
High Affinity Binding of N2-Modified Guanine Derivatives Significantly Disrupts the Ligand Binding Pocket of the Guanine Riboswitch


Abstract
:1. Introduction
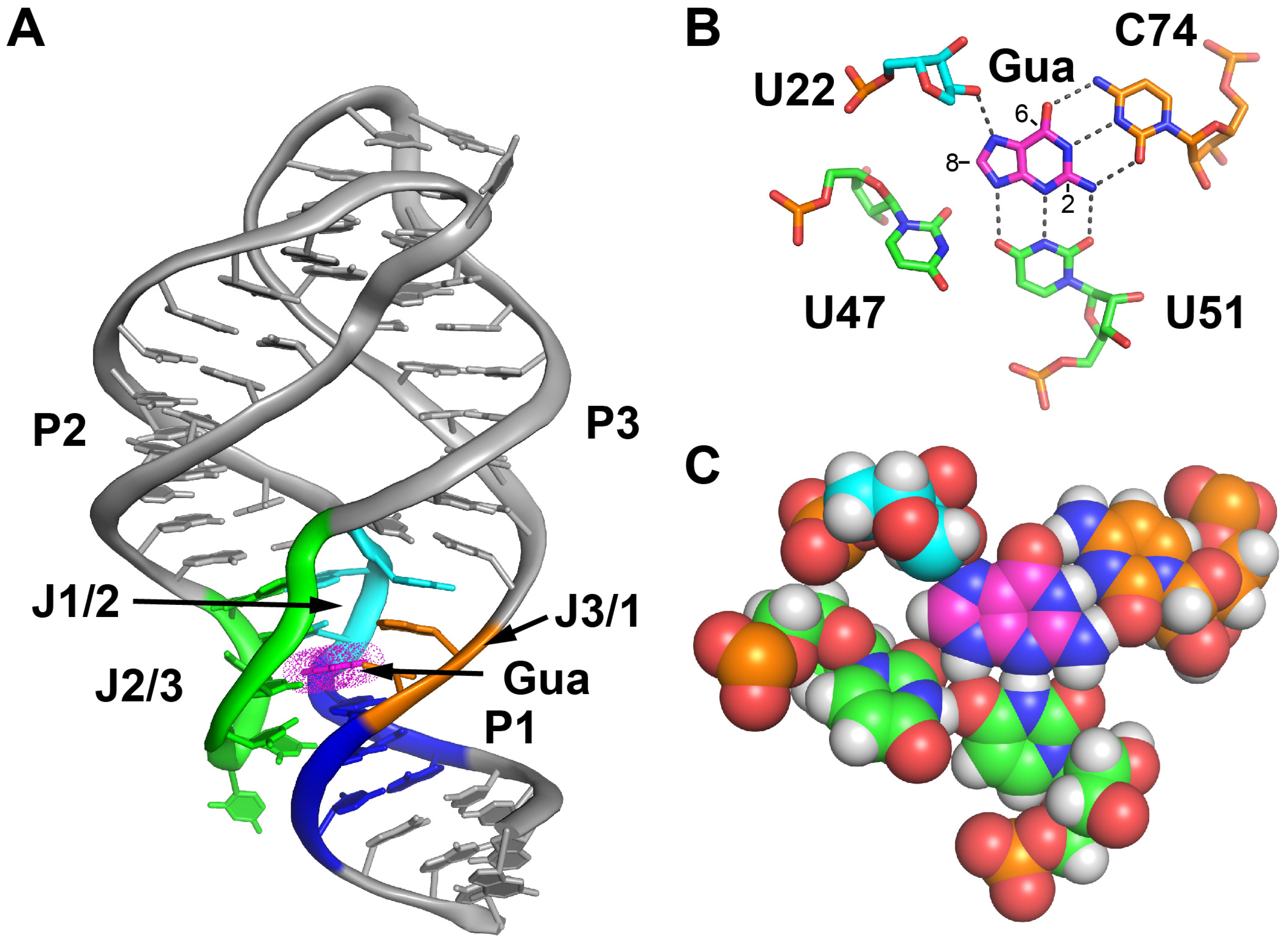
2. Results
2.1. C8-Modified Guanine Derivatives Minimally Perturb the Binding Pocket
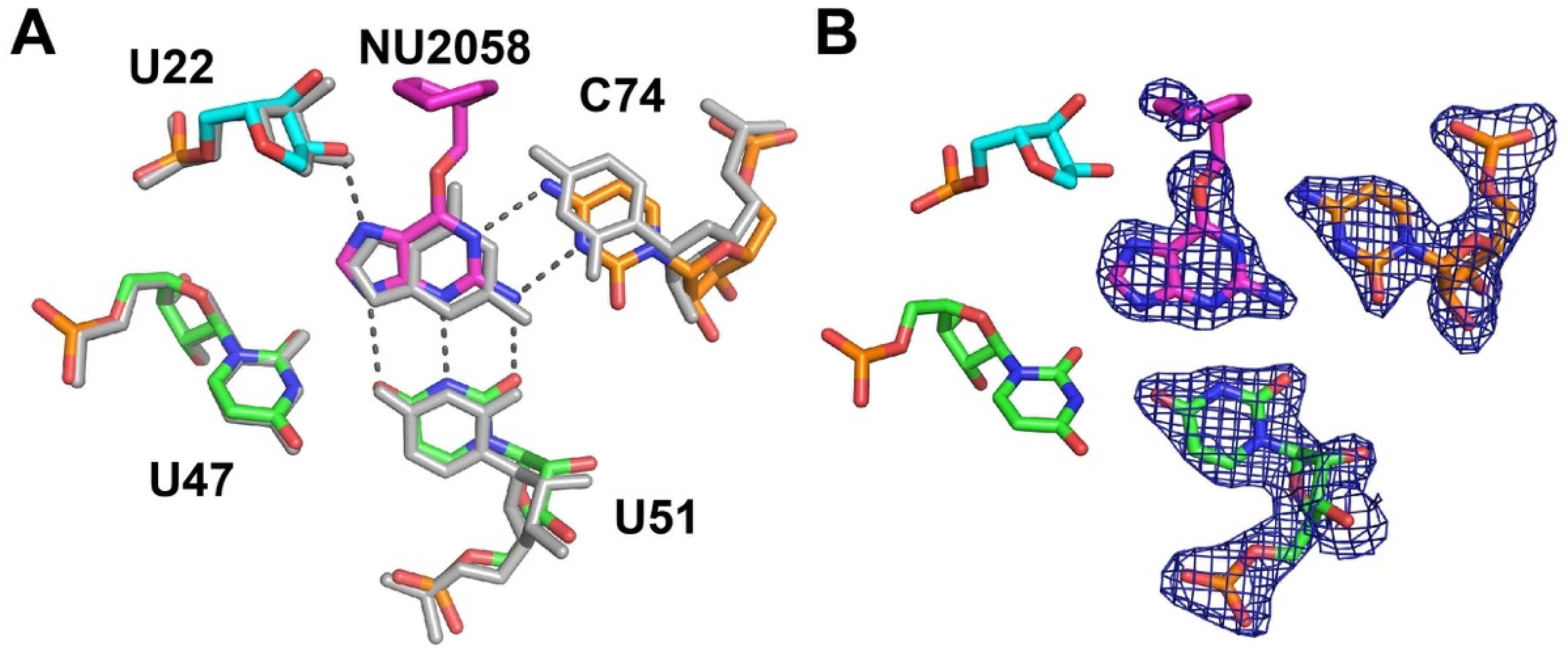
2.2. C6-Modified Guanine Derivatives Shift C74 Towards the Minor Groove
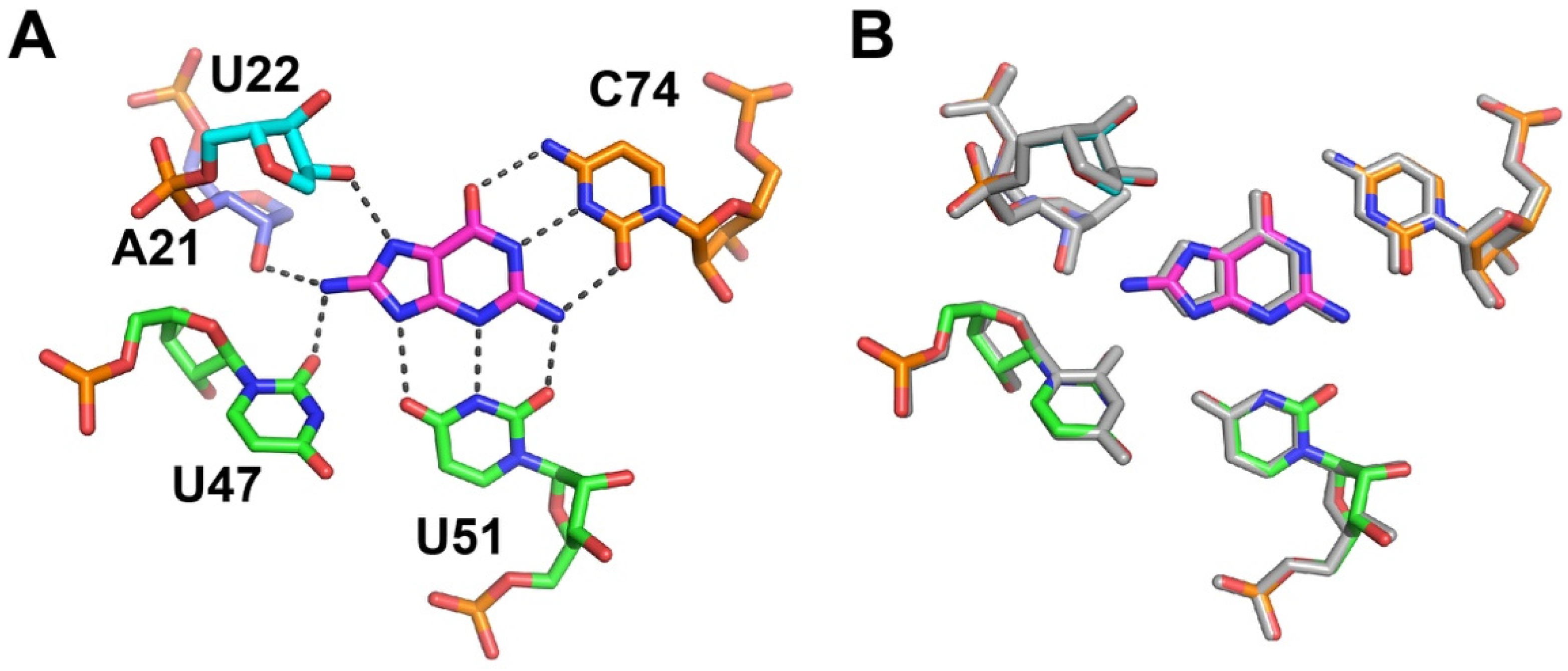
2.3. C2-Modified Guanine Derivatives Displace C74 from the Binding Pocket
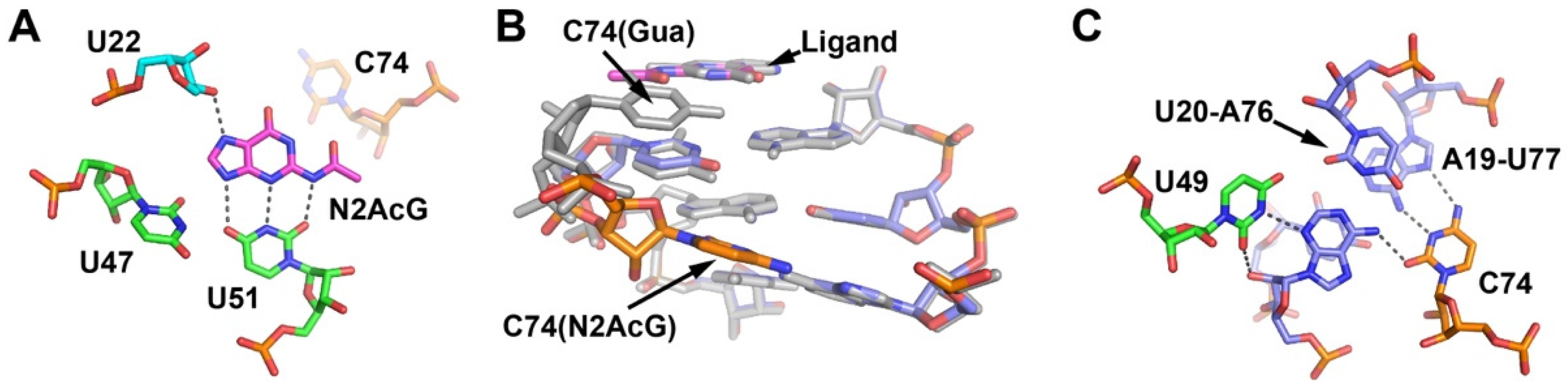
2.4. Cytosine at Position 74 Is Required for High Affinity C2-Modifed Purine Binding
2.5. The Third Junction-Roximal Base Pair in P1 Influences Binding Affinity of an N2-Modified Guanine Analog
2.6. Combined C2 and C6 Modifications Are Tolerated
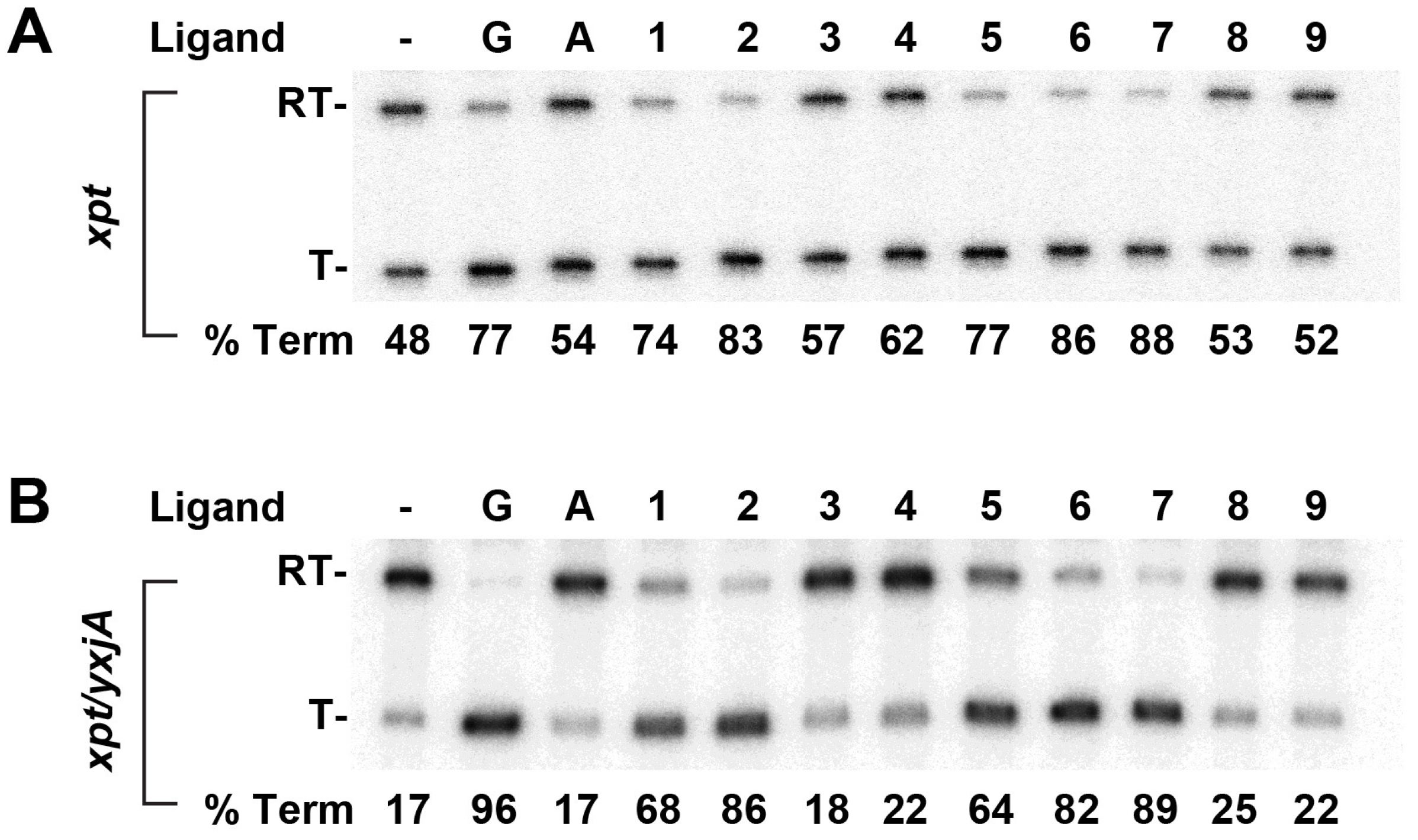
2.7. Modified Purines Support Regulatory Switching
3. Discussion
4. Materials and Methods
4.1. Synthesis of RNA for ITC and X-ray Crystallography
4.2. Crystallization of GR RNA-Ligand Complexes
4.3. Structure Determination and Model Refinement
4.4. Isothermal Titration Calorimetry
4.5. Single-Turnover In Vitro Transcription Assays
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Blount, K.F.; Breaker, R.R. Riboswitches as antibacterial drug targets. Nat. Biotechnol. 2006, 24, 1558–1564. [Google Scholar] [CrossRef] [PubMed]
- Connelly, C.M.; Moon, M.H.; Schneekloth, J.S. The Emerging Role of RNA as a Therapeutic Target for Small Molecules. Cell Chem. Biol. 2016, 23, 1077–1090. [Google Scholar] [CrossRef] [PubMed]
- Deigan, K.E.; Ferre-D’Amare, A.R. Riboswitches: Discovery of drugs that target bacterial gene-regulatory RNAs. Acc. Chem. Res. 2011, 44, 1329–1338. [Google Scholar] [CrossRef] [Green Version]
- Garst, A.D.; Edwards, A.L.; Batey, R.T. Riboswitches: Structures and mechanisms. Cold Spring Harb. Perspect. Biol. 2011, 3, a003533. [Google Scholar] [CrossRef]
- Breaker, R.R. Riboswitches and the RNA world. Cold Spring Harb. Perspect. Biol. 2012, 4, a003566. [Google Scholar] [CrossRef] [Green Version]
- Barrick, J.E.; Breaker, R.R. The distributions, mechanisms, and structures of metabolite-binding riboswitches. Genome Biol. 2007, 8, R239. [Google Scholar] [CrossRef] [Green Version]
- McCown, P.J.; Corbino, K.A.; Stav, S.; Sherlock, M.E.; Breaker, R.R. Riboswitch diversity and distribution. RNA 2017, 23, 995–1011. [Google Scholar] [CrossRef]
- Warner, K.D.; Hajdin, C.E.; Weeks, K.M. Principles for targeting RNA with drug-like small molecules. Nat. Rev. Drug Discov. 2018, 17, 547–558. [Google Scholar] [CrossRef]
- Disney, M.D.; Dwyer, B.G.; Childs-Disney, J.L. Drugging the RNA World. Cold Spring Harb. Perspect. Biol. 2018, 10, a034769. [Google Scholar] [CrossRef] [Green Version]
- Blount, K.F.; Megyola, C.; Plummer, M.; Osterman, D.; O’Connell, T.; Aristoff, P.; Quinn, C.; Chrusciel, R.A.; Poel, T.J.; Schostarez, H.J.; et al. Novel riboswitch-binding flavin analog that protects mice against Clostridium difficile infection without inhibiting cecal flora. Antimicrob. Agents Chemother. 2015, 59, 5736–5746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vicens, Q.; Mondragon, E.; Reyes, F.E.; Coish, P.; Aristoff, P.; Berman, J.; Kaur, H.; Kells, K.W.; Wickens, P.; Wilson, J.; et al. Structure-Activity Relationship of Flavin Analogues That Target the Flavin Mononucleotide Riboswitch. ACS Chem. Biol. 2018, 13, 2908–2919. [Google Scholar] [CrossRef] [PubMed]
- Serganov, A.; Yuan, Y.R.; Pikovskaya, O.; Polonskaia, A.; Malinina, L.; Phan, A.T.; Hobartner, C.; Micura, R.; Breaker, R.R.; Patel, D.J. Structural basis for discriminative regulation of gene expression by adenine- and guanine-sensing mRNAs. Chem. Biol. 2004, 11, 1729–1741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.N.; Roth, A.; Breaker, R.R. Guanine riboswitch variants from Mesoplasma florum selectively recognize 2’-deoxyguanosine. Proc. Natl. Acad. Sci. USA 2007, 104, 16092–16097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mandal, M.; Boese, B.; Barrick, J.E.; Winkler, W.C.; Breaker, R.R. Riboswitches control fundamental biochemical pathways in Bacillus subtilis and other bacteria. Cell 2003, 113, 577–586. [Google Scholar] [CrossRef] [Green Version]
- Mandal, M.; Breaker, R.R. Adenine riboswitches and gene activation by disruption of a transcription terminator. Nat. Struct. Mol. Biol. 2004, 11, 29–35. [Google Scholar] [CrossRef]
- Batey, R.T.; Gilbert, S.D.; Montange, R.K. Structure of a natural guanine-responsive riboswitch complexed with the metabolite hypoxanthine. Nature 2004, 432, 411–415. [Google Scholar] [CrossRef]
- Noeske, J.; Richter, C.; Grundl, M.A.; Nasiri, H.R.; Schwalbe, H.; Wohnert, J. An intermolecular base triple as the basis of ligand specificity and affinity in the guanine- and adenine-sensing riboswitch RNAs. Proc. Natl. Acad. Sci. USA 2005, 102, 1372–1377. [Google Scholar] [CrossRef] [Green Version]
- Edwards, A.L.; Batey, R.T. A structural basis for the recognition of 2’-deoxyguanosine by the purine riboswitch. J. Mol. Biol. 2009, 385, 938–948. [Google Scholar] [CrossRef] [Green Version]
- Matyjasik, M.M.; Batey, R.T. Structural basis for 2’-deoxyguanosine recognition by the 2’-dG-II class of riboswitches. Nucleic Acids Res. 2019, 47, 10931–10941. [Google Scholar] [CrossRef]
- Pikovskaya, O.; Polonskaia, A.; Patel, D.J.; Serganov, A. Structural principles of nucleoside selectivity in a 2’-deoxyguanosine riboswitch. Nat. Chem. Biol. 2011, 7, 748–755. [Google Scholar] [CrossRef] [Green Version]
- Gilbert, S.D.; Reyes, F.E.; Edwards, A.L.; Batey, R.T. Adaptive ligand binding by the purine riboswitch in the recognition of guanine and adenine analogs. Structure 2009, 17, 857–868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilbert, S.D.; Mediatore, S.J.; Batey, R.T. Modified pyrimidines specifically bind the purine riboswitch. J. Am. Chem Soc. 2006, 128, 14214–14215. [Google Scholar] [CrossRef] [PubMed]
- Mulhbacher, J.; Brouillette, E.; Allard, M.; Fortier, L.C.; Malouin, F.; Lafontaine, D.A. Novel riboswitch ligand analogs as selective inhibitors of guanine-related metabolic pathways. PLoS Pathog. 2010, 6, e1000865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ster, C.; Allard, M.; Boulanger, S.; Lamontagne Boulet, M.; Mulhbacher, J.; Lafontaine, D.A.; Marsault, E.; Lacasse, P.; Malouin, F. Experimental treatment of Staphylococcus aureus bovine intramammary infection using a guanine riboswitch ligand analog. J. Dairy Sci. 2013, 96, 1000–1008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daldrop, P.; Reyes, F.E.; Robinson, D.A.; Hammond, C.M.; Lilley, D.M.; Batey, R.T.; Brenk, R. Novel Ligands for a Purine Riboswitch Discovered by RNA-Ligand Docking. Chem. Biol. 2011, 18, 324–335. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.N.; Blount, K.F.; Puskarz, I.; Lim, J.; Link, K.H.; Breaker, R.R. Design and antimicrobial action of purine analogues that bind Guanine riboswitches. ACS Chem. Biol 2009, 4, 915–927. [Google Scholar] [CrossRef] [Green Version]
- Yan, L.H.; Le Roux, A.; Boyapelly, K.; Lamontagne, A.M.; Archambault, M.A.; Picard-Jean, F.; Lalonde-Seguin, D.; St-Pierre, E.; Najmanovich, R.J.; Fortier, L.C.; et al. Purine analogs targeting the guanine riboswitch as potential antibiotics against Clostridioides difficile. Eur. J. Med. Chem. 2018, 143, 755–768. [Google Scholar] [CrossRef]
- Krajewski, S.S.; Isoz, I.; Johansson, J. Antibacterial and antivirulence effect of 6-N-hydroxylaminopurine in Listeria monocytogenes. Nucleic Acids Res. 2017, 45, 1914–1924. [Google Scholar]
- Meibohm, B.; Derendorf, H. Basic concepts of pharmacokinetic/pharmacodynamic (PK/PD) modelling. Int. J. Clin. Pharm. 1997, 35, 401–413. [Google Scholar]
- Leavitt, S.; Freire, E. Direct measurement of protein binding energetics by isothermal titration calorimetry. Curr. Opin. Struct. Biol. 2001, 11, 560–566. [Google Scholar] [CrossRef]
- Salim, N.N.; Feig, A.L. Isothermal titration calorimetry of RNA. Methods 2009, 47, 198–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoddard, C.D.; Widmann, J.; Trausch, J.J.; Marcano-Velazquez, J.G.; Knight, R.; Batey, R.T. Nucleotides adjacent to the ligand-binding pocket are linked to activity tuning in the purine riboswitch. J. Mol. Biol. 2013, 425, 1596–1611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, T.G.; Bentley, J.; Arris, C.E.; Boyle, F.T.; Curtin, N.J.; Endicott, J.A.; Gibson, A.E.; Golding, B.T.; Griffin, R.J.; Hardcastle, I.R.; et al. Structure-based design of a potent purine-based cyclin-dependent kinase inhibitor. Nat. Struct. Biol. 2002, 9, 745–749. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, S.D.; Stoddard, C.D.; Wise, S.J.; Batey, R.T. Thermodynamic and kinetic characterization of ligand binding to the purine riboswitch aptamer domain. J. Mol. Biol. 2006, 359, 754–768. [Google Scholar] [CrossRef] [PubMed]
- Artsimovitch, I.; Henkin, T.M. In vitro approaches to analysis of transcription termination. Methods 2009, 47, 37–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marcano-Velazquez, J.G.; Batey, R.T. Structure-guided mutational analysis of gene regulation by the Bacillus subtilis pbuE adenine-responsive riboswitch in a cellular context. J. Biol. Chem. 2015, 290, 4464–4475. [Google Scholar] [CrossRef] [Green Version]
- Trausch, J.J.; Ceres, P.; Reyes, F.E.; Batey, R.T. The structure of a tetrahydrofolate-sensing riboswitch reveals two ligand binding sites in a single aptamer. Structure 2011, 19, 1413–1423. [Google Scholar] [CrossRef] [Green Version]
- Wostenberg, C.; Ceres, P.; Polaski, J.T.; Batey, R.T. A Highly Coupled Network of Tertiary Interactions in the SAM-I Riboswitch and Their Role in Regulatory Tuning. J. Mol. Biol. 2015, 427, 3473–3490. [Google Scholar] [CrossRef] [Green Version]
- Mulhbacher, J.; Lafontaine, D.A. Ligand recognition determinants of guanine riboswitches. Nucleic Acids Res. 2007, 35, 5568–5580. [Google Scholar] [CrossRef] [Green Version]
- Ceres, P.; Garst, A.D.; Marcano-Velazquez, J.G.; Batey, R.T. Modularity of select riboswitch expression platforms enables facile engineering of novel genetic regulatory devices. ACS Synth. Biol. 2013, 2, 463–472. [Google Scholar] [CrossRef] [Green Version]
- Stoddard, C.D.; Gilbert, S.D.; Batey, R.T. Ligand-dependent folding of the three-way junction in the purine riboswitch. RNA 2008, 14, 675–684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stagno, J.R.; Liu, Y.; Bhandari, Y.R.; Conrad, C.E.; Panja, S.; Swain, M.; Fan, L.; Nelson, G.; Li, C.; Wendel, D.R.; et al. Structures of riboswitch RNA reaction states by mix-and-inject XFEL serial crystallography. Nature 2017, 541, 242–246. [Google Scholar] [CrossRef] [PubMed]
- Stagno, J.R.; Bhandari, Y.R.; Conrad, C.E.; Liu, Y.; Wang, Y.X. Real-time crystallographic studies of the adenine riboswitch using an X-ray free-electron laser. Febs J. 2017, 284, 3374–3380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, M.; Bulusu, G.; Mitra, A. MD simulations of ligand-bound and ligand-free aptamer: Molecular level insights into the binding and switching mechanism of the add A-riboswitch. RNA 2009, 15, 1673–1692. [Google Scholar] [CrossRef] [Green Version]
- Wickiser, J.K.; Cheah, M.T.; Breaker, R.R.; Crothers, D.M. The kinetics of ligand binding by an adenine-sensing riboswitch. Biochemistry 2005, 44, 13404–13414. [Google Scholar] [CrossRef] [PubMed]
- Wickiser, J.K.; Winkler, W.C.; Breaker, R.R.; Crothers, D.M. The speed of RNA transcription and metabolite binding kinetics operate an FMN riboswitch. Mol. Cell 2005, 18, 49–60. [Google Scholar] [CrossRef]
- Howe, J.A.; Wang, H.; Fischmann, T.O.; Balibar, C.J.; Xiao, L.; Galgoci, A.M.; Malinverni, J.C.; Mayhood, T.; Villafania, A.; Nahvi, A.; et al. Selective small-molecule inhibition of an RNA structural element. Nature 2015, 526, 672–677. [Google Scholar] [CrossRef]
- Nelson, J.W.; Plummer, M.S.; Blount, K.F.; Ames, T.D.; Breaker, R.R. Small Molecule Fluoride Toxicity Agonists. Chem. Biol. 2015, 22, 527–534. [Google Scholar] [CrossRef] [Green Version]
- Edwards, A.L.; Garst, A.D.; Batey, R.T. Determining structures of RNA aptamers and riboswitches by X-ray crystallography. Methods Mol. Biol. 2009, 535, 135–163. [Google Scholar]
- Kao, C.; Rüdisser, S.; Zheng, M. A simple and efficient method to transcribe RNAs with reduced 3′ heterogeneity. Methods 2001, 23, 201–205. [Google Scholar] [CrossRef]
- Kao, C.; Zheng, M.; Rüdisser, S. A simple and efficient method to reduce nontemplated nucleotide addition at the 3 terminus of RNAs transcribed by T7 RNA polymerase. RNA 1999, 5, 1268–1272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minor, W.; Cymborowski, M.; Otwinowski, Z.; Chruszcz, M. HKL-3000: The integration of data reduction and structure solution--from diffraction images to an initial model in minutes. Acta Cryst. D Biol. Cryst. 2006, 62, 859–866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, P.D.; Afonine, P.V.; Bunkoczi, G.; Chen, V.B.; Echols, N.; Headd, J.J.; Hung, L.W.; Jain, S.; Kapral, G.J.; Grosse Kunstleve, R.W.; et al. The Phenix software for automated determination of macromolecular structures. Methods 2011, 55, 94–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, P.D.; Pannu, N.S.; Read, R.J.; Brunger, A.T. Extending the limits of molecular replacement through combined simulated annealing and maximum-likelihood refinement. Acta Cryst. D Biol. Cryst. 1999, 55, 181–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilbert, S.D.; Batey, R.T. Monitoring RNA-ligand interactions using isothermal titration calorimetry. Methods Mol. Biol. 2009, 540, 97–114. [Google Scholar]
- Turnbull, W.B.; Daranas, A.H. On the value of c: Can low affinity systems be studied by isothermal titration calorimetry? J. Am. Chem. Soc. 2003, 125, 14859–14866. [Google Scholar] [CrossRef]
- Ceres, P.; Trausch, J.J.; Batey, R.T. Engineering modular ‘ON’ RNA switches using biological components. Nucleic Acids Res. 2013, 41, 10449–10461. [Google Scholar] [CrossRef]
- Trausch, J.J.; Batey, R.T. A disconnect between high-affinity binding and efficient regulation by antifolates and purines in the tetrahydrofolate riboswitch. Chem. Biol. 2014, 21, 205–216. [Google Scholar] [CrossRef] [Green Version]
- Prodromou, C.; Pearl, L.H. Recursive PCR: A novel technique for total gene synthesis. Protein Eng. Des. Sel. 1992, 5, 827–829. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligand | n | KD (nM) | Krel 1 | ∆H (kJ/mol) | c Value |
|---|---|---|---|---|---|
| guanine | 1.0 ± 0.1 | 2.1 ± 0.7 | 1 | −190 ± 10 | 1400 ± 300 |
| hypoxanthine | 1.1 ± 0.1 | 240 ± 30 | 120 | −120 ± 10 | 180 ± 10 |
| C8-Modified Ligands | |||||
| 8-aminohypoxanthine | 1.0 ± 0.1 | 14,000 ± 1000 | 6600 | −290 ± 10 | 0.37 ± 0.02 |
| 8-aminoguanine | 1.2 ± 0.4 | 36 ± 13 | 17 | −70 ± 10 | 930 ± 370 |
| 8-hydroxyguanine | 1.1 ± 0.1 | 10 ± 4 | 5.0 | −110 ± 10 | 1900 ± 600 |
| C6-Modified Ligands | |||||
| 6-chloroguanine | 0.90 ± 0.02 | 340 ± 30 | 170 | −120 ± 10 | 130 ± 20 |
| O6-methylguanine | 0.82 ± 0.10 | 6400 ± 400 | 3100 | −120 ± 10 | 9.0 ± 1.9 |
| N6-cyclopropyl-9H-purine-2,6-diamine | 1.1 ± 0.1 | 25,000 ± 3000 | 12,000 | −110 ± 10 | 2.0 ± 0.2 |
| O6-benzylguanine | NA | No binding | NA | NA | NA |
| NU2058 | 1.2 ± 0.1 | 17,000 ± 1000 | 8000 | −2200 ± 600 | 0.31 ± 0.01 |
| C2-Modified Ligands | |||||
| N2-acetylguanine | 1.1 ± 0.1 | 300 ± 10 | 140 | −160 ± 10 | 100 ± 10 |
| N2-isobutyrylguanine | 1.1 ± 0.1 | 7.4 ± 0.5 | 3.6 | −210 ± 10 | 4300 ± 300 |
| N2-pivaloylguanine | 0.97 ± 0.05 | 230,000 ± 10,000 | 110,000 | −1300 ± 100 | 0.11 ± 0.01 |
| N2-methylguanine | 0.99 ± 0.03 | 17,000 ± 1000 | 8300 | −100 ± 10 | 5.2 ± 0.2 |
| N2-phenoxyacetyl guanine | 0.80 ± 0.06 | 8.8 ± 0.5 | 4.3 | −180 ± 10 | 1700 ± 100 |
| 4,6,7,8-tetrahydro-8-hydroxy-6-methylprimido[1,2-a]purin-10(3H)-one | 1.0 ± 0.2 | 79,000 ± 1000 | 38,000 | −58 ± 8 | 0.25 ± 0.01 |
| pyrimido[1,2-a]purin-10(1H)-one | NA | No binding | NA | NA | NA |
| C2-,C6-Modified Ligand | |||||
| NU6102 | 1.1 ± 0.1 | 5600 ± 600 | 2700 | −100 ± 10 | 1.2 ± 0.3 |
| Mutant | n | KD (nM) | Krel 1 | ∆H (kJ/mol) | c Value |
|---|---|---|---|---|---|
| WT | 0.80 ± 0.06 | 8.8 ± 0.5 | 1 | −180 ± 10 | 1700 ± 100 |
| C74U | 1.2 ± 0.3 | 7500 ± 800 | 850 | −96 ± 27 | 3.8 ± 0.4 |
| C74G | 1.0 ± 0.2 | 42,000 ± 2000 | 4800 | −110 ± 90 | 1.8 ± 0.2 |
| C74A | 1.0 ± 0.1 | 85,000 ± 25,000 | 9700 | −25 ± 3 | 0.36 ± 0.09 |
| A19C U77G | 1.3 ± 0.2 | 2.2 ± 0.6 | 0.25 | −110 ± 20 | 3200 ± 1000 |
| A19G U77C | 0.99 ± 0.02 | 4.9 ± 1.2 | 0.56 | −180 ± 10 | 3600 ± 1000 |
| A19G U77 | 1.0 ± 0.1 | 5.4 ± 1.2 | 0.61 | −190 ± 10 | 2800 ± 700 |
| A19U U77A | 1.0 ± 0.1 | 7.8 ± 1.6 | 0.89 | −170 ± 10 | 2300 ± 400 |
| A19C C74U U77G | 1.1 ± 0.1 | 4100 ± 400 | 470 | −190 ± 10 | 7.5 ± 0.5 |
| A19G C74U U77C | 1.0 ± 0.1 | 44,000 ± 4000 | 5000 | −150 ± 10 | 0.50 ± 0.04 |
| A19U C74U U77A | 1.0 ± 0.1 | 66,000 ± 5000 | 7500 | −120 ± 10 | 0.46 ± 0.03 |
| Ligand | n | KD (nM) | Krel 1 | ∆H (kJ/mol) | c Value |
|---|---|---|---|---|---|
| guanine | 1.0 ± 0.1 | 0.85 ± 0.31 | 1 | −130 ± 10 | 5700 ± 1800 |
| N2-isobutrylguanine | 0.98 ± 0.03 | 4.5 ± 2.0 | 5.3 | −130 ± 10 | 2600 ± 900 |
| N2-phenoxyacetyl guanine | 1.3 ± 0.2 | 2.2 ± 0.6 | 2.6 | −110 ± 20 | 3200 ± 1000 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matyjasik, M.M.; Hall, S.D.; Batey, R.T. High Affinity Binding of N2-Modified Guanine Derivatives Significantly Disrupts the Ligand Binding Pocket of the Guanine Riboswitch. Molecules 2020, 25, 2295. https://doi.org/10.3390/molecules25102295
Matyjasik MM, Hall SD, Batey RT. High Affinity Binding of N2-Modified Guanine Derivatives Significantly Disrupts the Ligand Binding Pocket of the Guanine Riboswitch. Molecules. 2020; 25(10):2295. https://doi.org/10.3390/molecules25102295
Chicago/Turabian StyleMatyjasik, Michal M., Simone D. Hall, and Robert T. Batey. 2020. "High Affinity Binding of N2-Modified Guanine Derivatives Significantly Disrupts the Ligand Binding Pocket of the Guanine Riboswitch" Molecules 25, no. 10: 2295. https://doi.org/10.3390/molecules25102295