
Unsymmetrical Bisquinolines with High Potency against P. falciparum Malaria


Abstract
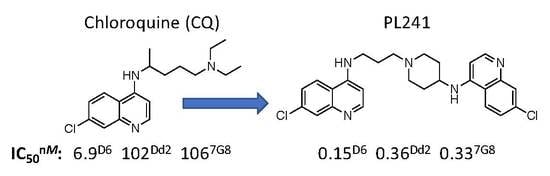
:
1. Introduction
2. Results

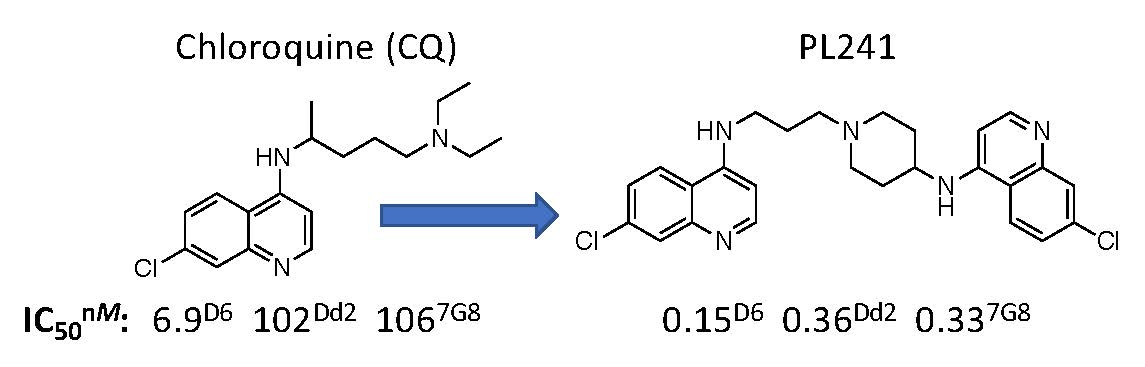{kind=link}
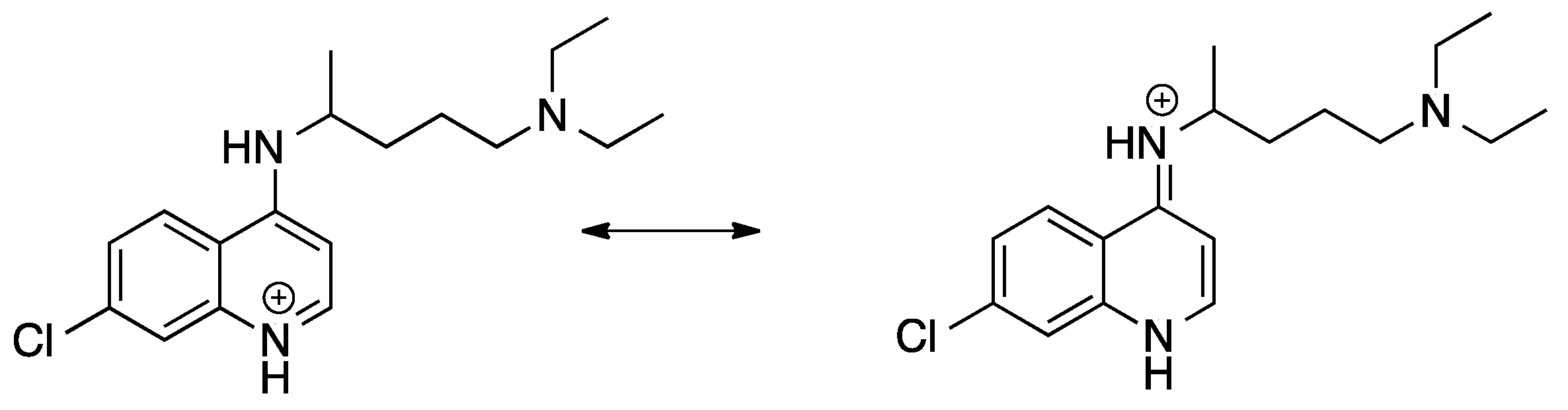{kind=link}
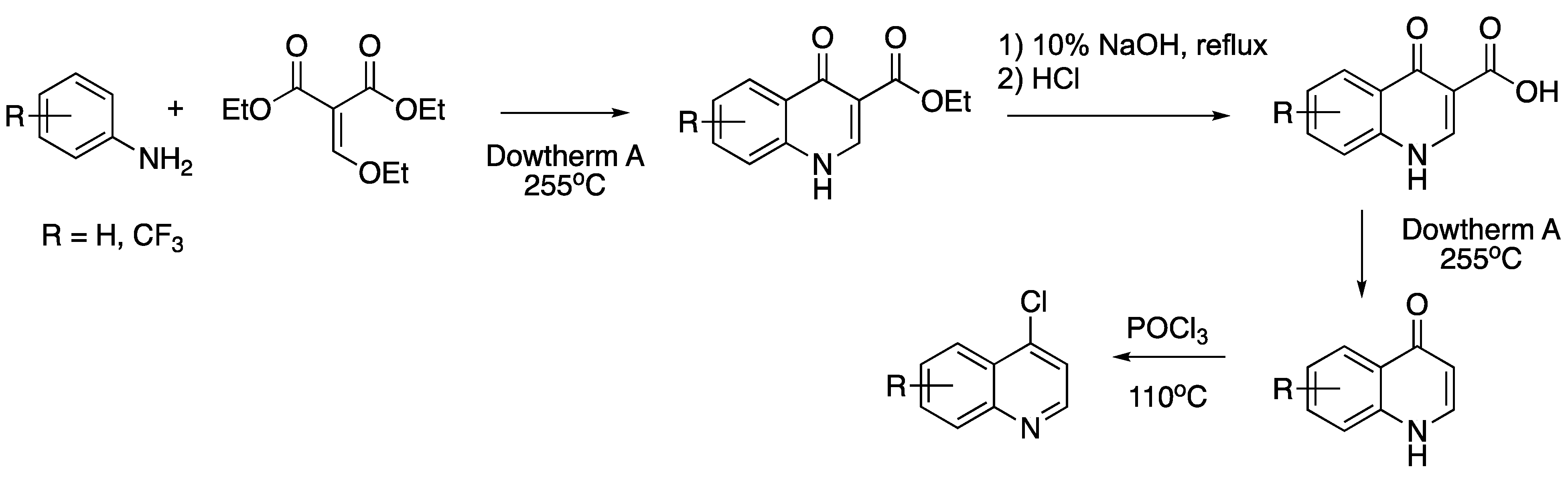{kind=link}
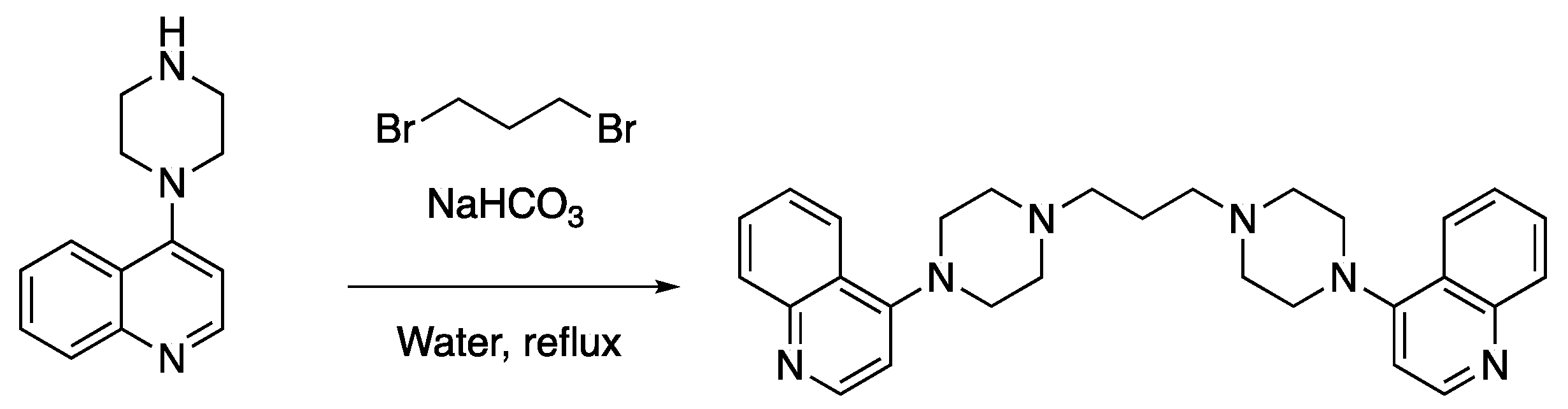{kind=link}
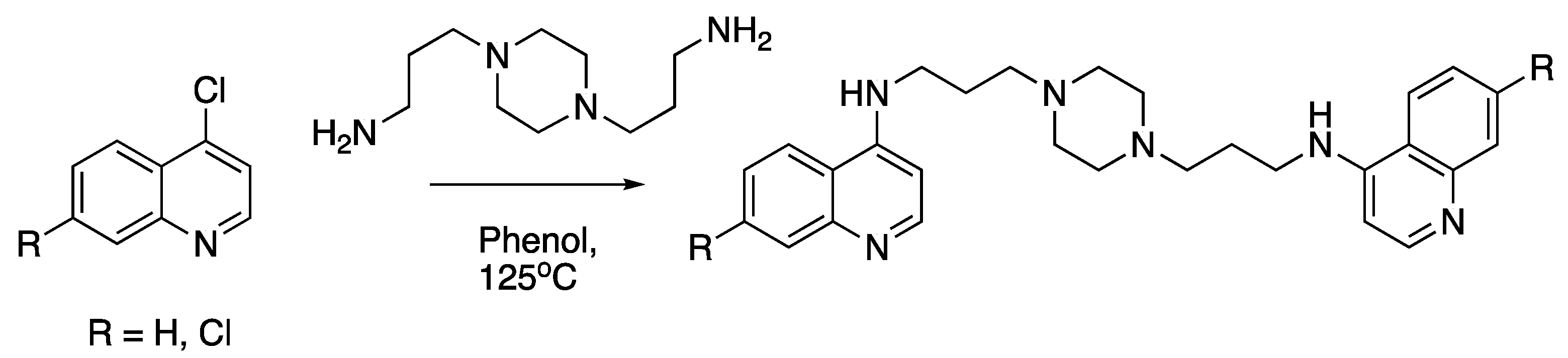{kind=link}
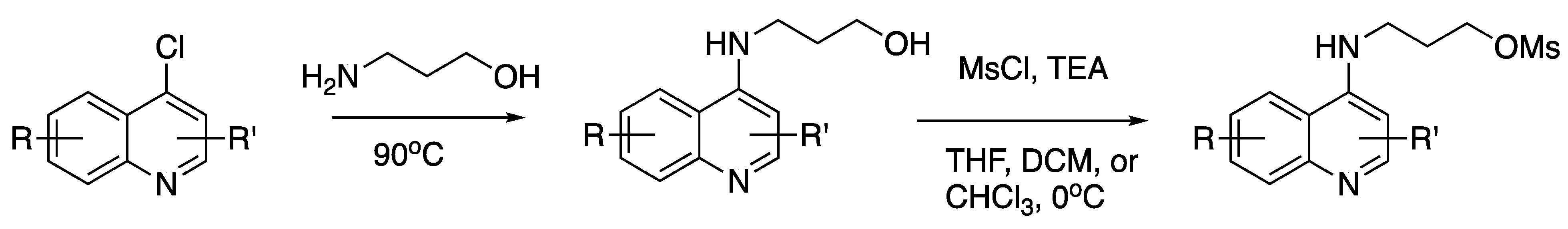{kind=link}
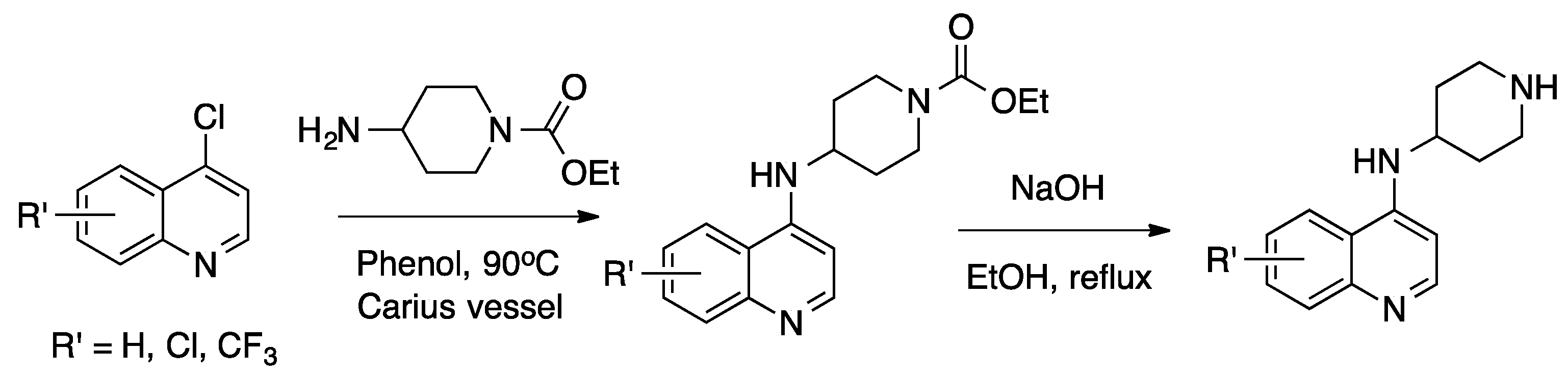{kind=link}
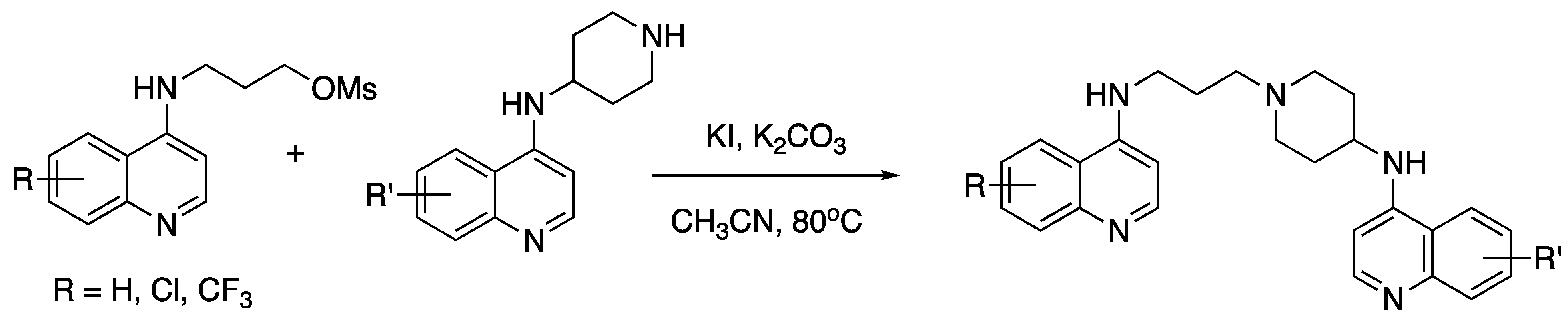{kind=link}
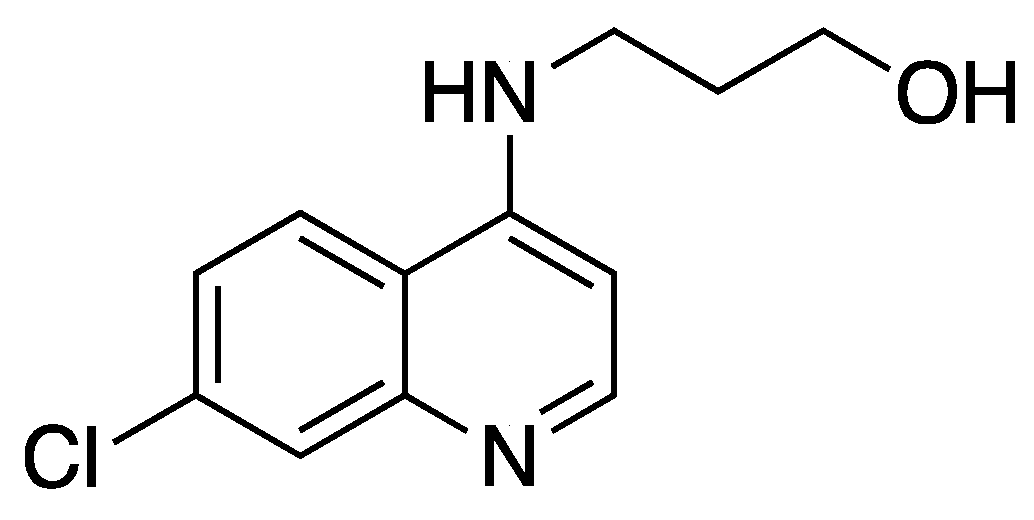{kind=link}
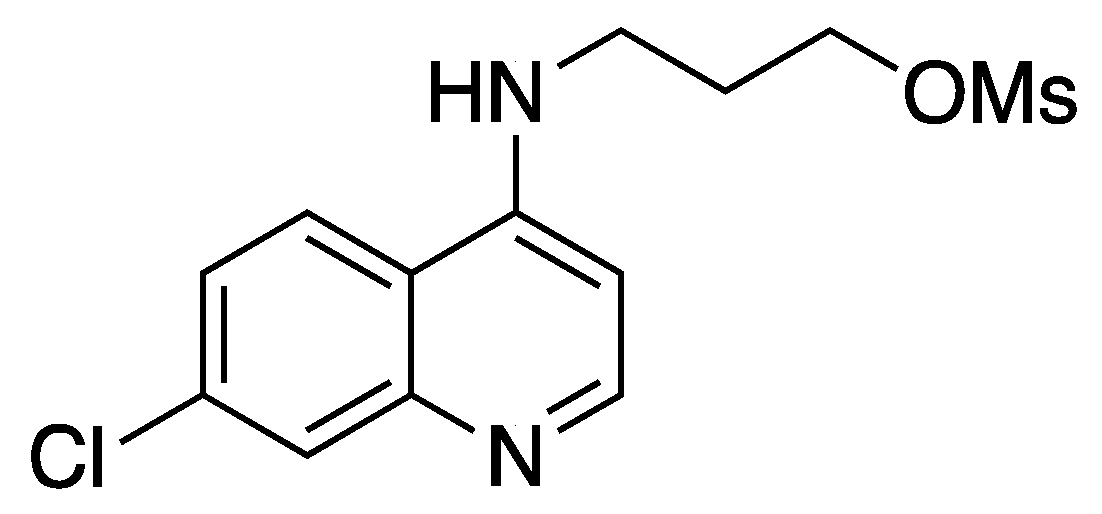{kind=link}
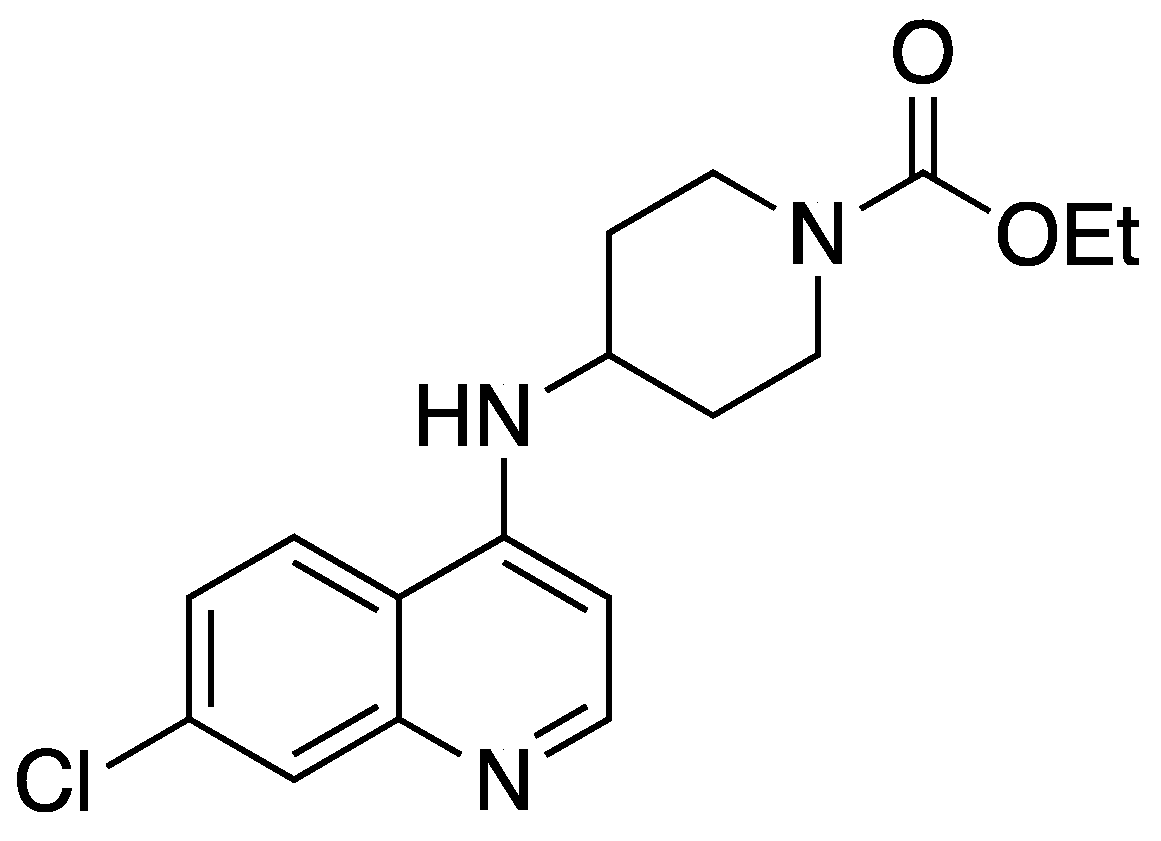{kind=link}
{kind=link}
{kind=link}
| Compound | Activity (IC50; nM) | Cytotoxicity | Structure | ||
|---|---|---|---|---|---|
| D6 | Dd2 | 7G8 | (LC50; nM) Mouse Spleen Lymphocytes | ||
| CQ | 6.9 | 102 | 106 | 12,400 |  |
| PPQ | 0.7 | 1.5 |  | ||
| 1 | 1.3 | 4.1 | 7.4 |  | |
| 2 [46] | 2.4 | 3.7 | 1.5 | 1100 |  |
| 3 | 1.5 | 5.0 | 1600 |  | |
| 4 (DM1157) [46,47] | 0.2 | 2.2 | 1.8 | 6500 |  |
| 5 [45] | 0.4 | 0.7 | 0.1 |  | |
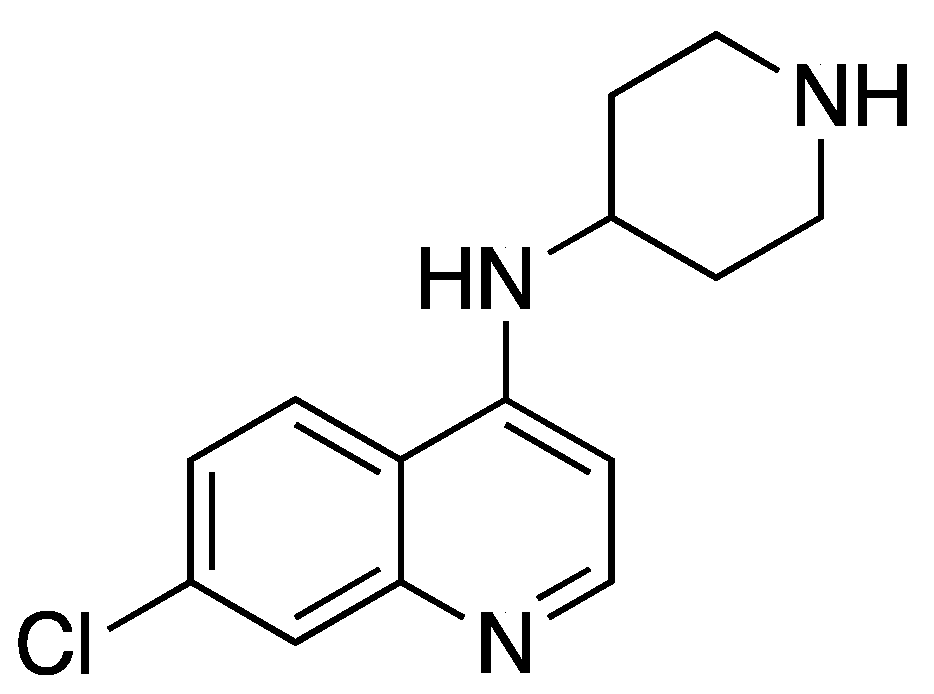
| 6 | 0.15 | 0.36 | 0.33 | 190 |  |
| 7 | 4.9 | 9.8 | 25 |  | |
| 8 | 0.68 | 2.1 | 0.63 |  | |
| 9 | 0.68 | 2.1 | 0.41 |  | |
| 10 | 15 | 89 | 106 |  | |
| 11 | 3.0 | <2.5 | <2.5 |  | |
| 12 | 4.5 | <2.5 | <2.5 |  | |
| 13 | 33 | 56 | 100 |  | |
3. Discussion
4. Materials and Methods
4.1. General Synthetic Methods
4.2. Characterization of Products
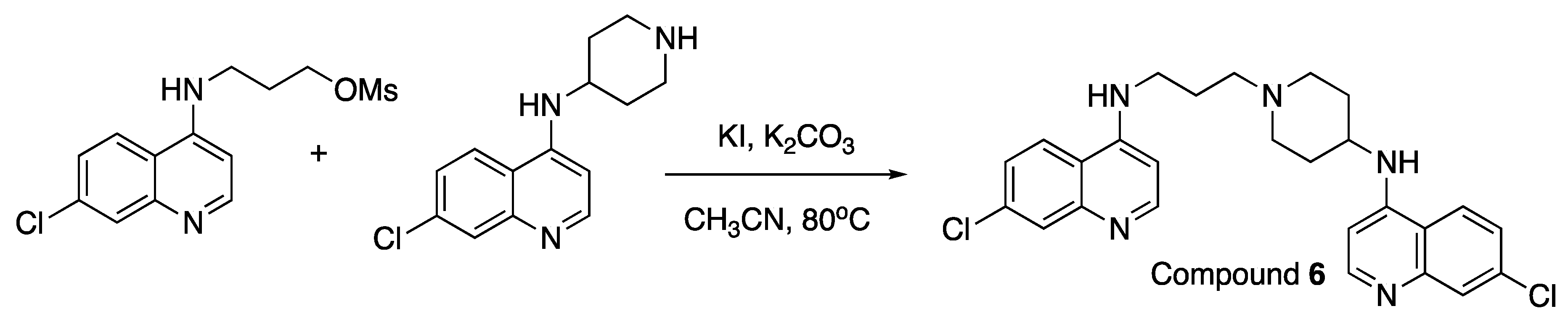
Example Synthesis: Compound 6
4.3. In Vitro Studies on Inhibition of P. falciparum Parasite Growth
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- World Health Organization (WHO). World Malaria Report 2016; World Health Organization Press: Geneva, Switzerland, 2016. [Google Scholar]
- World Health Organization (WHO). World Malaria Report 2019; World Health Organization Press: Geneva, Switzerland, 2019. [Google Scholar]
- Menard, D.; Dondorp, A. Antimalarial Drug Resistance: A Threat to Malaria Elimination. Cold Spring Harb. Perspect. Med. 2017, 7. [Google Scholar] [CrossRef] [Green Version]
- Nsanzabana, C. Resistance to Artemisinin Combination Therapies (ACTs): Do Not Forget the Partner Drug! Trop. Med. Infect. Dis. 2019, 4, 26. [Google Scholar] [CrossRef] [Green Version]
- Ross, L.S.; Fidock, D.A. Elucidating Mechanisms of Drug-Resistant Plasmodium falciparum. Cell Host Microbe 2019, 26, 35–47. [Google Scholar] [CrossRef] [Green Version]
- Keating, G.M. Dihydroartemisinin/Piperaquine: A review of its use in the treatment of uncomplicated Plasmodium falciparum malaria. Drugs 2012, 72, 937–961. [Google Scholar] [CrossRef]
- World Health Organization. Guidelines for the Treatment of Malaria, 3rd ed.; World Health Organization: Geneva, Switzerland, 2015. [Google Scholar]
- Au, B. Nouveaux Dérivés de la Quinoléine et Leur Préparation. France Patent FR84902, 14 June 1962. [Google Scholar]
- Benazet, F. Plasmodium berghei and prolonged-action antimalarials. Ann. Soc. Belges. Med. Trop. Parasitol. Mycol. 1965, 45, 459–472. [Google Scholar]
- Schneider, J.; Bouvry, M.; Le Quellec, J. Plasmodium berghei and chemotherapy. Ann. Soc. Belges. Med. Trop. Parasitol. Mycol. 1965, 45, 435–449. [Google Scholar]
- Brevent. Nouveaux dérivés de la quinoléine et leur préparation. Belgium Patent BE633453, 1963. [Google Scholar]
- Chen, C. Development of antimalarial drugs and their application in China: A historical review. Infect. Dis. Poverty 2014, 3, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, T.M.; Hung, T.Y.; Sim, I.K.; Karunajeewa, H.A.; Ilett, K.F. Piperaquine: A resurgent antimalarial drug. Drugs 2005, 65, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Schlitzer, M. Malaria chemotherapeutics part I: History of antimalarial drug development, currently used therapeutics, and drugs in clinical development. Chem. Med. Chem. 2007, 2, 944–986. [Google Scholar] [CrossRef] [PubMed]
- Warhurst, D.C.; Craig, J.C.; Adagu, I.S.; Guy, R.K.; Madrid, P.B.; Fivelman, Q.L. Activity of piperaquine and other 4-aminoquinoline antiplasmodial drugs against chloroquine-sensitive and resistant blood-stages of Plasmodium falciparum. Role of beta-haematin inhibition and drug concentration in vacuolar water- and lipid-phases. Biochem. Pharmacol. 2007, 73, 1910–1926. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Qu, F.Y.; Zhou, Y.C. Field observations on the antimalarial piperaquine. Chin. Med. J. 1982, 95, 281–286. [Google Scholar] [PubMed]
- Kondaparla, S.; Agarwal, P.; Srivastava, K.; Puri, S.K.; Katti, S.B. Design, synthesis and in vitro antiplasmodial activity of some bisquinolines against chloroquine-resistant strain. Chem. Biol. Drug Des. 2017, 89, 901–906. [Google Scholar] [CrossRef]
- Naude, B.; Brzostowski, J.A.; Kimmel, A.R.; Wellems, T.E. Dictyostelium discoideum expresses a malaria chloroquine resistance mechanism upon transfection with mutant, but not wild-type, Plasmodium falciparum transporter PfCRT. J. Biol. Chem. 2005, 280, 25596–25603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vennerstrom, J.L.; Ellis, W.Y.; Ager, A.L., Jr.; Andersen, S.L.; Gerena, L.; Milhous, W.K. Bisquinolines. 1. N,N-bis(7-chloroquinolin-4-yl)alkanediamines with potential against chloroquine-resistant malaria. J. Med. Chem. 1992, 35, 2129–2134. [Google Scholar] [CrossRef] [PubMed]
- Basco, L.K.; Ringwald, P. In vitro activities of piperaquine and other 4-aminoquinolines against clinical isolates of Plasmodium falciparum in Cameroon. Antimicrob. Agents Chemother. 2003, 47, 1391–1394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, Y.; Zhang, J.; Geng, J.; Xu, S.; Deng, S.; Zeng, W.; Wang, Z.; Ngassa Mbenda, H.G.; Zhang, J.; Li, N.; et al. Longitudinal surveillance of drug resistance in Plasmodium falciparum isolates from the China-Myanmar border reveals persistent circulation of multidrug resistant parasites. Int. J. Parasitol. Drugs Drug Resist. 2018, 8, 320–328. [Google Scholar] [CrossRef]
- Lon, C.; Manning, J.E.; Vanachayangkul, P.; So, M.; Sea, D.; Se, Y.; Gosi, P.; Lanteri, C.; Chaorattanakawee, S.; Sriwichai, S.; et al. Efficacy of two versus three-day regimens of dihydroartemisinin-piperaquine for uncomplicated malaria in military personnel in northern Cambodia: An open-label randomized trial. PLoS ONE 2014, 9, e93138. [Google Scholar] [CrossRef]
- van der Pluijm, R.W.; Imwong, M.; Chau, N.H.; Hoa, N.T.; Thuy-Nhien, N.T.; Thanh, N.V.; Jittamala, P.; Hanboonkunupakarn, B.; Chutasmit, K.; Saelow, C.; et al. Determinants of dihydroartemisinin-piperaquine treatment failure in Plasmodium falciparum malaria in Cambodia, Thailand, and Vietnam: A prospective clinical, pharmacological, and genetic study. Lancet Infect. Dis. 2019, 19, 952–961. [Google Scholar] [CrossRef] [Green Version]
- Cowell, A.N.; Winzeler, E.A. The genomic architecture of antimalarial drug resistance. Brief. Funct. Genom. 2019, 18, 314–328. [Google Scholar] [CrossRef] [Green Version]
- Briolant, S.; Henry, M.; Oeuvray, C.; Amalvict, R.; Baret, E.; Didillon, E.; Rogier, C.; Pradines, B. Absence of association between piperaquine in vitro responses and polymorphisms in the pfcrt, pfmdr1, pfmrp, and pfnhe genes in Plasmodium falciparum. Antimicrob. Agents Chemother. 2010, 54, 3537–3544. [Google Scholar] [CrossRef] [Green Version]
- Eastman, R.T.; Dharia, N.V.; Winzeler, E.A.; Fidock, D.A. Piperaquine resistance is associated with a copy number variation on chromosome 5 in drug-pressured Plasmodium falciparum parasites. Antimicrob. Agents Chemother. 2011, 55, 3908–3916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hao, M.; Jia, D.; Li, Q.; He, Y.; Yuan, L.; Xu, S.; Chen, K.; Wu, J.; Shen, L.; Sun, L.; et al. In vitro sensitivities of Plasmodium falciparum isolates from the China-Myanmar border to piperaquine and association with polymorphisms in candidate genes. Antimicrob. Agents Chemother. 2013, 57, 1723–1729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pascual, A.; Madamet, M.; Bertaux, L.; Amalvict, R.; Benoit, N.; Travers, D.; Cren, J.; Taudon, N.; Rogier, C.; Parzy, D.; et al. In vitro piperaquine susceptibility is not associated with the Plasmodium falciparum chloroquine resistance transporter gene. Malar. J. 2013, 12, 431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ross, L.S.; Dhingra, S.K.; Mok, S.; Yeo, T.; Wicht, K.J.; Kumpornsin, K.; Takala-Harrison, S.; Witkowski, B.; Fairhurst, R.M.; Ariey, F.; et al. Emerging Southeast Asian PfCRT mutations confer Plasmodium falciparum resistance to the first-line antimalarial piperaquine. Nat. Commun. 2018, 9, 3314. [Google Scholar] [CrossRef] [Green Version]
- Akhigbe, J.; Luciano, M.; Zeller, M.; Bruckner, C. Mono- and Bisquinoline-Annulated Porphyrins from Porphyrin beta,beta’-Dione Oximes. J. Org. Chem. 2015, 80, 499–511. [Google Scholar] [CrossRef]
- Basco, L.K.; Andersen, S.L.; Milhous, W.K.; Le Bras, J.; Vennerstrom, J.L. In vitro activity of bisquinoline WR268,668 against African clones and isolates of Plasmodium falciparum. Am. J. Trop. Med. Hyg. 1994, 50, 200–205. [Google Scholar] [CrossRef]
- Elslager, E.F. Progress in malaria chemotherapy. 1. Repository antimalarial drugs. Prog. Drug. Res. 1969, 13, 170–216. [Google Scholar] [CrossRef]
- Raynes, K. Bisquinoline antimalarials: Their role in malaria chemotherapy. Int. J. Parasitol. 1999, 29, 367–379. [Google Scholar] [CrossRef]
- Raynes, K.; Foley, M.; Tilley, L.; Deady, L.W. Novel bisquinoline antimalarials. Synthesis, antimalarial activity, and inhibition of haem polymerisation. Biochem. Pharmacol. 1996, 52, 551–559. [Google Scholar] [CrossRef]
- Ridley, R.G.; Matile, H.; Jaquet, C.; Dorn, A.; Hofheinz, W.; Leupin, W.; Masciadri, R.; Theil, F.P.; Richter, W.F.; Girometta, M.A.; et al. Antimalarial activity of the bisquinoline trans-N1,N2-bis (7-chloroquinolin-4-yl)cyclohexane-1,2-diamine: Comparison of two stereoisomers and detailed evaluation of the S,S enantiomer, Ro 47–7737. Antimicrob. Agents Chemother. 1997, 41, 677–686. [Google Scholar] [CrossRef] [Green Version]
- van Heerden, L.; Cloete, T.T.; Breytenbach, J.W.; de Kock, C.; Smith, P.J.; Breytenbach, J.C.; N’Da, D.D. Synthesis and in vitro antimalarial activity of a series of bisquinoline and bispyrrolo[1,2a]quinoxaline compounds. Eur. J. Med. Chem. 2012, 55, 335–345. [Google Scholar] [CrossRef] [PubMed]
- Vennerstrom, J.L.; Ager, A.L., Jr.; Dorn, A.; Andersen, S.L.; Gerena, L.; Ridley, R.G.; Milhous, W.K. Bisquinolines. 2. Antimalarial N,N-bis(7-chloroquinolin-4-yl)heteroalkanediamines. J. Med. Chem. 1998, 41, 4360–4364. [Google Scholar] [CrossRef]
- Girault, S.; Grellier, P.; Berecibar, A.; Maes, L.; Lemiere, P.; Mouray, E.; Davioud-Charvet, E.; Sergheraert, C. Antiplasmodial activity and cytotoxicity of bis-, tris-, and tetraquinolines with linear or cyclic amino linkers. J. Med. Chem. 2001, 44, 1658–1665. [Google Scholar] [CrossRef] [PubMed]
- Girault, S.; Grellier, P.; Berecibar, A.; Maes, L.; Mouray, E.; Lemiere, P.; Debreu, M.A.; Davioud-Charvet, E.; Sergheraert, C. Antimalarial, antitrypanosomal, and antileishmanial activities and cytotoxicity of bis(9-amino-6-chloro-2-methoxyacridines): Influence of the linker. J. Med. Chem. 2000, 43, 2646–2654. [Google Scholar] [CrossRef] [PubMed]
- Ryckebusch, A.; Deprez-Poulain, R.; Maes, L.; Debreu-Fontaine, M.A.; Mouray, E.; Grellier, P.; Sergheraert, C. Synthesis and in vitro and in vivo antimalarial activity of N1-(7-chloro-4-quinolyl)-1,4-bis(3-aminopropyl)piperazine derivatives. J. Med. Chem. 2003, 46, 542–557. [Google Scholar] [CrossRef] [PubMed]
- Dorn, A.; Vippagunta, S.R.; Matile, H.; Bubendorf, A.; Vennerstrom, J.L.; Ridley, R.G. A Comparison and Analysis of Several Ways to Promote Haematin (Haem) Polymerisation and an Assessment of Its Initiation In Vitro. Biochem. Pharmacol. 1998, 55, 737–747. [Google Scholar] [CrossRef]
- Dorn, A.; Vippagunta, S.R.; Matile, H.; Jaquet, C.; Vennerstrom, J.L.; Ridley, R.G. An Assessment of Drug-Haematin Binding as a Mechanism for Inhibition of Haematin Polymerisation by Quinoline Antimalarials. Biochem. Pharmacol. 1998, 55, 727–736. [Google Scholar] [CrossRef]
- Karle, J.M.; Bhattacharjee, A.K.; Vennerstrom, J.L. Crystal structure of the potent bisquinoline antimalarial agent (+/−)-trans-N-1,N-2-bis(7-chloroquinolin-4-yl) cyclohexane-1,2-diamine dimethanesulfonate salt hydrate in relation to its biological properties. J. Chem. Crystallogr. 2002, 32, 133–139. [Google Scholar] [CrossRef]
- Ridley, R.G.; Dorn, A.; Vippagunta, S.R.; Vennerstrom, J.L. Haematin (haem) polymerization and its inhibition by quinoline antimalarials. Ann. Trop. Med. Parasitol. 1997, 91, 559–566. [Google Scholar] [CrossRef]
- Brevent. Nouveaux dérivés de la quinoléine et leur préparation. Belgium Patent BE612207, 2 July 1962. [Google Scholar]
- Burgess, S.J.; Kelly, J.X.; Shomloo, S.; Wittlin, S.; Brun, R.; Liebmann, K.; Peyton, D.H. Synthesis, structure-activity relationship, and mode-of-action studies of antimalarial reversed chloroquine compounds. J. Med. Chem. 2010, 53, 6477–6489. [Google Scholar] [CrossRef] [Green Version]
- Peyton, D.H. Reversed chloroquine molecules as a strategy to overcome resistance in malaria. Curr. Top. Med. Chem. 2012, 12, 400–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burgess, S.J. Design and Synthesis of Antimalarial Drugs Based on a Chloroquine Scaffold, Eds; ProQuest Limited Liability Company, Portland State University: Portland, OR, USA, 2008. [Google Scholar]
- Schönhöfer, F. Über die Bedeutung der chinoiden Bindung in Chinolinverbindungen für die Malariawirkung. Hoppe-Seyler’s Zeitschrift für Physiologische Chemie 1942, 274, 1–8. [Google Scholar] [CrossRef]
- Wiselogle, F.Y. A Survey of Antimalarial Drugs, 1941–1945; J. W. Edwards: Ann Arbor, MI, USA, 1946; p. 2500. [Google Scholar]
- Andersag, H. Antimalariamittel aus der Gruppe halogensubstituierter Chinolinverbindungen. Chem. Ber. 1948, 81, 499–507. [Google Scholar] [CrossRef]
- Coatney, G.R.; Cooper, W.C.; Eddy, N.B.; Greenberg, J. Survey of antimalarial agents: Chemotherapy of Plasmodium gallinaceum infections; toxicity; correlation of structure and action. Public Health Monogr. 1953, 9, 1–322. [Google Scholar]
- Mietzsch, F. Trends of progress in chemotherapy. Klin. Wochenschr. 1951, 29, 125–135. [Google Scholar] [CrossRef]
- Dann, O.; Steuding, W.; Lisson, K.G.; Seidel, H.R.; Fink, E.; Nickel, P. Antimalarial 6-aminoquinolines XV. 6- and 4-aminoquinolines with a tertiary basic alkylated amino group. Gegen Malaria wirksame 6-Aminochinoline. Arzneimittel-Forsch. 1982, 32, 1219–1223. [Google Scholar]
- Egan, T.J.; Hunter, R.; Kaschula, C.H.; Marques, H.M.; Misplon, A.; Walden, J. Structure-function relationships in aminoquinolines: Effect of amino and chloro groups on quinoline-hematin complex formation, inhibition of beta-hematin formation, and antiplasmodial activity. J. Med. Chem. 2000, 43, 283–291. [Google Scholar] [CrossRef]
- Pisciotta, J.M.; Coppens, I.; Tripathi, A.K.; Scholl, P.F.; Shuman, J.; Bajad, S.; Shulaev, V.; Sullivan, D.J., Jr. The role of neutral lipid nanospheres in Plasmodium falciparum haem crystallization. Biochem. J. 2007, 402, 197–204. [Google Scholar] [CrossRef] [Green Version]
- Egan, T.J. Haemozoin formation. Mol. Biochem. Parasitol. 2008, 157, 127–136. [Google Scholar] [CrossRef]
- Ambele, M.A.; Sewell, B.T.; Cummings, F.R.; Smith, P.J.; Egan, T.J. Synthetic Hemozoin (beta-Hematin) Crystals Nucleate at the Surface of Neutral Lipid Droplets that Control Their Sizes. Cryst. Growth. Des. 2013, 13, 4442–4452. [Google Scholar] [CrossRef] [Green Version]
- Andrews, S.; Burgess, S.J.; Skaalrud, D.; Kelly, J.X.; Peyton, D.H. Reversal agent and linker variants of reversed chloroquines: Activities against Plasmodium falciparum. J. Med. Chem. 2010, 53, 916–919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madrid, P.B.; Sherrill, J.; Liou, A.P.; Weisman, J.L.; Derisi, J.L.; Guy, R.K. Synthesis of ring-substituted 4-aminoquinolines and evaluation of their antimalarial activities. Bioorg. Med. Chem. Lett. 2005, 15, 1015–1018. [Google Scholar] [CrossRef] [PubMed]
- Barlin, G.B.; Nguyen, T.M.T.; Kotecka, B.; Rieckmann, K.H. Potential Antimalarials. XVII. Di- and Mono-Mannich Bases of 2(and 4)-[2(and 8)-Trifluoromethylquinolin-4-ylamino]phenol. Aust. J. Chem. 1993, 46, 21–29. [Google Scholar] [CrossRef]
- Barlin, G.B.; Tan, W.L. Potential Antimalarials. V. 4-(7′-Trifluoromethylquinolin-4′-Ylamino)Phenols, 4-[2′,7′ and 2’,8′-Bis(Trifluoromethyl)Quinolin-4′-Ylamino]Phenols and N4-Substituted 2,7-(and 2,8-)Bis(Trifluoromethyl)-Quinolin-4-Amines. Aust. J. Chem. 1985, 38, 1827–1835. [Google Scholar] [CrossRef]
- Fielding, A.J.; Lukinovic, V.; Evans, P.G.; Alizadeh-Shekalgourabi, S.; Bisby, R.H.; Drew, M.G.B.; Male, V.; Del Casino, A.; Dunn, J.F.; Randle, L.E.; et al. Modulation of Antimalarial Activity at a Putative Bisquinoline Receptor In Vivo Using Fluorinated Bisquinolines. Chemistry 2017, 23, 6811–6828. [Google Scholar] [CrossRef] [Green Version]
- Gemma, S.; Campiani, G.; Butini, S.; Joshi, B.P.; Kukreja, G.; Coccone, S.S.; Bernetti, M.; Persico, M.; Nacci, V.; Fiorini, I.; et al. Combining 4-aminoquinoline- and clotrimazole-based pharmacophores toward innovative and potent hybrid antimalarials. J. Med. Chem. 2009, 52, 502–513. [Google Scholar] [CrossRef] [Green Version]
- Kgokong, J.L.; Matsabisa, G.M.; Smithand, P.P.; Breytenbach, J.C. N,N-Bis(trifluoromethylquinolin-4-yl)diamino alkanes: Synthesis and antimalarial activity. Med. Chem. 2008, 4, 438–445. [Google Scholar] [CrossRef]
- Vippagunta, S.R.; Dorn, A.; Matile, H.; Bhattacharjee, A.K.; Karle, J.M.; Ellis, W.Y.; Ridley, R.G.; Vennerstrom, J.L. Structural specificity of chloroquine-hematin binding related to inhibition of hematin polymerization and parasite growth. J. Med. Chem. 1999, 42, 4630–4639. [Google Scholar] [CrossRef]
- Blackie, M.A.; Beagley, P.; Croft, S.L.; Kendrick, H.; Moss, J.R.; Chibale, K. Metallocene-based antimalarials: An exploration into the influence of the ferrocenyl moiety on in vitro antimalarial activity in chloroquine-sensitive and chloroquine-resistant strains of Plasmodium falciparum. Bioorg. Med. Chem. 2007, 15, 6510–6516. [Google Scholar] [CrossRef]
- De, D.; Krogstad, F.M.; Cogswell, F.B.; Krogstad, D.J. Aminoquinolines that circumvent resistance in Plasmodium falciparum in vitro. Am. J. Trop. Med. Hyg. 1996, 55, 579–583. [Google Scholar] [CrossRef]
- Kaschula, C.H.; Egan, T.J.; Hunter, R.; Basilico, N.; Parapini, S.; Taramelli, D.; Pasini, E.; Monti, D. Structure-activity relationships in 4-aminoquinoline antiplasmodials. The role of the group at the 7-position. J. Med. Chem. 2002, 45, 3531–3539. [Google Scholar] [CrossRef] [PubMed]
- Perez, B.C.; Teixeira, C.; Albuquerque, I.S.; Gut, J.; Rosenthal, P.J.; Gomes, J.R.; Prudencio, M.; Gomes, P. N-cinnamoylated chloroquine analogues as dual-stage antimalarial leads. J. Med. Chem. 2013, 56, 556–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pesic, D.; Starcevic, K.; Toplak, A.; Herreros, E.; Vidal, J.; Almela, M.J.; Jelic, D.; Alihodzic, S.; Spaventi, R.; Peric, M. Design, synthesis, and in vitro activity of novel 2’-O-substituted 15-membered azalides. J. Med. Chem. 2012, 55, 3216–3227. [Google Scholar] [CrossRef] [PubMed]
- Terzic, N.; Konstantinovic, J.; Tot, M.; Burojevic, J.; Djurkovic-Djakovic, O.; Srbljanovic, J.; Stajner, T.; Verbic, T.; Zlatovic, M.; Machado, M.; et al. Reinvestigating Old Pharmacophores: Are 4-Aminoquinolines and Tetraoxanes Potential Two-Stage Antimalarials? J. Med. Chem. 2016, 59, 264–281. [Google Scholar] [CrossRef]
- Warhurst, D.C.; Gould, S. The chemotherapy of rodent malaria XXXIII. The activity of chloroquine and related blood schizontocides and of some analogues in drug-induced pigment clumping. Ann. Trop. Med. Parasitol. 1982, 76, 257–264. [Google Scholar] [CrossRef]
- De, D.; Krogstad, F.M.; Byers, L.D.; Krogstad, D.J. Structure-activity relationships for antiplasmodial activity among 7-substituted 4-aminoquinolines. J. Med. Chem. 1998, 41, 4918–4926. [Google Scholar] [CrossRef]
- Claisen, L. Untersuchungen über die Oxymethylenverbindungen. (Zweite Abhandlung.). Justus Liebig’s Annalen der Chemie 1897, 297, 1–98. [Google Scholar] [CrossRef] [Green Version]
- Price, C.C.; Roberts, R.M. The synthesis of 4-hydroxyquinolines; through ethoxymethylene malonic ester. J. Am. Chem. Soc. 1946, 68, 1204–1208. [Google Scholar] [CrossRef]
- Gould, R.G.; Jacobs, W.A. The Synthesis of Certain Substituted Quinolines and 5,6-Benzoquinolines. J. Am. Chem. Soc. 1939, 61, 2890–2895. [Google Scholar] [CrossRef]
- Abel, M.D.; Luu, H.T.; Micetich, R.G.; Nguyen, D.Q.; Oreski, A.B.; Tempest, M.L.; Daneshtalab, M. Synthesis of azolylalkylquinolines with cytotoxic activity. J. Heterocyclic. Chem. 1996, 33, 415–420. [Google Scholar] [CrossRef]
- Ghosh, B.; Antonio, T.; Zhen, J.; Kharkar, P.; Reith, M.E.; Dutta, A.K. Development of (S)-N6-(2-(4-(isoquinolin-1-yl)piperazin-1-yl)ethyl)-N6-propyl-4,5,6,7-tetrahydro benzo[d]-thiazole-2,6-diamine and its analogue as a D3 receptor preferring agonist: Potent in vivo activity in Parkinson’s disease animal models. J. Med. Chem. 2010, 53, 1023–1037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Surrey, A.R.; Cutler, R.A. The Role of Phenol in the Reaction of 4,7-Dichloroquinoline with Novol Diamine. J. Am. Chem. Soc. 1951, 73, 2623–2626. [Google Scholar] [CrossRef]
- Iwasaki, N.; Sakaguchi, J.; Ohashi, T.; Takahara, E.; Ogawa, N.; Yasuda, S.; Koshinaka, E.; Kato, H.; Ito, Y.; Sawanishi, H. Amphoteric drugs. I. Synthesis and antiallergic activity of [4-(diphenylmethoxy)piperidino]-, [4-(diphenylmethyl)piperazinyl]- and [4-(diphenylmethylene)piperidino]alkanoic acid derivatives. Chem. Pharm. Bull. (Tokyo) 1994, 42, 2276–2284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burgess, S.J.; Selzer, A.; Kelly, J.X.; Smilkstein, M.J.; Riscoe, M.K.; Peyton, D.H. A chloroquine-like molecule designed to reverse resistance in Plasmodium falciparum. J. Med. Chem. 2006, 49, 5623–5625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smilkstein, M.; Sriwilaijaroen, N.; Kelly, J.X.; Wilairat, P.; Riscoe, M. Simple and inexpensive fluorescence-based technique for high-throughput antimalarial drug screening. Antimicrob. Agents Chemother. 2004, 48, 1803–1806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Sample Availability: Not available. |












© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liebman, K.M.; Burgess, S.J.; Gunsaru, B.; Kelly, J.X.; Li, Y.; Morrill, W.; Liebman, M.C.; Peyton, D.H. Unsymmetrical Bisquinolines with High Potency against P. falciparum Malaria. Molecules 2020, 25, 2251. https://doi.org/10.3390/molecules25092251
Liebman KM, Burgess SJ, Gunsaru B, Kelly JX, Li Y, Morrill W, Liebman MC, Peyton DH. Unsymmetrical Bisquinolines with High Potency against P. falciparum Malaria. Molecules. 2020; 25(9):2251. https://doi.org/10.3390/molecules25092251
Chicago/Turabian StyleLiebman, Katherine M., Steven J. Burgess, Bornface Gunsaru, Jane X. Kelly, Yuexin Li, Westin Morrill, Michael C. Liebman, and David H. Peyton. 2020. "Unsymmetrical Bisquinolines with High Potency against P. falciparum Malaria" Molecules 25, no. 9: 2251. https://doi.org/10.3390/molecules25092251