.; Pritchard, S.E.; Garnett, M.C.; Amidon, G.L.; Gowland, P.A.; Spiller, R.C.; Amidon, G.E.; Marciani, L. Quantification of gastrointestinal liquid volumes and distribution following a 240 mL dose of water in the fasted state. Mol. Pharm. 2014, 11, 3039–3047. [Google Scholar] [CrossRef] [PubMed]
- Tsume, Y.; Langguth, P.; Garcia-Arieta, A.; Amidon, G.L. In silico prediction of drug dissolution and absorption with variation in intestinal pH for BCS class II weak acid drugs: Ibuprofen and ketoprofen. Biopharm. Drug Dispos. 2012, 33, 366–377. [Google Scholar] [CrossRef] [Green Version]
- Heikkilä, T.; Karjalainen, M.; Ojala, K.; Partola, K.; Lammert, F.; Augustijns, P.; Urtti, A.; Yliperttula, M.; Peltonen, L.; Hirvonen, J. Equilibrium drug solubility measurements in 96-well plates reveal similar drug solubilities in phosphate buffer pH 6.8 and human intestinal fluid. Int. J. Pharm. 2011, 405, 132–136. [Google Scholar] [CrossRef]
- Augustijns, P.; Wuyts, B.; Hens, B.; Annaert, P.; Butler, J.; Brouwers, J. A review of drug solubility in human intestinal fluids: Implications for the prediction of oral absorption. Eur. J. Pharm. Sci. 2014, 57, 322–332. [Google Scholar] [CrossRef]
- Walravens, J.; Brouwers, J.; Spriet, I.; Tack, J.; Annaert, P.; Augustijns, P. Effect of pH and comedication on gastrointestinal absorption of posaconazole: Monitoring of intraluminal and plasma drug concentrations. Clin. Pharm. 2011, 50, 725–734. [Google Scholar] [CrossRef]
- Pavliv, L.; Voss, B.; Rock, A. Pharmacokinetics, safety, and tolerability of a rapid infusion of i.v. ibuprofen in healthy adults. Am. J. Health Syst. Pharm. 2011, 68, 47–51. [Google Scholar] [CrossRef]
- Locatelli, I.; Mrhar, A.; Bogataj, M. Gastric emptying of pellets under fasting conditions: A mathematical model. Pharm. Res. 2009, 26, 1607–1617. [Google Scholar] [CrossRef]
- Yu, L.X.; Amidon, G.L. A compartmental absorption and transit model for estimating oral drug absorption. Int. J. Pharm. 1999, 186, 119–125. [Google Scholar] [CrossRef]
- Fadda, H.M.; Merchant, H.A.; Arafat, B.T.; Basit, A.W. Physiological bicarbonate buffers: Stabilisation and use as dissolution media for modified release systems. Int. J. Pharm. 2009, 382, 56–60. [Google Scholar] [CrossRef] [PubMed]
- Al-Gousous, J.; Tsume, Y.; Fu, M.; Salem, I.I.; Langguth, P. Unpredictable Performance of pH-Dependent Coatings Accentuates the Need for Improved Predictive in vitro Test Systems. Mol. Pharm. 2017, 14, 4209–4219. [Google Scholar] [CrossRef]
- Al-Gousous, J.; Amidon, G.L.; Langguth, P. Toward Biopredictive Dissolution for Enteric Coated Dosage Forms. Mol. Pharm. 2016, 13, 1927–1936. [Google Scholar] [CrossRef] [PubMed]
- Karkossa, F.; Klein, S. Individualized in vitro and in silico methods for predicting in vivo performance of enteric-coated tablets containing a narrow therapeutic index drug. Eur. J. Pharm. Biopharm. 2019, 135, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Pepin, X.J.H.; Sanderson, N.J.; Blanazs, A.; Grover, S.; Ingallinera, T.G.; Mann, J.C. Bridging in vitro dissolution and in vivo exposure for acalabrutinib. Part I. Mechanistic modelling of drug product dissolution to derive a P-PSD for PBPK model input. Eur. J. Pharm. Biopharm. 2019, 142, 421–434. [Google Scholar] [CrossRef] [PubMed]
- Al-Gousous, J.; Salehi, N.; Amidon, G.E.; Ziff, R.M.; Langguth, P.; Amidon, G.L. Mass Transport Analysis of Bicarbonate Buffer: Effect of the CO2-H2CO3 Hydration-Dehydration Kinetics in the Fluid Boundary Layer and the Apparent Effective p Ka Controlling Dissolution of Acids and Bases. Mol. Pharm. 2019, 16, 2626–2635. [Google Scholar] [CrossRef]
- Al-Gousous, J.; Sun, K.X.; McNamara, D.P.; Hens, B.; Salehi, N.; Langguth, P.; Bermejo, M.; Amidon, G.E.; Amidon, G.L. Mass Transport Analysis of the Enhanced Buffer Capacity of the Bicarbonate-CO2 Buffer in a Phase-Heterogenous System: Physiological and Pharmaceutical Significance. Mol. Pharm. 2018, 15, 5291–5301. [Google Scholar] [CrossRef]
- Krieg, B.J.; Taghavi, S.M.; Amidon, G.L.; Amidon, G.E. In vivo predictive dissolution: Transport analysis of the CO2, bicarbonate in vivo buffer system. J. Pharm. Sci. 2014, 103, 3473–3490. [Google Scholar] [CrossRef] [Green Version]
- Cristofoletti, R.; Dressman, J.B. FaSSIF-V3, but not compendial media, appropriately detects differences in the peak and extent of exposure between reference and test formulations of ibuprofen. Eur. J. Pharm. Biopharm. 2016, 105, 134–140. [Google Scholar] [CrossRef]
- Cristofoletti, R.; Hens, B.; Patel, N.; Esteban, V.V.; Schmidt, S.; Dressman, J. Integrating Drug- and Formulation-Related Properties with Gastrointestinal Tract Variability Using a Product-Specific Particle Size Approach: Case Example Ibuprofen. J. Pharm. Sci. 2019, 108, 3842–3847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofmann, M.; Thieringer, F.; Nguyen, M.A.; Månsson, W.; Galle, P.R.; Langguth, P. A novel technique for intraduodenal administration of drug suspensions/solutions with concurrent pH monitoring applied to ibuprofen formulations. Eur. J. Pharm. Biopharm. 2019, 136, 192–202. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, H.C.; Stanescu, I.; Frampton, C.; Salem, I.I.; Beasley, C.P.H.; Robson, R. Pharmacokinetics and Bioavailability of a Fixed-Dose Combination of Ibuprofen and Paracetamol after Intravenous and Oral Administration. Clin. Drug Investig. 2015, 35, 625–632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, A.; Jackson, T.; Tsume, Y.; Koenigsknecht, M.; Wysocki, J.; Marciani, L.; Amidon, G.L.; Frances, A.; Baker, J.R.; Hasler, W.; et al. Mechanistic Fluid Transport Model to Estimate Gastrointestinal Fluid Volume and Its Dynamic Change Over Time. AAPS J. 2017, 19, 1682–1690. [Google Scholar] [CrossRef]
- Amidon, G.L.; Lennernäs, H.; Shah, V.P.; Crison, J.R. A Theoretical Basis for a Biopharmaceutic Drug Classification: The Correlation of in vitro Drug Product Dissolution and in vivo Bioavailability. Pharm. Res. 1995, 12, 413–420. [Google Scholar] [CrossRef] [Green Version]
- Heimbach, T.; Suarez-Sharp, S.; Kakhi, M.; Holmstock, N.; Olivares-Morales, A.; Pepin, X.; Sjögren, E.; Tsakalozou, E.; Seo, P.; Li, M.; et al. Dissolution and Translational Modeling Strategies Toward Establishing an in vitro-In vivo Link—A Workshop Summary Report. AAPS J. 2019, 21, 29. [Google Scholar] [CrossRef]
- Suarez-Sharp, S.; Li, M.; Duan, J.; Shah, H.; Seo, P. Regulatory Experience with in vivo in vitro Correlations (IVIVC) in New Drug Applications. AAPS J. 2016, 18, 1379–1390. [Google Scholar] [CrossRef]
- Suarez-Sharp, S.; Cohen, M.; Kesisoglou, F.; Abend, A.; Marroum, P.; Delvadia, P.; Kotzagiorgis, E.; Li, M.; Nordmark, A.; Bandi, N.; et al. Applications of Clinically Relevant Dissolution Testing: Workshop Summary Report. AAPS J. 2018, 20, 93. [Google Scholar] [CrossRef]



















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Input Parameter | Value/Selection Dynamic Settings | Value/Selection Default Settings | Reference |
|---|---|---|---|
| Physicochemical Properties | |||
| Molecular weight (g/mol) | 206.29 | 206.29 | ADMET Predictor 9.0 |
| pKa (acidic) | 4.54 | 4.54 | ADMET Predictor 9.0 |
| Octanol/water partition coefficient (logP) | 3.65 | 3.65 | ADMET Predictor 9.0 |
| Biopharmaceutics Properties | |||
| Human effective permeability (Peff) (× 10−4 cm/s) | 4.1 | 4.1 | [27] |
| Particle size radius (um) | 62 | 62 | In-house data |
| Dose volume (mL) | 250 | 250 | [13] |
| pH at reference solubility | 6.2 | 6.2 | [28] |
| Solubility at reference pH (mg/mL) | 1.99 | 1.99 | [28] |
| Solubility in Fasted state human gastric fluid (FaHGF) (mg/mL)—pH 1.46 | 0.0048 | 0.0048 | [29] |
| Solubility in Fasted state human intestinal fluid (FaHIF) (duodenum) (mg/mL)—pH 3.74 | 0.0102 | 0.0102 | [29] |
| Solubility in FaHIF (jejunum) (mg/mL)—pH 4.6 | 1.2 | 1.2 | [30] |
| Distribution and Clearance | |||
| Pharmacokinetic model | Two-compartmental | Two-compartmental | [31] |
| Clearance (L/h) | 4.05 | 4.05 | [31] |
| K10 (1/h) | 1.16 | 1.16 | [31] |
| K12 (1/h) | 4.55 | 4.55 | [31] |
| K21 (1/h) | 3.46 | 3.46 | [31] |
| Advanced Compartmental and AbsorptionTransit model (ACAT™) Model Parameters | |||
| Gastric transit time (h) | 2.04 | 0.25 | [13] |
| Dynamic fluid volume model | Based on 100% of the volumes measured in human MRI study after drinking a glass of water (240 mL) | Default static values under the physiology tab ‘Human—Physiological—Fasted’ | [26] |
| Dynamic pH model | Based on average pH values derived from gastric, duodenal and jejunal aspirated fluids after oral administration of 800 mg of ibuprofen | Default static values under the physiology tab ‘Human—Physiological—Fasted’ | [13] |
| Plasma | InVivo_GIS | InVivo_GISPlus |
|---|---|---|
| Cmax | 48.3 | 19.8 |
| Tmax | 50.8 | 15.7 |
| AUC | 11.0 | 13.1 |
| Duodenum | InVivo_GIS | InVivo_GISPlus |
| Cmax | 50.8 | 15.7 |
| Tmax | 62.0 | 27.4 |
| AUC | 82.8 | 88.3 |
| Jejunum | InVivo_GIS | InVivo_GISPlus |
| Cmax | 46.91 | 25.47 |
| Tmax | 50.83 | 15.75 |
| AUC | 78.68 | 28.19 |
| Parameter | KEMPT (1/h) | K_TD (1/h) | K_TJ (1/h) | K_Diss mL/(ug*h) | V1 (mL)—Duodenal | V2 (mL)—Jejunal |
|---|---|---|---|---|---|---|
| Average | 0.84 | 1.99 | 0.13 | 0.10 | 155.11 | 70.64 |
| SD | 1.27 | 3.01 | 0.25 | 0.36 | 128.44 | 80.48 |
| CV% | 151.84 | 150.83 | 196.68 | 361.28 | 82.81 | 113.93 |
| B003V1 | 0.39 | 0.20 | 0.130 | 0.101 | 80.4 | 20.0 |
| B004V1 | 0.08 | 0.59 | 0.059 | 1.48 × 10−3 | 73.9 | 18.5 |
| B004V2 | 0.02 | 1.86 | 0.209 | 5.05 × 10−3 | 195.4 | 209.2 |
| B005V1 | 0.51 | 0.22 | 0.010 | 1.61 × 10−4 | 15.3 | 2.5 |
| B005V2 | 0.18 | 0.45 | 0.224 | 3.62 × 10−4 | 350.5 | 84.5 |
| B006V1 | 0.95 | 1.84 | 0.042 | 3.51 × 10−4 | 123.2 | 64.6 |
| B017V1 | 0.16 | 0.41 | 0.081 | 5.75 × 10−3 | 62.8 | 10.2 |
| B017V2 | 0.07 | 0.08 | 0.036 | 1.11 × 10−3 | 77.6 | 19.5 |
| B042V1 | 0.13 | 10.26 | 0.000 | 7.31 × 10−2 | 29.0 | 5.2 |
| B042V2 | 0.05 | 0.19 | 0.086 | 1.52 × 10−3 | 150.0 | 80.6 |
| B049V1 | 0.05 | 0.19 | 0.086 | 1.52 × 10−3 | 262.3 | 10.6 |
| B049V2 | 0.05 | 0.19 | 0.086 | 1.52 × 10−3 | 150.0 | 80.6 |
| B052V1 | 2.79 | 2.99 | 0.019 | 6.96 × 10−4 | 19.0 | 27.6 |
| B053V1 | 1.24 | 1.79 | 0.039 | 3.48 × 10−4 | 137.8 | 215.8 |
| B055V1 | 4.05 | 0.47 | 0.100 | 9.00 × 10−5 | 126.6 | 50.0 |
| B055V2 | 3.65 | 0.07 | 0.001 | 4.33 × 10−4 | 119.6 | 93.4 |
| B063V1 | 1.00 × 10−3 | 4.12 | 1.140 | 1.96 × 10−3 | 115.9 | 56.3 |
| B065V1 | 0.20 | 2.51 | 0.092 | 0.121 | 500.0 | 283.1 |
| B065V2 | 1.28 | 9.48 | 0.002 | 1.59 | 357.7 | 10.1 |
| Median | 0.18 | 0.47 | 8.1 × 10−2 | 1.52 × 10−3 | 123.24 | 49.99 |
| Average Subject | 0.45 | 0.47 | 7.9 × 10−2 | 9.34 × 10−4 | 136.53 | 56.89 |
| GI Compartment | Volume (mL) | pH | Transit Time (h) |
|---|---|---|---|
| 1. Stomach | 48.92 | 1.3 | 0.25 |
| 2. Duodenum | 44.57 | 6 | 0.26 |
| 3. Jejunum 1 | 166.6 | 6.2 | 0.94 |
| 4. Jejunum 2 | 131 | 6.4 | 0.75 |
| 5. Ileum 1 | 102 | 6.6 | 0.58 |
| 6. Ileum 2 | 75.35 | 6.9 | 0.42 |
| 7. Ileum 3 | 53.57 | 7.4 | 0.29 |
| 8. Caecum | 50.49 | 6.4 | 4.48 |
| 9. Ascending Colon | 53.55 | 6.8 | 13.44 |
| Pharmacokinetic (PK) Parameters | Observed Pharmacokinetic Data | Simulated Pharmacokinetic Data |
|---|---|---|
| Plasma Cmax (µg/mL) | 41.7 | 55.5 |
| Plasma Tmax (h) | 3.00 | 1.31 |
| Plasma AUC0-∞ (µg·h/mL) | 259 | 189 |
| Plasma AUC0-28h (µg·h/mL) | 255 | 189 |
| Pharmacokinetic (PK) Parameters | Observed Pharmacokinetic Data | Simulated Pharmacokinetic Data |
|---|---|---|
| Plasma Cmax (µg/mL) | 41.7 | 32.5 |
| Plasma Tmax (h) | 3.0 | 3.0 |
| Plasma AUC0-∞ (µg·h/mL) | 259 | 189 |
| Plasma AUC0-28h (µg·h/mL) | 255 | 189 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bermejo, M.; Hens, B.; Dickens, J.; Mudie, D.; Paixão, P.; Tsume, Y.; Shedden, K.; Amidon, G.L. A Mechanistic Physiologically-Based Biopharmaceutics Modeling (PBBM) Approach to Assess the In Vivo Performance of an Orally Administered Drug Product: From IVIVC to IVIVP. Pharmaceutics 2020, 12, 74. https://doi.org/10.3390/pharmaceutics12010074
Bermejo M, Hens B, Dickens J, Mudie D, Paixão P, Tsume Y, Shedden K, Amidon GL. A Mechanistic Physiologically-Based Biopharmaceutics Modeling (PBBM) Approach to Assess the In Vivo Performance of an Orally Administered Drug Product: From IVIVC to IVIVP. Pharmaceutics. 2020; 12(1):74. https://doi.org/10.3390/pharmaceutics12010074
Chicago/Turabian StyleBermejo, Marival, Bart Hens, Joseph Dickens, Deanna Mudie, Paulo Paixão, Yasuhiro Tsume, Kerby Shedden, and Gordon L. Amidon. 2020. "A Mechanistic Physiologically-Based Biopharmaceutics Modeling (PBBM) Approach to Assess the In Vivo Performance of an Orally Administered Drug Product: From IVIVC to IVIVP" Pharmaceutics 12, no. 1: 74. https://doi.org/10.3390/pharmaceutics12010074