Inhibition of Pseudomonas aeruginosa Biofilm Formation with Surface Modified Polymeric Nanoparticles

 ,
,
Abstract
:1. Introduction
2. Results
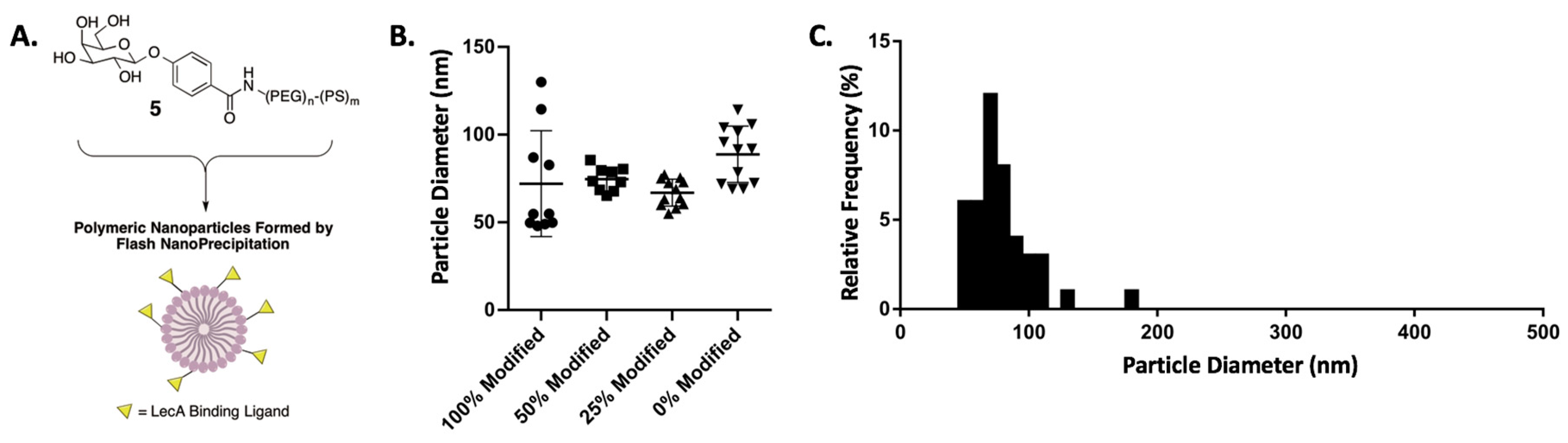
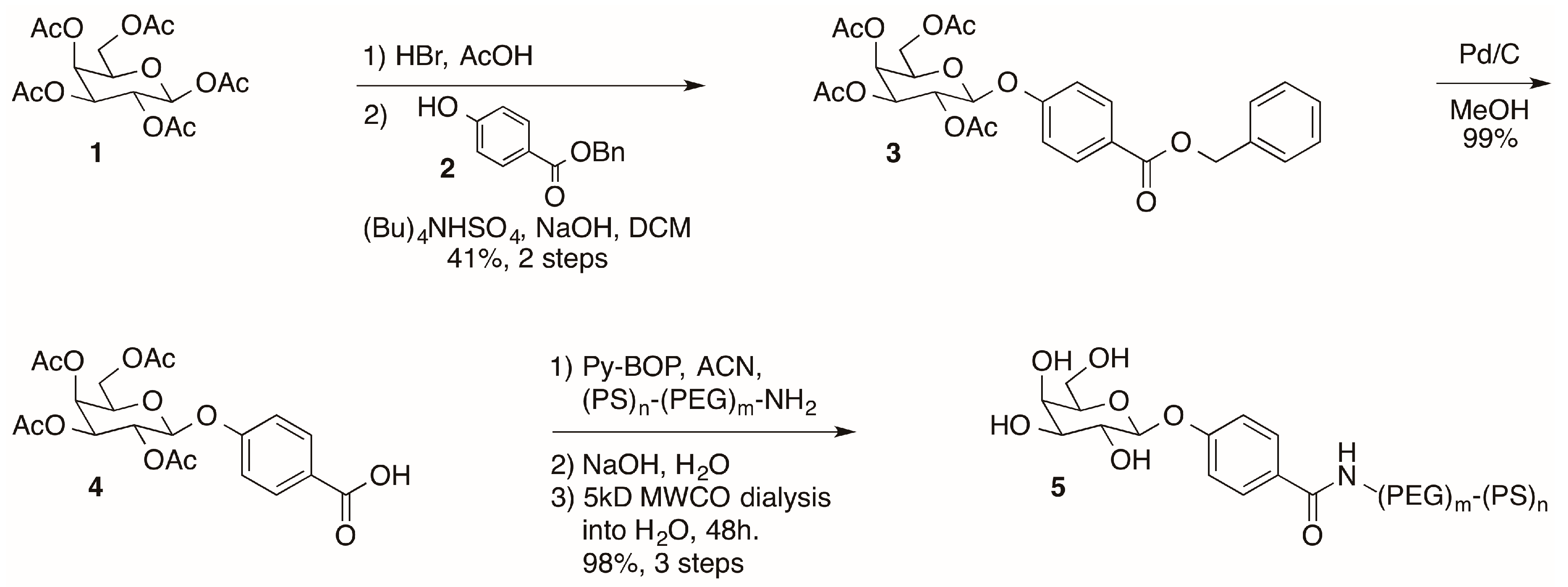
2.1. Synthesis of Galactose-Modified Di-block Co-polymer
2.2. Nanoparticle Preparation
2.3. Inhibition of LecA-Mediated Hemagglutination
2.4. Inhibition of P. aeruginosa PAO1 Biofilm Formation
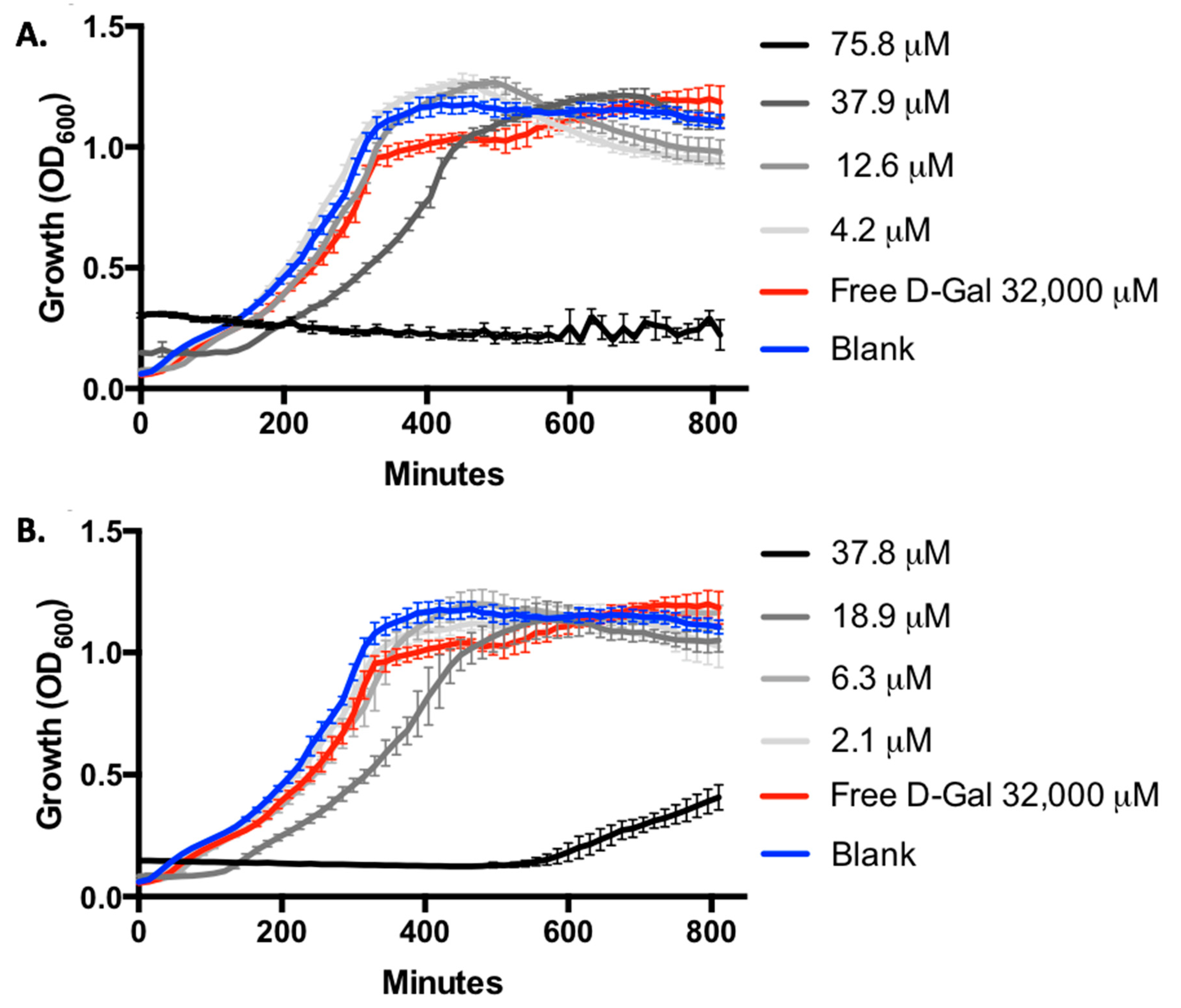
2.5. Evaluation of Growth Inhibition
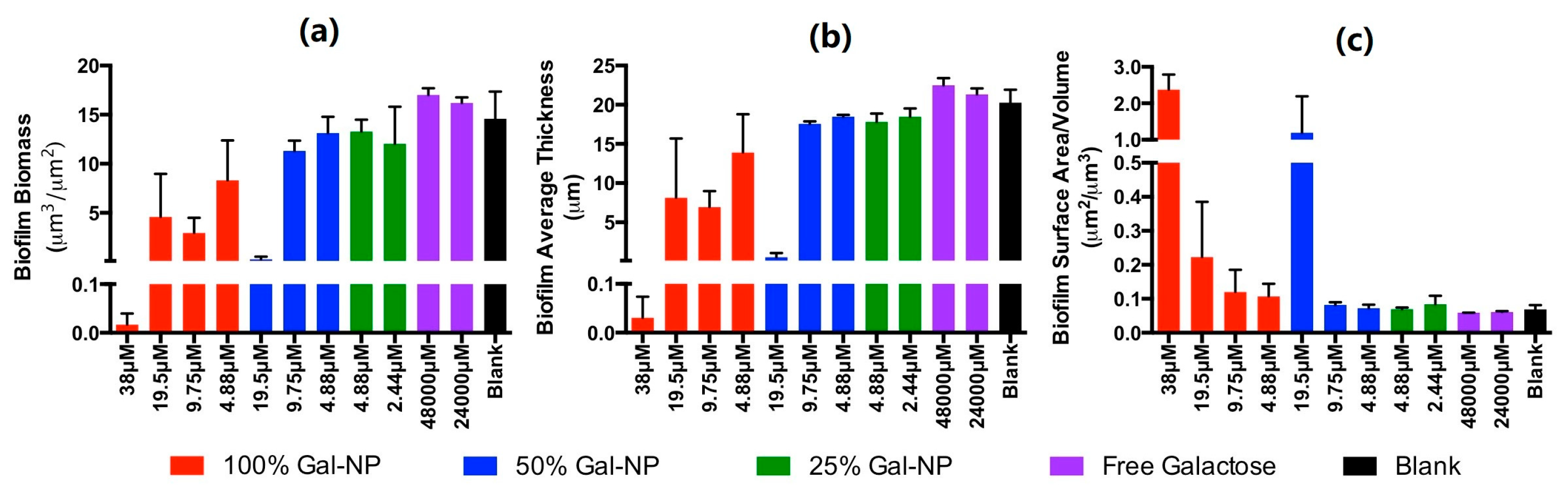
2.6. Evaluation of Biofilm Inhibition and Morphology
3. Discussion
4. Conclusions
5. Methods and Materials
5.1. General Experimental
5.2. Synthesis of D-Galactose-Modified Polymers
5.3. Nanoparticle Assembly
5.4. Hemagglutination Assay
5.5. Crystal Violet Biofilm Inhibition Assay
5.6. Growth Inhibition Assay
5.7. Fluorescent Confocal Microscopy
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fischbach, M.A.; Walsh, C.T. Antibiotics for emerging pathogens. Science 2009, 325, 1089–1093. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, E.B.; Tam, V.H. Impact of multidrug-resistant Pseudomonas aeruginosainfection on patient outcomes. Expert Rev. Pharmacoeconom. Outcomes Res. 2010, 10, 441–451. [Google Scholar] [CrossRef] [PubMed]
- Brown, E.D.; Wright, G.D. Antibacterial drug discovery in the resistance era. Nature 2016, 529, 336–343. [Google Scholar] [CrossRef]
- Sun, H.-Y. Pneumonia Due to Pseudomonas aeruginosa. Chest 2011, 139, 1172. [Google Scholar] [CrossRef]
- Wright, G.D. The antibiotic resistome: The nexus of chemical and genetic diversity. Nat. Rev. Microbiol. 2007, 5, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Lister, P.D.; Wolter, D.J.; Hanson, N.D. Antibacterial-resistant Pseudomonas aeruginosa: Clinical impact and complex regulation of chromosomally encoded resistance mechanisms. Clin. Microbiol. Rev. 2009, 22, 582–610. [Google Scholar] [CrossRef]
- Centers for Disease Control and Prevention. Antibiotic Resistance Threats in the United States; US Department of Health and Human Services: Washington, DC, USA, 2013. Available online: https://www.cdc.gov/drugresistance/biggest_threats.html (accessed on 4 April 2019).
- Oliver, A. High frequency of hypermutable Pseudomonas aeruginosa in cystic fibrosis lung infection. Science 2000, 288, 1251–1253. [Google Scholar] [CrossRef] [PubMed]
- Deretic, V.; Schurr, M.J.; Boucher, J.C.; Martin, D.W. Conversion of Pseudomonas aeruginosa to mucoidy in cystic fibrosis: Environmental stress and regulation of bacterial virulence by alternative sigma factors. J. Bacteriol. 1994, 176, 2773–2780. [Google Scholar] [CrossRef]
- Wagner, S.; Sommer, R.; Hinsberger, S.; Lu, C.; Hartmann, R.W.; Empting, M.; Titz, A. Novel strategies for the treatment of Pseudomonas aeruginosa infections. J. Med. Chem. 2016, 59, 5929–5969. [Google Scholar] [CrossRef] [PubMed]
- Hadinoto, K.; Cheow, W.S. Nano-antibiotics in chronic lung infection therapy against Pseudomonas aeruginosa. Colloids Surf. B Biointerf. 2014, 116, 772–785. [Google Scholar] [CrossRef] [PubMed]
- Grishin, A.V.; Krivozubov, M.S.; Karyagina, A.S.; Gintsburg, A.L. Pseudomonas Aeruginosa Lectins as targets for novel antibacterials. Acta Nat. 2015, 7, 29–41. [Google Scholar]
- Winzer, K.; Falconer, C.; Garber, N.C.; Diggle, S.P. The Pseudomonas aeruginosa lectins PA-IL and PA-IIL are controlled by quorum sensing and by RpoS. J. Bacteriol. 2000, 182, 6401–6411. [Google Scholar] [CrossRef]
- Mattmann, M.E.; Blackwell, H.E. Small molecules that modulate quorum sensing and control virulence in Pseudomonas aeruginosa. J. Org. Chem. 2010, 75, 6737–6746. [Google Scholar] [CrossRef]
- Tielker, D.; Hacker, S.; Loris, R.; Strathmann, M.; Wingender, J.; Wilhelm, S.; Rosenau, F.; Jaeger, K.-E. Pseudomonas aeruginosa lectin LecB is located in the outer membrane and is involved in biofilm formation. Microbiology 2005, 151, 1313–1323. [Google Scholar] [CrossRef]
- Imberty, A.; Wimmerova, M.; Mitchell, E.P. Structures of the lectins from Pseudomonas aeruginosa: Insights into the molecular basis for host glycan recognition. Microbes Infect. 2004, 6, 221–228. [Google Scholar] [CrossRef]
- Chemani, C.; Imberty, A.; de Bentzmann, S.; Pierre, M.; Wimmerova, M.; Guery, B.P.; Faure, K. Role of lecA and lecB lectins in Pseudomonas aeruginosa-induced lung injury and effect of carbohydrate ligands. Infect. Immun. 2009, 77, 2065–2075. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.D.; Yang, S.S.; Wilson, B.K.; McManus, S.A.; Chen, C.V.H.H.; Prud’homme, R.K. Nanoparticle targeting of gram-positive and gram-negative bacteria for magnetic-based separations of bacterial pathogens. Appl. Nanosci. 2017, 7, 83–93. [Google Scholar] [CrossRef]
- D'addio, S.M.; Prud'homme, R.K. Controlling drug nanoparticle formation by rapid precipitation. Adv. Drug Deliv. Rev. 2011, 63, 417–426. [Google Scholar] [CrossRef]
- Cecioni, S.; Imberty, A.; Vidal, S. Glycomimetics versus multivalent glycoconjugates for the design of high affinity lectin ligands. Chem. Rev. 2015, 115, 525–561. [Google Scholar] [CrossRef]
- Peeters, E.; Nelis, H.J.; Coenye, T. Comparison of multiple methods for quantification of microbial biofilms grown in microtiter plates. J. Microbiol. Methods 2008, 72, 157–165. [Google Scholar] [CrossRef]
- Coenye, T.; Nelis, H.J. In vitro and in vivo model systems to study microbial biofilm formation. J. Microbiol. Methods 2010, 83, 89–105. [Google Scholar] [CrossRef] [PubMed]
- Drescher, K.; Shen, Y.; Bassler, B.L. Biofilm streamers cause catastrophic disruption of flow with consequences for environmental and medical systems. Proc. Nat. Acad. Sci. USA 2013, 110, 4345–4350. [Google Scholar] [CrossRef]
- O’Brien, K.T.; Noto, J.G.; Nichols-O’Neill, L.; Perez, L.J. Potent irreversible inhibitors of LasR quorum sensing in Pseudomonas aeruginosa. ACS Med. Chem. Lett. 2015, 6, 162–167. [Google Scholar] [CrossRef] [PubMed]
- Rahme, L.G.; Stevens, E.J.; Wolfort, S.F.; Shao, J.; Tompkins, R.G.; Ausubel, F.M. Common virulence factors for bacterial pathogenicity in plants and animals. Science 1995, 268, 1899–1902. [Google Scholar] [CrossRef] [PubMed]
- Capilato, J.N.; Philippi, S.V.; Reardon, T.; McConnell, A.; Oliver, D.C.; Warren, A.; Adams, J.; Wu, C.; Perez, L.J. Development of a novel series of non-natural triaryl agonists and antagonists of the Pseudomonas aeruginosa LasR quorum sensing receptor. Bioorg. Med. Chem. 2017, 25, 153–165. [Google Scholar] [CrossRef]
- Titz, A. Carbohydrate-based anti-virulence compounds against chronic Pseudomonas aeruginosa Infections with a focus on small molecules. Top. Med. Chem. 2014, 12, 169–186. [Google Scholar]
- Imberty, A.; Chabre, Y.M.; Roy, R. Glycomimetics and glycodendrimers as high affinity microbial anti-adhesins. Chem. Eur. J. 2008, 14, 7490–7499. [Google Scholar] [CrossRef]
- Bajolet-Laudinat, O.; Girod-de Bentzmann, S.; Tournier, J.M.; Madoulet, C.; Plotkowski, M.C.; Chippaux, C.; Puchelle, E. Cytotoxicity of Pseudomonas aeruginosa internal lectin PA-I to respiratory epithelial cells in primary culture. Infect. Immun. 1994, 62, 4481–4487. [Google Scholar]
- Carlmark, A.; Hawker, C.; Hult, A.; Malkoch, M. New methodologies in the construction of dendritic materials. Chem. Soc. Rev. 2009, 38, 352–362. [Google Scholar] [CrossRef]
- Grayson, S.M.; Frechet, J. Convergent dendrons and dendrimers: From synthesis to applications. Chem. Rev. 2001, 101, 3819–3868. [Google Scholar] [CrossRef]
- Singh, R.; Lillard, J.W. Nanoparticle-based targeted drug delivery. Exp. Mol. Pathol. 2009, 86, 215–223. [Google Scholar] [CrossRef] [PubMed]
- Bogart, L.K.; Pourroy, G.; Murphy, C.J.; Puntes, V.; Pellegrino, T.; Rosenblum, D.; Peer, D.; Lévy, R. Nanoparticles for imaging, sensing, and therapeutic intervention. ACS Nano 2014, 8, 3107–3122. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Radovic-Moreno, A.F.; Wu, J.; Langer, R.; Shi, J. Nanomedicine in the management of microbial infection—Overview and perspectives. Nano Today 2014, 9, 478–498. [Google Scholar] [CrossRef]
- Huh, A.J.; Kwon, Y.J. “Nanoantibiotics”: A new paradigm for treating infectious diseases using nanomaterials in the antibiotics resistant era. J. Control. Release 2011, 156, 128–145. [Google Scholar] [CrossRef]
- Kalhapure, R.S.; Suleman, N.; Mocktar, C.; Seedat, N.; Govender, T. Nanoengineered drug delivery systems for enhancing antibiotic therapy. J. Pharm. Sci. 2015, 104, 872–905. [Google Scholar] [CrossRef] [PubMed]
- Pelgrift, R.Y.; Friedman, A.J. Nanotechnology as a therapeutic tool to combat microbial resistance. Adv. Drug Deliv. Rev. 2013, 65, 1803–1815. [Google Scholar] [CrossRef]
- Lu, H.D.; Spiegel, A.C.; Hurley, A.; Perez, L.J.; Maisel, K.; Ensign, L.M.; Hanes, J.; Bassler, B.L.; Semmelhack, M.F.; Prud’homme, R.K. Modulating Vibrio cholerae quorum-sensing-controlled communication using autoinducer-loaded nanoparticles. Nano Lett. 2015, 15, 2235–2241. [Google Scholar] [CrossRef] [PubMed]
- D’Addio, S.M.; Baldassano, S.; Shi, L.; Cheung, L.; Adamson, D.H.; Bruzek, M.; Anthony, J.E.; Laskin, D.L.; Sinko, P.J.; Prud’homme, R.K. Optimization of cell receptor-specific targeting through multivalent surface decoration of polymeric nanocarriers. J. Control. Release 2013, 168, 41–49. [Google Scholar] [CrossRef]
- O'Toole, G.A. Microtiter dish biofilm formation assay. J. Vis. Exp. 2011, 47, 2437. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Zhu, Z.; Qian, H.; Wohl, A.R.; Beaman, C.J.; Hoye, T.R.; Macosko, C.W. A Simple Confined Impingement Jets Mixer for Flash Nanoprecipitation. J. Pharm. Sci. 2012, 101, 4018–4023. [Google Scholar] [CrossRef] [PubMed]
- Müsken, M.; Di Fiore, S.; Römling, U.; Häussler, S. A 96-well-plate–based optical method for the quantitative and qualitative evaluation of Pseudomonas aeruginosa biofilm formation and its application to susceptibility testing. Nat. Protoc. 2010, 5, 1460–1469. [Google Scholar] [CrossRef] [PubMed]
- Heydorn, A.; Nielsen, A.T.; Hentzer, M.; Sternberg, C.; Givskov, M.; Ersbøll, B.K.; Molin, S. Quantification of biofilm structures by the novel computer program COMSTAT. Microbiology 2000, 146, 2395–2407. [Google Scholar] [CrossRef] [PubMed]
- Vorregaard, M. Comstat2—A Modern 3D Image Analysis Environment for Biofilms, in Informatics and Mathematical Modelling. Master’s Thesis, Technical University of Denmark, Kongens Lyngby, Denmark, 31 January 2008. Available online: www.comstat.dk (accessed on 23 April 2019).







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formulation | Active Stabilizer | Stabilizer | Inert Core | NP Properties | ||||
|---|---|---|---|---|---|---|---|---|
| Block co-polymer | Conc. (mg/mL) | Block co-polymer | Conc. (mg/mL) | Filler | Conc. (mg/mL) | Z-diameter (nm) 1 | PDI 2 | |
| 0% | PS-b-PEG-Gal | 0.00 | PS-b-PEG | 0.40 | VitE | 0.40 | 88.83 ± 4.64 | 0.0327 |
| 25% Gal-NP | PS-b-PEG-Gal | 0.10 | PS-b-PEG | 0.30 | VitE | 0.40 | 73.78 ± 2.23 | 0.0132 |
| 50% Gal-NP | PS-b-PEG-Gal | 0.20 | PS-b-PEG | 0.20 | VitE | 0.40 | 74.74 ± 2.26 | 0.0083 |
| 100% Gal-NP | PS-b-PEG-Gal | 0.40 | PS-b-PEG | 0.00 | VitE | 0.40 | 81.47 ± 12.75 | 0.2693 |
| Entry | Formulation | Hemagglutination Assay MIC (uM) 1 | Relative Potency 4 |
|---|---|---|---|
| 1 | D-galactose | 3125.0 | 1.0 |
| 2 | 100%-Modified D-galactose-NP | 6.31 | 495.2 |
| 3 | 50%-Modified D-galactose-NP | 3.15 | 992.1 |
| 4 | 25%-Modified D-galactose-NP | none 2 | - |
| 5 | 100%-Modified mannose-NP | none 3 | - |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Flockton, T.R.; Schnorbus, L.; Araujo, A.; Adams, J.; Hammel, M.; Perez, L.J. Inhibition of Pseudomonas aeruginosa Biofilm Formation with Surface Modified Polymeric Nanoparticles. Pathogens 2019, 8, 55. https://doi.org/10.3390/pathogens8020055
Flockton TR, Schnorbus L, Araujo A, Adams J, Hammel M, Perez LJ. Inhibition of Pseudomonas aeruginosa Biofilm Formation with Surface Modified Polymeric Nanoparticles. Pathogens. 2019; 8(2):55. https://doi.org/10.3390/pathogens8020055
Chicago/Turabian StyleFlockton, Tyler R., Logan Schnorbus, Agustin Araujo, Jill Adams, Maryjane Hammel, and Lark J. Perez. 2019. "Inhibition of Pseudomonas aeruginosa Biofilm Formation with Surface Modified Polymeric Nanoparticles" Pathogens 8, no. 2: 55. https://doi.org/10.3390/pathogens8020055