Commercially available reagents were purchased from Fluka (Sydney, NSW, Australia), Aldrich (Sydney, NSW, Australia), Acros Organics (Morris Plains, NJ, USA), Alfa Aesar (Lancashire, UK) and Lancaster (Lancashire, UK) and purified if necessary. The synthetic procedures have been reported for all compounds as general methods and appropriate references have been given for known compounds. 1H (300 MHz) and 13C-NMR (75 MHz) spectra were obtained in the designated solvents on a DPX 300 spectrometer (Bruker, Sydney, NSW, Australia). Melting points were measured using a Mel-Temp melting point apparatus and are uncorrected. Infrared spectra were recorded on Avatar Series FT-IR spectrophotometer as KBr disks (Thermo Nicolet, Waltham, MA, USA). Ultraviolet spectra were measured using a Cary 100 spectrophotometer (Varian, Santa Clara, CA, USA) in the designated solvents and data reported as wavelength (λ) in nm and adsorption coefficient (ε) in cm−1M−1. High-resolution [ESI] mass spectra were recorded by the UNSW Bioanalytical Mass Spectrometry Facility, on an Orbitrap LTQ XL (Thermo Scientific, Waltham, MA, USA) ion trap mass spectrometer using a nanospray (nano-electrospray) ionization source.
3.1.6. GP-6 General Procedure for the Synthesis of Dihydropyranoindoles:
A solution of alkyne indole ethers (1.04 mmol) in chlorobenzene (20 mL) was heated under reflux until TLC analysis showed consumption of the starting indole (12 h). The heating was discontinued and the solvent was evaporated under reduced pressure. The crude product was purified using flash column chromatography (SiO2), eluted with 30% dichloromethane/n-hexane, to give the dihydropyranoindole.
Methyl 2-azido-3-(3,4-dibenzyloxyphenyl)-propenoate (13) The title compound was prepared as described in GP-2 from 3,4-dibenzyloxybenzaldehyde (12) (2.95 g, 9.3 mmol) and methyl azidoacetate (10.69 g, 93 mmol) in anhydrous methanol (30 mL) to give the product (2.27 g, 59%) as a pale yellow granular solid; m.p. 114–116 °C; IR (KBr): vmax 2917, 2119, 1701, 1683, 1590, 1508, 1432, 1379, 1233, 1201, 1130, 999, 802, 728 cm−1; UV-vis (CH3CN): λmax325 (24,500); 1H-NMR: (300 MHz, CDCl3): δ 3.91 (s, 3H, CO2Me), 5.25 (s, 4H, 2 × O-CH2), 6.83 (s, 1H, CH=C), 7.35–7.52 (m, 12H, ArH), 7.64 (d, J = 2.2 Hz, 1H, H2′); 13C-NMR: (75.6 MHz, CDCl3): δ 52.7 (CO2Me), 70.9 (CH2), 71.3 (CH2), 113.8 (C2′), 116.6 (C5′), 123.3 (CH=C), 125.4 (C6′), 125.7 (ArC), 126.6 (ArC), 127.1 (2 × ArC), 127.2 (2 × ArC), 127.8 (ArC), 127.9 (ArC), 128.5 (2 × ArC), 128.6 (2 × ArC), 136.8 (C1′), 137.1 (CH=C), 148.3 (C4′), 150.1 (C3′), 164.1 (CO2Me); HRMS (+ESI): Found m/z 438.1439 [M + Na]+, C24H21N3O4Na requires 438.1439.
Methyl 5,6-dibenzyloxyindole-2-carboxylate (14) The title compound was prepared as described in GP-3 from methyl 2-azido-3-(3′,4′-dibenzyloxyphenyl)propenoate (13) (2.76 g, 6.66 mmol) in xylene (50 mL) to give the product (1.84 g, 71%) as a yellow granular solid; m.p. 148–150 °C; IR (KBr): vmax 3309, 2942, 2871, 2113, 1680, 1627, 1519, 1488, 1452, 1353, 1288, 1246, 1208, 1144, 1000, 918, 905, 839, 794 cm−1; UV-vis (CH3CN): λmax 208 nm (ε 72,400 cm−1 M−1), 320 (31,700); 1H-NMR (300 MHz, CDCl3): δ 3.93 (s, 3H, CO2Me), 5.21 (s, 2H, O-CH2), 5.24 (s, 2H, O-CH2), 7.00 (s, 1H, H4), 7.01 (s, 1H, H7), 7.23 (d, J = 2.1 Hz, 1H, H3), 7.33–7.51 (m, 10H, ArH), 8.73 (bs, 1H, NH); 13C-NMR (75.6 MHz, CDCl3): 51.8 (CO2Me), 71.3 (CH2), 72.0 (CH2), 97.0 (C7), 107.0 (C4), 108.8 (C3), 121.0 (ArC), 126.0 (C2), 127.0 (ArC), 127.2 (2 × ArC), 127.3 (ArC), 127.7 (2 × ArC), 127.8 (ArC), 128.4 (ArC), 128.5 (ArC), 132.4 (ArC), 137.0 (ArC), 137.4 (2 × ArC), 145.8 (C5), 149.9 (C6), 162.1 (CO2Me); HRMS (+ESI): Found m/z 410.1364 [M + Na]+, C24H21NO4Na requires 410.1363.
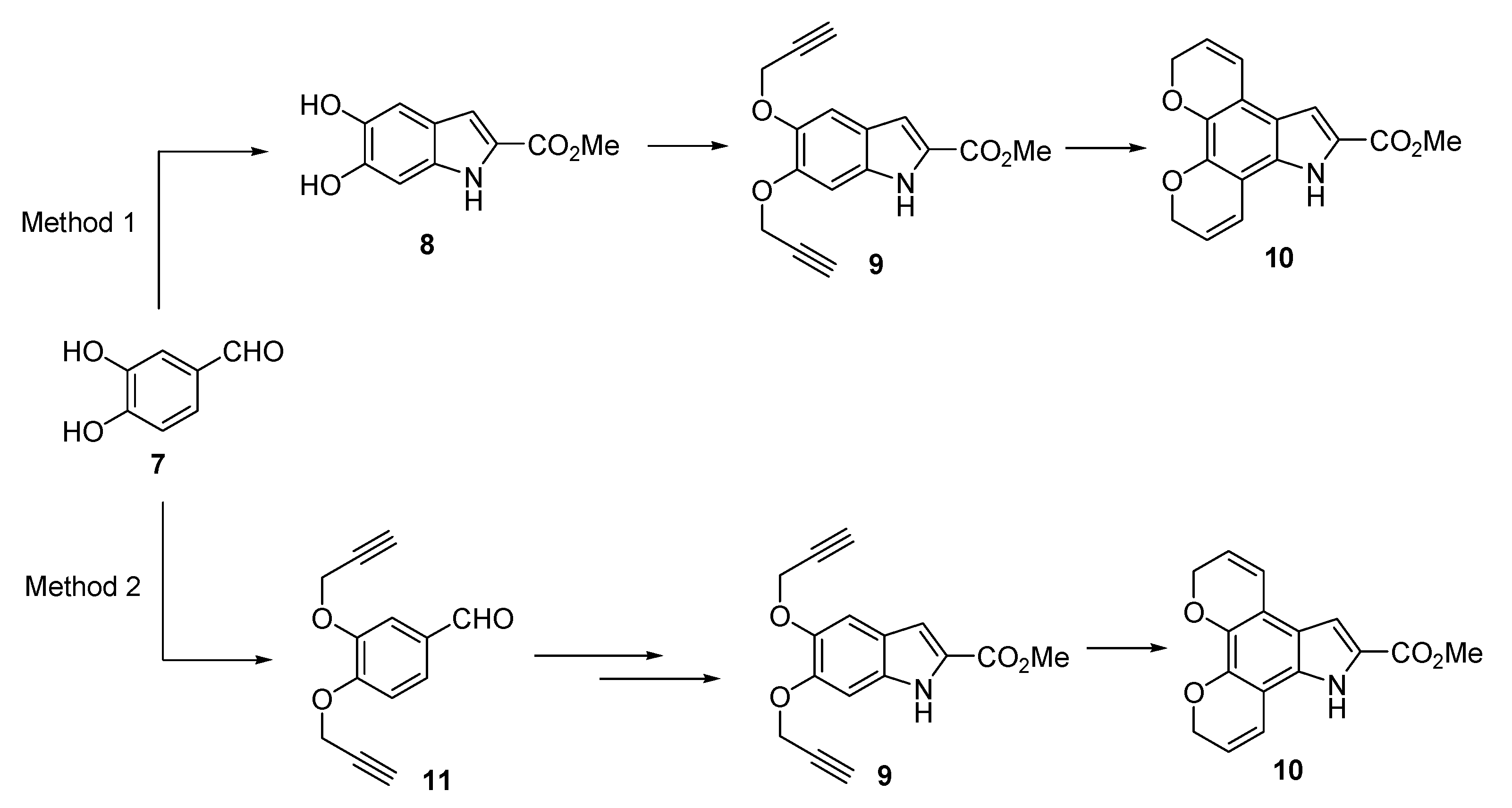
Methyl 5,6-dihydroxyindole-2-carboxylate (8) The title compound was prepared as described in GP-4 from methyl 5,6-dibenzyloxyindole-2-carboxylate (14) (0.387 g, 1.0 mmol) and 5% Pd/C catalyst (40 mg) in methanol/THF mixture (15 mL) to give the product (159 mg, 77%) as yellow solid; m.p. 256–258 °C; IR (KBr): vmax 3437, 3315, 2953, 2107, 1653, 1632, 1531, 1506, 1437, 1311, 1283, 1230, 1198, 1139, 992, 937, 849, 825, 767 cm−1; UV-vis (CH3CN): λmax 203 nm (ε 42,100 cm−1 M−1), 318 (27,000);1H-NMR (300MHz, d6-DMSO): δ 3.82 (s, 3H, CO2Me), 6.79 (d, J = 0.8 Hz, 1H, H4), 6.88 (s, 1H, H3), 6.90 (d, J = 0.8 Hz, 1H, H7), 8.84 and 9.17 (bs, each 1H, OH), 11.28 (bs, 1H, NH); 13C-NMR (75.6 MHz, CDCl3): 52.0 (CO2Me), 97.3 (C7), 105.4 (C4), 108.4 (C3), 120.3 (ArC), 125.0 (C2), 133.2 (ArC), 142.3 (C5), 146.7 (C6), 162.5 (CO2Me); HRMS (+ESI): Found m/z 230.0423 [M + Na]+, C10H9NO4Na requires 230.0424.
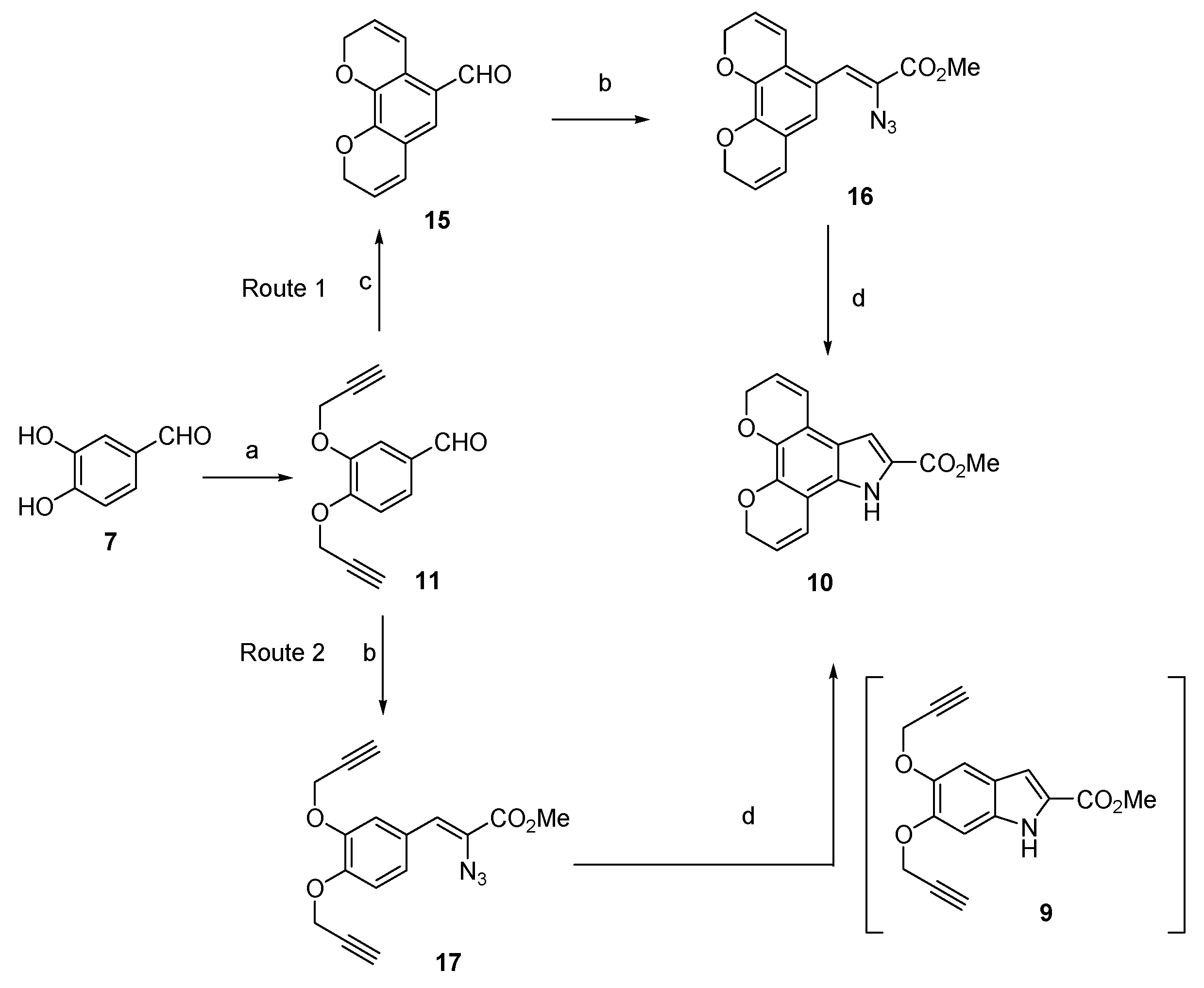
Methyl 5,6-bis(prop-2-yn-1-yloxy)-1H-indole-2-carboxylate (9) The title compound was prepared as described in GP-5 from methyl 5,6-dihydroxyindole-2-carboxylate (8) (993 mg, 4.8 mmol), potassium carbonate (1.32 g, 9.6 mmol) and propargyl bromide (1.36 g, 10.6 mmol) in acetone to give the product (828 mg, 61%) as a yellow solid;m.p. 170–172 °C; IR (KBr): νmax 3332, 3285, 2939, 2925, 2865, 2292, 2108, 1720, 1684, 1625, 1520, 1480, 1435, 1380, 1365, 1280, 1247, 1203, 1150 1024, 986, 892, 819, 805, 759, 720, 672 cm−1; UV-vis (CH3CN): λmax 209 nm (ε 28,300 cm−1 M−1), 313 (18,000); 1H-NMR (300 MHz, CDCl3): δ 2.55 (t, J = 1.6 Hz, 1H, CH≡C), 2.57 (t, J = 1.6 Hz, 1H, CH≡C), 3.95 (s, 3H, CO2Me), 4.82 (d, J = 2.4 Hz, 2H, O-CH2), 4.85 (d, J = 2.4 Hz, 2H, O-CH2), 7.10 (d, J = 0.7 Hz, 1H, H7), 7.16 (q, J = 0.9 Hz, 1H, H3), 7.31 (s, 1H, H4), 8.86 (bs, 1H, NH), 13C-NMR (75.6 MHz, CDCl3): δ51.8 (CO2Me), 57.0 (O-CH2), 57.5 (O-CH2), 75.7 (C≡CH), 75.7 (C≡CH), 78.6 (C≡CH), 79.1 (C≡CH), 97.2 (C7), 107.2 (C4), 108.9 (C3), 121.3 (aryl C), 126.4 (C2), 132.4 (aryl C), 144.2 (C4), 148.2 (C5), 162.1 (CO2Me); HRMS (+ESI): Found m/z 306.0736 [M + Na]+, C16H13NO4Na requires 306.0737.
2,9-Dihydropyrano[3,2-h]chromene-5-carbaldehyde (15) The title compound was prepared as described in GP-6 from 3,4-bis(prop-2-yn-1-yloxy)benzaldehyde (11) (214 mg, 1.04 mmol) in chlorobenzene (20 mL) to give the product (77 mg, 35%) as a white solid; m.p. 102–104 °C; IR (KBr): νmax 1680, 1570, 1490, 1440, 1340, 1301, 1205, 1102, 995, 945, 900, 898, 861, 745 cm−1; UV-vis (CH3CN): λmax 225 nm (ε 27,400 cm−1 M−1), 268 (18,800), 305 (17,500); 1H-NMR (300 MHz, CDCl3): δ 4.40 (dd, J = 3.0, 1.4 Hz, 2H, O-CH2), 4.79 (dd, J = 3.0, 1.4 Hz, 2H, O-CH2), 6.97 (d, J = 8.7 Hz, 2H, H4 and H7), 7.37 (s, 1H, H6), 7.40–7.42 (m, 2H, H3 and H8), 9.80 (s, 1H, CHO); 13C-NMR (75.6 MHz, CDCl3): δ71.3 (O-CH2), 71.5 (O-CH2), 119.6 (C6), 120.6 (aryl C), 121.6 (C4), 121.8 (C7), 125.3 (aryl C), 126.5 (C3), 126.9 (C8), 127.1 (C5), 147.9 (aryl C), 154.3 (aryl C), 190.7 (CHO); HRMS (+ESI): Found m/z 237.0533 [M + Na]+, C13H10O3Na requires 237.0528.
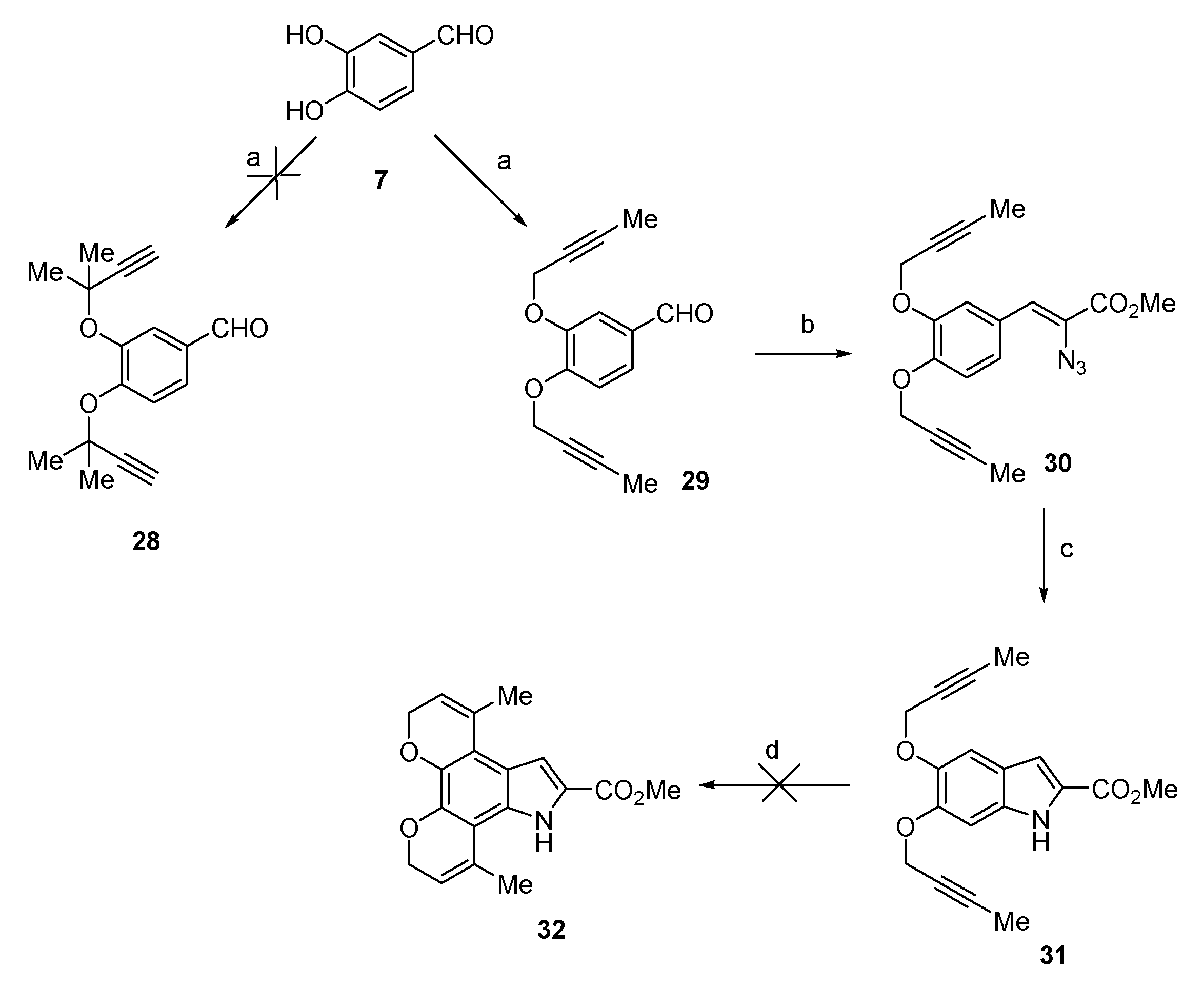
3,4-Bis(but-2-yn-1-yloxy)benzaldehyde (29) The title compound was prepared as described in GP-5 from 3,4-dihydroxybenzaldehyde (7) (660 mg, 4.8 mmol), potassium carbonate (1.32 g, 9.6 mmol) and 1-bromobut-2-yne (1.40 g, 10.6 mmol) in acetone to give the product (1.01 g, 87%) as a white solid; m.p. 86–88 °C; IR (KBr): νmax 1688, 1582, 1499, 1431, 1375, 1248, 1201, 1125, 987, 920, 857, 797, 729 cm−1; UV-vis (CH3CN): λmax 228 nm (ε 31,500 cm−1 M−1), 272 (22,200), 303 (14,800); 1H-NMR (300 MHz, CDCl3): δ 1.85–1.87 (m, 6H, 2 × C-CH3), 4.79 (q, J = 2.3 Hz, 2H, O-CH2), 4.82 (q, J = 2.3 Hz, 2H, O-CH2), 7.17 (d, J = 8.2 Hz, 1H, H5), 7.52 (dd, J = 8.2, 1.9 Hz, 1H, H6), 7.57 (d, J = 1.9 Hz, 1H, H2), 9.89 (s, 1H, CHO); 13C-NMR (75.6 MHz, CDCl3): δ 3.6 (C-CH3), 3.7 (C-CH3), 57.2 (2 × O-CH2), 73.1 (C-CH3), 73.3 (C-CH3), 84.6 (C≡C(CH3)), 84.9 (C≡C(CH3)), 112.0 (C5), 112.7 (C2), 126.4 (C6), 130.3 (aryl C), 147.9 (C4′), 152.9 (C3′), 190.8 (CHO); HRMS (+ESI): Found m/z 265.0840 [M + Na]+, C15H14O3Na requires 265.0835.
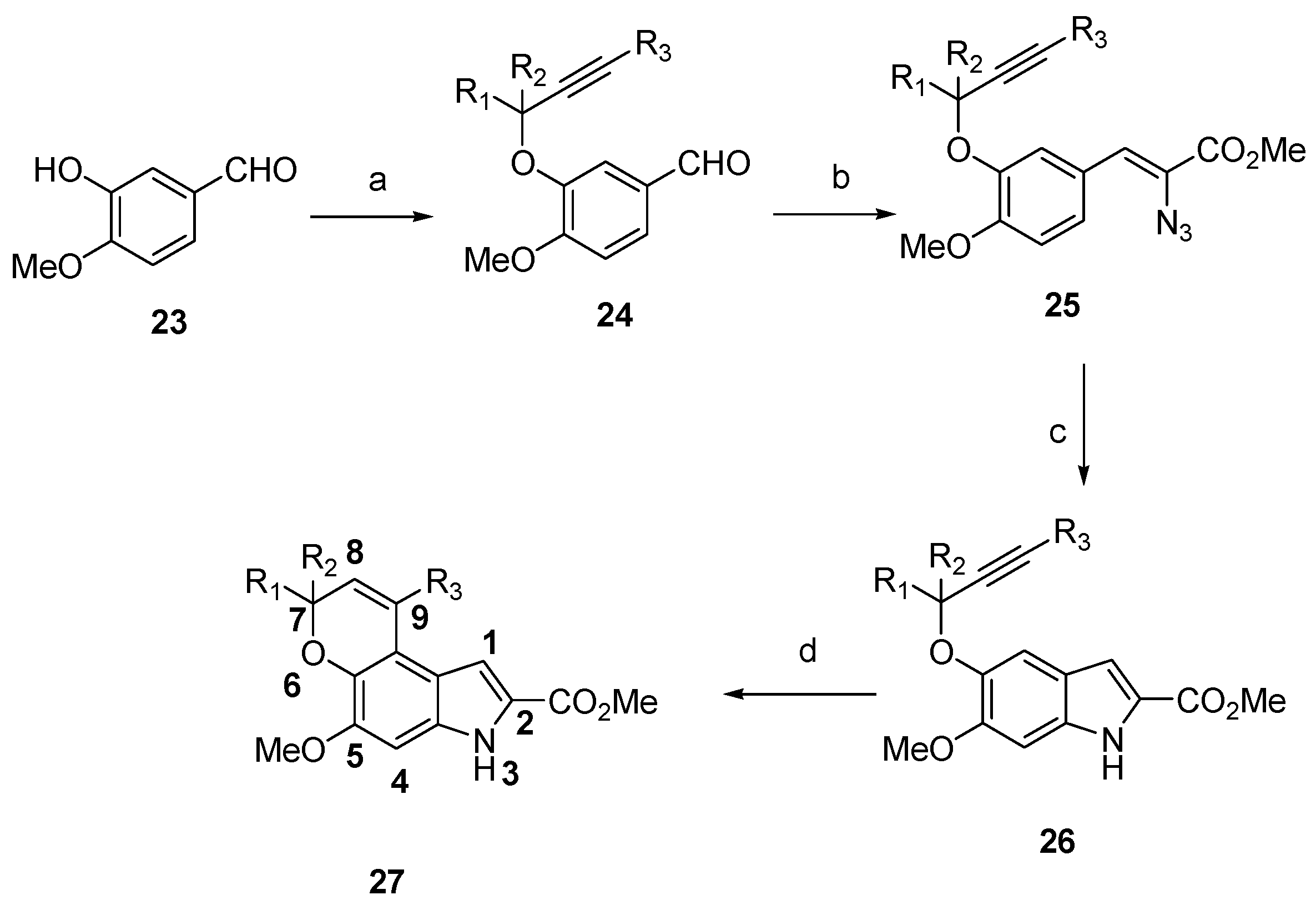
4-Methoxy-3-((3-phenylprop-2-yn-1-yl)oxy)benzaldehyde (24d) The title compound was prepared as described in GP-5 from 3-hydroxy-4-methoxybenzaldehyde 23 (730 mg, 4.8 mmol), potassium carbonate (662 mg, 4.8 mmol) and 3-phenylprop-2-yn-1-yl-4-methylbenzenesulfonate (1.66 g, 5.8 mmol) in acetone to give the product (1.17 g, 92%) as a white solid; m.p. 112–114 °C; IR (KBr): νmax 1673, 1579, 1506, 1430, 1378, 1256, 1221, 1165, 1130, 1012, 931, 874, 810, 754, 686 cm−1; UV-vis (CH3CN): λmax 232 nm (ε 45,500 cm−1 M−1), 272 (21,500), 302 (14,800); 1H-NMR (300 MHz, CDCl3): δ 4.00 (s, 3H, OMe), 5.08 (s, 2H, O-CH2), 7.03 (d, J = 8.2 Hz, 1H, H5), 7.30–7.33 (m, 3H, ArH), 7.34–7.45 (m, 2H, ArH), 7.54 (dd, J = 8.2, 1.9 Hz, 1H, H6), 7.68 (d, J = 1.9 Hz, 1H, H2), 9.90 (s, 1H, CHO); 13C-NMR (75.6 MHz, CDCl3): δ 56.2 (OMe), 57.5 (O-CH2), 83.0 (C≡C(Ph)), 88.0 (C-Ph), 110.9 (C5), 112.1 (C2), 122.1 (aryl C), 127.1 (2 × aryl CH), 127.8 (aryl CH), 128.8 (C6), 130.0 (aryl C), 131.9 (2 × aryl CH), 147.5 (C4), 155.0 (C3), 190.7 (CHO); HRMS (+ESI): Found m/z 289.0837 [M + Na]+, C17H14O3Na requires 289.0835.
Methyl (Z)-2-azido-3-(3,4-bis(prop-2-yn-1-yloxy)phenyl) acrylate (17) The title compound was prepared as described in GP-2 from 3,4-bis(prop-2-yn-1-yloxy)benzaldehyde (11) (558 mg, 2.6 mmol) and methyl azidoacetate (2.99 g, 26.0 mmol) in anhydrous methanol (30 mL) to give the product (582 mg, 63%) as a pale yellow granular solid; m.p. 114–116 °C; IR (KBr): νmax 2954, 2123, 1709, 1594, 1506, 1433, 1378, 1239, 1133, 1081, 1009, 793, 745, 683 cm−1; UV-vis (CH3CN): λmax 327 nm (ε 19,100 cm−1 M−1); 1H-NMR (300 MHz,CDCl3): δ 2.57 (t, J = 2.4 Hz, 1H, CH≡C), 2.59 (t, J = 2.4 Hz, 1H, CH≡C), 3.92 (s, 3H, CO2Me), 4.83 (d, J = 2.4 Hz, 4H, 2 × O-CH2), 6.91 (s, 1H, CH=C), 7.09 (d, J = 8.5 Hz, 1H, H5′), 7.44 (dd, J = 8.5, 2.0 Hz, 1H, H6′), 7.77 (d, J = 2.0 Hz, 1H, H2′), 13C-NMR (75.6 MHz, CDCl3): δ 52.8 (CO2Me), 56.6 (OMe), 57.0 (2 × O-CH2), 76.1 (C≡CH), 76.2 (C≡CH), 77.2 (C≡CH), 77.4 (C≡CH), 113.8 (C5′), 116.6 (C2′), 123.9 (CH=C), 125.3 (C6′), 125.6 (aryl C), 146.9 (aryl C), 148.5 (aryl C), 164.1 (CO2Me); HRMS could not be determined due to the unstable properties of the compound
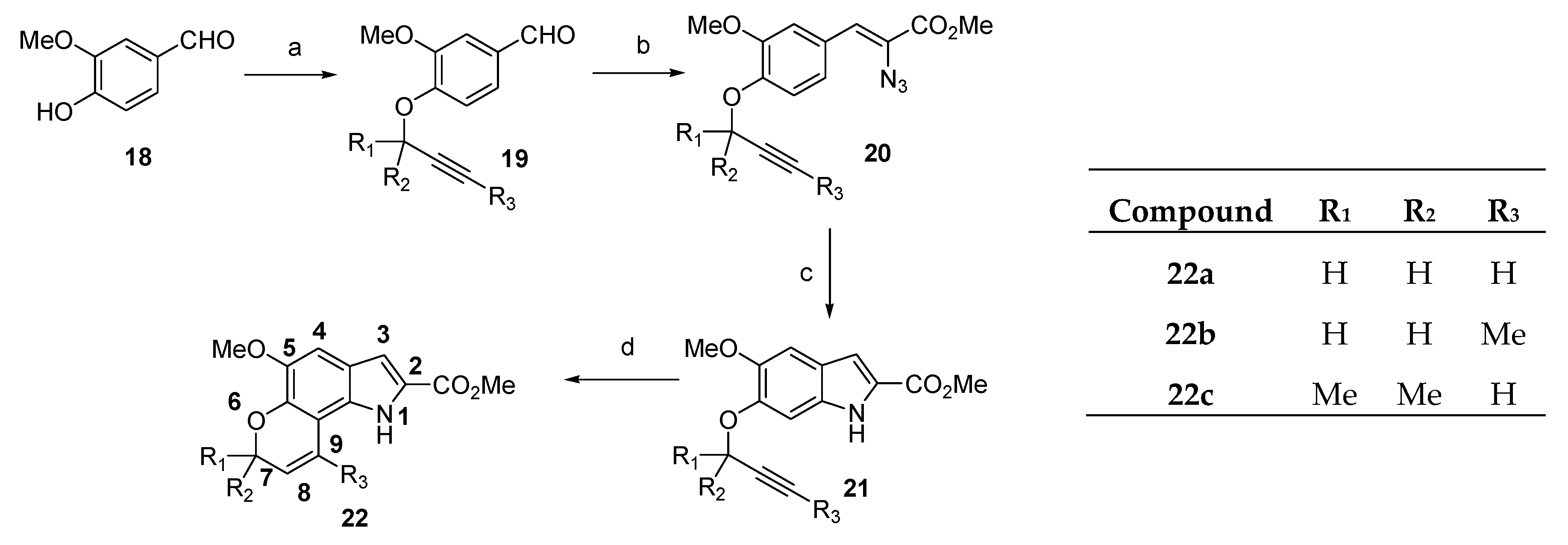
Methyl (Z)-2-azido-3-(4-methoxy-3-(prop-2-yn-1-yloxy)phenyl)acrylate (25a) The title compound was prepared as described in GP-2 from 4-methoxy-3-(prop-2-yn-1-yloxy)benzaldehyde (24a) (494 mg, 2.6 mmol) and methyl azidoacetate (2.99 g, 26.0 mmol) in anhydrous methanol (30 mL) to give the product (537 mg, 72%) as a pale yellow granular solid; m.p. 116–118 °C; IR (KBr): νmax 2951, 2101, 1698, 1592, 1507, 1432, 1379, 1254, 1214, 1139, 1084, 1016, 809, 745 cm−1; UV-vis (CH3CN): λmax 235 nm (ε 16,600 cm−1 M−1), 330 (27,100); 1H-NMR (300 MHz, CDCl3): δ 2.58 (t, J = 2.4 Hz, 1H, CH≡C), 3.94 (s, 3H, OMe), 3.94 (s, 3H, CO2Me), 4.83 (d, J = 2.4 Hz, 2H, O-CH2), 6.90 (s, 1H, CH=C), 6.93 (d, J = 8.5 Hz, 1H, H5′), 7.43 (dd, J = 8.5, 2.0 Hz, 1H, H6′), 7.76 (d, J = 2.0 Hz, 1H, H2′); 13C-NMR (75.6 MHz, CDCl3): δ 52.8 (CO2Me), 55.9 (OMe), 56.8 (O-CH2), 76.0 (C≡CH), 77.4 (C≡CH), 111.2 (C5′), 116.1 (C2′), 123.4 (CH=C), 125.6 (C6′), 125.0 (aryl C), 126.1 (CH=C), 146.3 (C4′), 150.8 (C3′), 164.1 (CO2Me); HRMS (+ESI): Found m/z 310.0800 [M + Na]+, C14H13N3O4Na requires 310.0798.
Methyl (Z)-2-azido-3-(3-methoxy-4-(prop-2-yn-1-yloxy) phenyl)acrylate (20a) The title compound was prepared as described in GP-2 from 3-methoxy-4-(prop-2-yn-1-yloxy)benzaldehyde (19a) (494 mg, 2.6 mmol) and methyl azidoacetate (2.99 g, 26.0 mmol) in anhydrous methanol (30 mL) to give the product (567 mg, 76%) as a pale yellow granular solid; m.p. 120–122 °C; IR (KBr): νmax 2920, 2122, 1704, 1592, 1509, 1450, 1345, 1243, 1206, 1141, 1008, 922, 804, 764 cm−1; UV-vis (CH3CN): λmax 323 nm (ε 17,500 cm−1 M−1); 1H-NMR (300 MHz, CDCl3): δ 2.56 (t, J = 2.4 Hz, 1H, CH≡C), 3.94 (s, 3H, OMe), 3.95 (s, 3H, CO2Me), 4.83 (d, J = 2.4 Hz, 2H, O-CH2), 6.91 ( s, 1H, CH=C), 7.06 (d, J = 8.5 Hz, 1H, H5′), 7.43 (dd, J = 8.5, 1.9 Hz, 1H, H6′), 7.56 (d, J = 1.9 Hz, 1H, H2′); 13C-NMR (75.6 MHz, CDCl3): δ 52.8 (CO2Me), 55.9 (OMe), 56.5 (O-CH2), 76.2 (C≡CH), 77.4 (C≡CH), 113.3 (C5′), 113.5 (C2′), 123.7 (CH=C), 124.4 (C6′), 125.5 (aryl C), 127.3 (CH=C), 147.9 (C3′), 149.1 (C4′), 164.1 (CO2Me); HRMS could not be determined due to the unstable properties of the compound.
Methyl (Z)-2-azido-3-(3,4-bis(but-2-yn-1-yl)oxy)phenyl) acrylate (30) The title compound was prepared as described in GP-2 from 3,4-bis(but-2-yn-1-yloxy)benzaldehyde (29) (629 mg, 2.6 mmol) and methyl azidoacetate (2.99 g, 26.0 mmol) in anhydrous methanol (30 mL) to give the product (555 mg, 63%) as a pale yellow granular solid; m.p. 124–126 °C; IR (KBr): νmax 2918, 2119, 1712, 1687, 1590, 1506, 1432, 1373, 1314, 1243, 1128, 992, 804, 745 cm−1; UV-vis (CH3CN): λmax 307 nm (ε 14,500 cm−1 M−1); 1H-NMR (300 MHz, CDCl3): δ 1.87 (t, J = 2.3 Hz, 6H, 2 × C-CH3), 3.93 (s, 3H, CO2Me), 4.79 (q, J = 2.3 Hz, 2H, O-CH2), 4.82 (q, J = 2.3 Hz, 2H, O-CH2), 6.91 (s, 1H CH=C), 7.05 (d, J = 8.5 Hz, 1H, H5′), 7.44 (dd, J = 8.5, 1.8 Hz, 1H, H6′), 7.73 (d, J = 2.0 Hz, 1H, H2′); 13C-NMR (75.6 MHz, CDCl3): δ 3.7 (2×C-CH3), 52.8 (CO2Me), 57.1 (OMe), 57.2 (O-CH2), 57.5 (O-CH2), 73.9 (2×C-CH3), 84.2 (C≡C(CH3)), 84.9 (C≡C(CH3)), 113.2 (C5′), 116.0 (C2′), 125.3 (CH=C), 125.8 (C6′), 126.5 (aryl C), 126.6 (CH=C), 147.9 (C4′), 148.8 (C3), 164.2 (CO2Me); HRMS could not be determined due to the unstable properties of the compound.
Methyl (Z)-2-azido-3-(3-(but-2-yn-1-yloxy)-4-methoxyphenyl) acrylate (25b) The title compound was prepared as described in GP-2 from 3-(but-2-yn-1-yloxy)-4-methoxybenzaldehyde (24b) (530 mg, 2.6 mmol) and methyl azidoacetate (2.9 g, 26.0 mmol) in anhydrous methanol (30 mL) to give the product (500 mg, 64%) as a pale yellow granular solid; m.p. 96–98 °C; IR (KBr): νmax 2951, 2101, 1698, 1592, 1507, 1431, 1379, 1254, 1139, 1016, 810, 745 cm−1; UV-vis (CH3CN): λmax 327 nm (ε 25,700 cm−1 M−1); 1H-NMR (300 MHz, CDCl3): δ 1.89 (t, J = 2.3 Hz, 3H, CH3), 3.93 (s, 3H, OMe), 3.94 (s, 3H, CO2Me), 4.78 (q, J = 2.3 Hz, 2H, O-CH2), 6.91 (d, J = 8.5 Hz, 1H, H5′), 6.92 (s, 1H, CH=C), 7.41 (dd, J = 8.5, 2.0 Hz, 1H, H6′), 7.73 (d, J = 2.0 Hz, 1H, H2′); 13C-NMR (75.6 MHz, CDCl3): δ 3.7 (C-CH3), 52.8 (CO2Me), 55.9 (OMe), 57.4 (O-CH2), 73.7 (C-CH3), 84.3 (C≡C(CH3)), 111.0 (C5′), 115.6 (C2′), 123.3 (CH=C), 125.6 (C6′), 125.9 (aryl C), 126.0 (CH=C), 146.7 (C4′), 150.7 (C3′), 164.2 (CO2Me); HRMS (+ESI): Found m/z 324.0944 [M + Na]+, C15H15N3O4Na requires 324.0955.
Methyl (Z)-2-azido-3-(4-(but-2-yn-1-yloxy)-3-methoxyphenyl)acrylate (20b) The title compound was prepared as described in GP-2 from 4-(but-2-yn-1-yloxy)-3-methoxybenzaldehyde (19b)(530 mg, 2.6 mmol) and methyl azidoacetate (2.99 g, 26.0 mmol) in anhydrous methanol (30 mL) to give the product (477 mg, 61%) as a pale yellow granular solid; m.p. 94–96 °C; IR (KBr): νmax 2920, 2120, 1702, 1592, 1509, 1434, 1377, 1244, 1140, 1011, 801, 763 cm−1; UV-vis (CH3CN): λmax 324 nm (ε 14,900 cm−1 M−1); 1H-NMR (300 MHz, CDCl3): δ 1.87 (t, J = 2.3 Hz, 3H, CH3), 3.94 (s, 3H, OMe), 3.94 (s, 3H, CO2Me), 4.77 (q, J = 2.3 Hz, 2H, O-CH2), 6.90 (s, 1H, CH=C), 7.05 (d, J = 8.5 Hz, 1H, H5′), 7.38 (dd, J = 8.5, 1.8, Hz, 1H, H6′), 7.53 (d, J = 1.8 Hz, 1H, H2′); 13CNMR (75.6 MHz, CDCl3): δ 3.7 (C-CH3), 52.8 (CO2Me), 55.9 (OMe), 57.1 (O-CH2), 73.5 (C-CH3), 84.4 (C≡C(CH3)), 112.9 (C5′), 113.3 (C2′), 123.4 (aryl C), 124.5 (CH=C), 125.8 (C6′), 126.8 (CH=C), 14.34 (C3′), 149.0 (C4′), 164.2 (CO2Me); HRMS (+ESI): Found m/z 324.0955 [M + Na]+, C15H15N3O4Na requires 324.0955.
Methyl (Z)-2-azido-3-(4-methoxy-3-((3-phenylprop-2-yn-1-yl) oxy)phenyl)acrylate (25d) The title compound was prepared as described in GP-2 from 4-methoxy-3-((3-phenylprop-2-yn-1-yl)oxy)benzaldehyde (24d) (692 mg, 2.6 mmol) and methyl azidoacetate (2.99 g, 26.0 mmol) in anhydrous methanol (30 mL) to give the product (632 mg, 67%) as a pale yellow granular solid; m.p. 118–120 °C; IR (KBr): νmax 2921, 2827, 2119, 1674, 1612, 1580, 1506, 1431, 1378, 1275, 1232, 1129, 1008, 966. 873, 809, 753, 686 cm−1; UV-vis (CH3CN): λmax 234 nm (ε 57,700 cm−1 M−1), 272 (22,600), 306 (21,800); 1H-NMR (300 MHz, CDCl3): δ 3.93 (s, 3H, OMe), 3.96 (s, 3H, CO2Me), 5.04 (d, J = 6.0 Hz, 2H, O-CH2), 6.91 (s, 1H, CH≡C), 6.92 (d, J = 8.0 Hz, 1H, H5′), 7.09 (dd, J = 8.0, 1.8 Hz, 1H, H6′), 7.27–7.33 (m, 3H, ArH), 7.43–7.46 (m, 2H, ArH), 7.85 (d, J = 1.8 Hz, 1H, H2′); 13C-NMR (75.6 MHz, CDCl3): δ 52.6 (CO2Me), 55.9 (OMe), 57.5 (O-CH2), 83.9 (C-Ph), 87.5 (C≡C(Ph)), 111.1 (C5′), 116.0 (C2′), 120.4 (aryl C), 125.7 (CH=C), 126.0 (C6′), 126.1 (aryl C), 128.3 (2×aryl CH), 128.6 (CH=C), 130.6 (aryl CH), 131.9 (2×aryl CH), 146.7 (C4′), 150.9 (C3′), 164.2 (CO2Me); HRMS (+ESI): Found m/z 386.1099 [M + Na]+, C20H17N3O4Na requires 386.1117.
Methyl (Z)-2-azido-3-(4-methoxy-3-((2-methylbut-3-yn-2-yl) oxy)phenyl)acrylate (25c) The title compound was prepared as described in GP-2 from 4-methoxy-3-((2-methylbut-3-yn-2-yl)oxy)benzaldehyde (24c) (567 mg, 2.6 mmol) and methyl azidoacetate (2.99 g, 26.0 mmol) in anhydrous methanol (30 mL) to give the product (507 mg, 62%) as a pale yellow granular solid; m.p. 122–124 °C; IR (KBr): νmax 2988, 2835, 2094, 1691, 1619, 1565, 1506, 1425, 1374, 1253, 1138, 1084, 1027, 962. 891, 799, 756, 687 cm−1; UV-vis (CH3CN): λmax 238 nm (ε 16,800 cm−1 M−1), 328 (35,400); 1H-NMR (300 MHz, CDCl3): δ 1.70 (s, 6H, 2 × C-CH3), 2.59 (d, J = 0.9 Hz, 1H, CH≡C), 3.87 (s, 3H, OMe), 3.92 (s, 3H, CO2Me), 6.91 (s, 1H, CH=C), 6.90 (d, J = 8.5 Hz, 1H, H5′), 7.45 (dd, J = 1.9, 8.5 Hz, 1H, H6′), 8.16 (d, J = 2.0 Hz, 1H, H2′);13C-NMR (75.6 MHz, CDCl3): δ 29.3 (2×C-CH3), 52.7 (CO2Me), 55.7 (OMe), 73.6 (C≡CH), 74.1 (C-(CH3)2), 86.1 (C≡CH), 111.5 (C5′), 123.2 (C2′), 124.8 (CH=C), 125.7 (C6′), 125.9 (aryl C), 127.4 (CH=C), 144.2 (C4′), 153.9 (C3′), 164.2 (CO2Me); HRMS (+ESI): Found m/z 338.1099 [M + Na]+, C16H17N3O4Na requires 338.1117.
Methyl (Z)-2-azido-3-(3-methoxy-4-((2-methylbut-3-yn-2-yl)oxy)phenyl)acrylate (20c) The title compound was prepared as described in GP-2 from 3-methoxy-4-((2-methylbut-3-yn-2-yl)oxy)benzaldehyde (19c) (567 mg, 2.6 mmol) and methyl azidoacetate (2.1 g, 26. mmol) in anhydrous methanol (30 mL) to give the product (524 mg, 62%) as a pale yellow granular solid; m.p. 116–118 °C; IR (KBr): νmax 2975, 2802, 2112, 1703, 1635, 1565, 1512, 1400, 1387, 1212, 1100, 1052, 998, 957, 888, 765, 745, 677 cm−1; UV-vis (CH3CN): λmax 328 nm (ε 17,100 cm−1 M−1); 1H-NMR (300 MHz, CDCl3): δ 1.72 (s, 6H, 2 × C-CH3), 2.60 (d, J = 0.9 Hz, 1H, CH≡C), 3.89 (s, 3H, OMe), 3.93 (s, 3H, CO2Me), 6.91 (s, 1H, CH=C), 7.33 (dd, J = 8.5, 1.9 Hz, 1H, H6′), 7.45 (d, J = 8.5 Hz, 1H, H5′), 7.52 (d, J = 1.9 Hz, 1H, H2′); 13C-NMR (75.6 MHz, CDCl3): δ 29.4 (2 × C-CH3), 52.8 (CO2Me), 55.9 (OMe), 73.9 (C≡CH), 73.9 (C-(CH3)2) 85.8 (C≡CH), 113.9 (C5′), 121.5 (C2′), 123.8 (CH=C), 123.9 (C6′), 125.7 (aryl C), 128.5 (CH=C), 146.1 (C3′), 152.0 (C4′), 164.1 (CO2Me); HRMS (+ESI): Found m/z 338.1104 [M + Na]+, C16H17N3O4Na requires 338.1117.
Methyl 6-methoxy-5-(prop-2-yn-1-yloxy)-1H-indole-2-carboxylate (26a) The title compound was prepared as described in GP-3 from methyl (Z)-2-azido-3-(3-(but-2-yn-1-yloxy)-4-methoxyphenyl)acrylate (25a) (596 mg, 2.08 mmol) in xylene (20 mL) to give the product (196 mg, 73%) as a yellow solid; m.p. 164–166 °C; IR (KBr): νmax 3325, 3259, 2953, 2925, 2123, 1671, 1624, 1520, 1480, 1440, 1367, 1320, 1249, 1208, 1185, 1146, 1005, 890, 833, 766, 694 cm−1; UV-vis (CH3CN): λmax 208 nm (ε 40,600 cm−1 M−1), 318 (28,700); 1H-NMR (300 MHz, CDCl3): δ 2.55 (t, J = 2.4 Hz, 1H, CH≡C), 3.95 (s, 3H, OMe), 3.96 (s, 3H, CO2Me), 4.82 (d, J = 2.4 Hz, 2H, O-CH2), 6.90 (d, J = 0.8 Hz, 1H, H4), 7.16 (q, J = 0.9 Hz, 1H, H3), 7.28 (s, 1H, H7), 8.91 (bs, 1H, NH), 13C-NMR (75.6 MHz, CDCl3): δ 51.8 (CO2Me), 56.0 (OMe), 57.3 (O-CH2), 75.7 (C≡CH), 78.7 (C≡CH), 94.0 (C7), 106.6 (C4), 109.0 (C3), 120.2 (aryl C), 125.9 (C2), 132.9 (aryl C), 143.7 (C6), 150.6 (C5), 162.3 (CO2Me); HRMS (+ESI): Found m/z 282.0725 [M + Na]+, C14H13NO3Na requires 282.0742.
Methyl 5-methoxy-6-(prop-2-yn-1-yloxy)-1H-indole-2-carboxylate (21a) The title compound was prepared as described in GP-3 from methyl (Z)-2-azido-3-(4-(but-2-yn-1-yloxy)-3-methoxyphenyl)acrylate (20a) (596 mg, 2.08 mmol) in xylene (20 mL) to give the product (183 mg, 68%) as a yellow solid; m.p. 152–154 °C; IR (KBr): νmax 3325, 3241, 2999, 2937, 2113, 1681, 1638, 1522, 1475, 1452, 1360, 1280, 1237, 1211, 1195, 1143, 1003, 924, 843, 819, 761, 675 cm−1; UV-vis (CH3CN): λmax 209 nm (ε 35,800 cm−1 M−1), 308 (21,400); 1H-NMR (300 MHz, CDCl3): δ 2.57 (t, J = 2.4 Hz, 1H, CH≡C), 3.95 (s, 3H, OMe), 3.96 (s, 3H, CO2Me), 4.85 (d, J = 2.4 Hz, 2H, O-CH2), 7.08 (d, J = 0.8 Hz, 1H, H4), 7.11 (s, 1H, H7), 7.15 (q, J = 0.9 Hz, 1H, H3), 8.87 (bs, 1H, NH); 13C-NMR (75.6 MHz, CDCl3): δ 51.8 (CO2Me), 56.2 (OMe), 56.9 (O-CH2), 76.1 (C≡CH), 77.4 (C≡CH), 96.8 (C7), 103.0 (C4), 108.7 (C3), 121.4 (aryl C), 126.1 (C2), 131.6 (aryl C), 146.6 (C5), 147.6 (C6), 162.2 (CO2Me); HRMS (+ESI): Found m/z 282.0726 [M + Na]+, C14H13NO3Na requires 282.0742.
Methyl 5,6-bis(but-2-yn-1-yloxy)-1H-indole-2-carboxylate (31) The title compound was prepared as described in GP-3 from methyl (Z)-2-azido-3-(3,4-bis(but-2-yn-1-yloxy)phenyl)acrylate (30) (704 mg, 2.08 mmol) in xylene (20 mL) to give the product 210 mg, 65%) as a yellow solid; m.p. 146–148 °C; IR (KBr): νmax 3332, 2912, 2294, 2120, 1682, 1644, 1519, 1434, 1379, 1244, 1204, 1145, 984, 891, 819, 765 cm−1; UV-vis (CH3CN): λmax 210 nm (ε 39,600 cm−1 M−1), 317 (24,900); 1H-NMR (300 MHz, CDCl3): δ 1.87 (q, J = 2.4 Hz, 6H, 2 × C-CH3), 3.96 (s, 3H, CO2Me), 4.76 (q, J = 2.4 Hz, 2H, O-CH2), 4.79 (q, J = 2.4 Hz, 2H, O-CH2), 7.08 (d, J = 0.7 Hz, 1H, H4), 7.16 (q, J = 0.9 Hz, 1H, H3), 7.25 (s, 1H, H7), 9.03 (bs, 1H, NH); 13C-NMR (75.6 MHz, CDCl3): δ 3.7 (2 × C-CH3), 51.8 (CO2Me), 57.4 and 57.8 (O-CH2), 73.9 (C-CH3), 74.2 (C-CH3), 83.8 (C≡C(CH3)), 84.2 (C≡C(CH3)), 96.5 (C7), 106.1 (C4), 108.9 (C3), 120.9 (aryl C), 126.0 (C2), 132.5 (aryl C), 144.4 (C5), 148.5 (C6), 162.3 (CO2Me); HRMS (+ESI): Found m/z 334.1046 [M + Na]+, C18H17NO4Na requires 334.1050.
Methyl 5-(but-2-yn-1-yloxy)-6-methoxy-1H-indole-2-carboxylate (26b) The title compound was prepared as described in GP-3 from methyl (Z)-2-azido-3-(3-(but-2-yn-1-yloxy)-4-methoxyphenyl)acrylate (25b) (626 mg, 2.08 mmol) in xylene (20 mL) to give the product (201 mg, 71%) as a yellow solid; m.p. 160–162 °C; IR (KBr): νmax 3324, 3258, 3009, 2918, 2795, 2123, 2108, 1672, 1600, 1520, 1490, 1425, 1400, 1385, 1346, 1249, 1208, 1185, 1146, 1005, 980, 890, 833, 766 cm−1; UV-vis (CH3CN): λmax 208 nm (ε 27,700 cm−1 M−1), 319 (18,900);1H-NMR (300 MHz, CDCl3): δ 1.88 (q, J = 2.4 Hz, 3H, C-CH3), 3.95 (s, 3H, OMe), 3.96 (s, 3H, CO2Me), 4.77 (q, J = 2.4 Hz, 2H, O-CH2), 6.88 (d, J = 0.7 Hz, 1H, H4), 7.16 (q, J = 0.9 Hz, 1H, H3), 7.23 (s, 1H, H7), 8.82 (bs, 1H, NH); 13C-NMR (75.6 MHz, CDCl3): δ 5.2 (C-CH3), 53.2 (CO2Me), 57.4 (OMe), 59.1 (O-CH2), 75.5 (C-CH3), 85.3 (C≡C), 95.2 (C7), 107.1 (C4), 110.4 (C3), 121.7 (aryl C), 127.1 (C2), 134.0 (aryl C), 145.5 (C6), 151.9 (C5), 163.7 (CO2Me); HRMS (+ESI): Found m/z 296.0884 [M + Na]+, C15H15NO4Na requires 296.0899.
Methyl 6-(but-2-yn-1-yloxy)-5-methoxy-1H-indole-2-carboxylate (21b) The title compound was prepared as described in GP-3 from methyl (Z)-2-azido-3-(4-(but-2-yn-1-yloxy)-3-methoxyphenyl)acrylate (20b) (626 mg, 2.08 mmol) in xylene (20 mL) to give the product (190 mg, 67%) as a yellow solid; m.p. 164–166 °C; IR (KBr): νmax 3324, 3224, 3005, 2936, 2835, 2215, 2108, 1680, 1584, 1495, 1455, 1433, 1362, 1285, 1243, 1228, 1146, 1045, 988, 946, 846, 819, 760, 672 cm−1; UV-vis (CH3CN): λmax 210 nm (ε 37,100 cm−1 M−1), 319 (22,300); 1H-NMR (300 MHz, CDCl3): δ 1.87 (q, J = 2.4 Hz, 3H, C-CH3), 3.94 (s, 3H, OMe), 3.96 (s, 3H, CO2Me), 4.80 (q, J = 2.4 Hz, 2H, O-CH2), 7.06 (d, J = 0.7 Hz, 1H, H4), 7.08 (s, 1H, H7), 7.14 (q, J = 0.9 Hz, 1H, H3), 9.01 (bs, 1H, NH); 13C-NMR (75.6 MHz, CDCl3): δ 3.7 (C-CH3), 51.8 (CO2Me), 56.1 (OMe), 57.4 (O-CH2), 73.8 (C-CH3), 84.3 (C≡C(CH3)), 96.2 (C7), 102.7 (C4), 108.7 (C3), 121.0 (aryl C), 125.8 (C2), 131.8 (aryl C), 146.1 (C5), 148.0 (C6), 162.3 (CO2Me); HRMS (+ESI): Found m/z 296.0883 [M + Na]+, C15H15NO4Na requires 296.0889.
Methyl 6-methoxy-5-((3-phenylprop-2-yn-1-yl)oxy)-1H-indole-2-carboxylate (26d) The title compound was prepared as described in GP-3 from methyl (Z)-2-azido-3-(4-methoxy-3-((3-phenylprop-2-yn-1-yl)oxy)phenyl)acrylate (25d) (754 mg, 2.08 mmol) in xylene (20 mL) to give the product (257 mg, 74%) as a yellow solid; m.p. 138–140 °C; IR (KBr): νmax 3326, 2984, 2924, 2740, 2113, 1675, 1645, 1520, 1480, 1439, 1381, 1247, 1195, 1145, 996, 845, 823, 761, 672 cm−1; UV-vis (CH3CN): λmax 203 nm (ε 58,900 cm−1 M−1), 318 (22,200); 1H-NMR (300 MHz, CDCl3): δ 3.95 (s, 3H, OMe), 3.98 (s, 3H, CO2Me), 5.04 (s, 2H, O-CH2), 6.90 (s, 1H, H4), 7.17 (q, J = 0.9 Hz, 1H, H3), 7.31 (s, 1H, H7), 7.32–7.36 (m, 3H, ArCH), 7.44–7.47 (m, 2H, ArCH), 8.84 (bs, 1H, NH); 13C-NMR (75.6 MHz, CDCl3): δ 51.8 (CO2Me), 56.0 (OMe), 58.2 (O-CH2), 84.1 (C≡C(Ph)), 87.4 (C-Ph), 93.9 (C7), 106.6 (C4), 109.0 (C3), 120.3 (aryl C), 122.4 (aryl C), 125.8 (C2), 128.2 (2 × aryl CH), 128.5 (aryl CH), 131.8 (2 × aryl CH), 132.8 (aryl C), 144.0 (C6), 150.7 (C5), 162.2 (CO2Me); HRMS (+ESI): Found m/z 336.1221 [M + H]+, C20H18NO4 requires 336.1236.
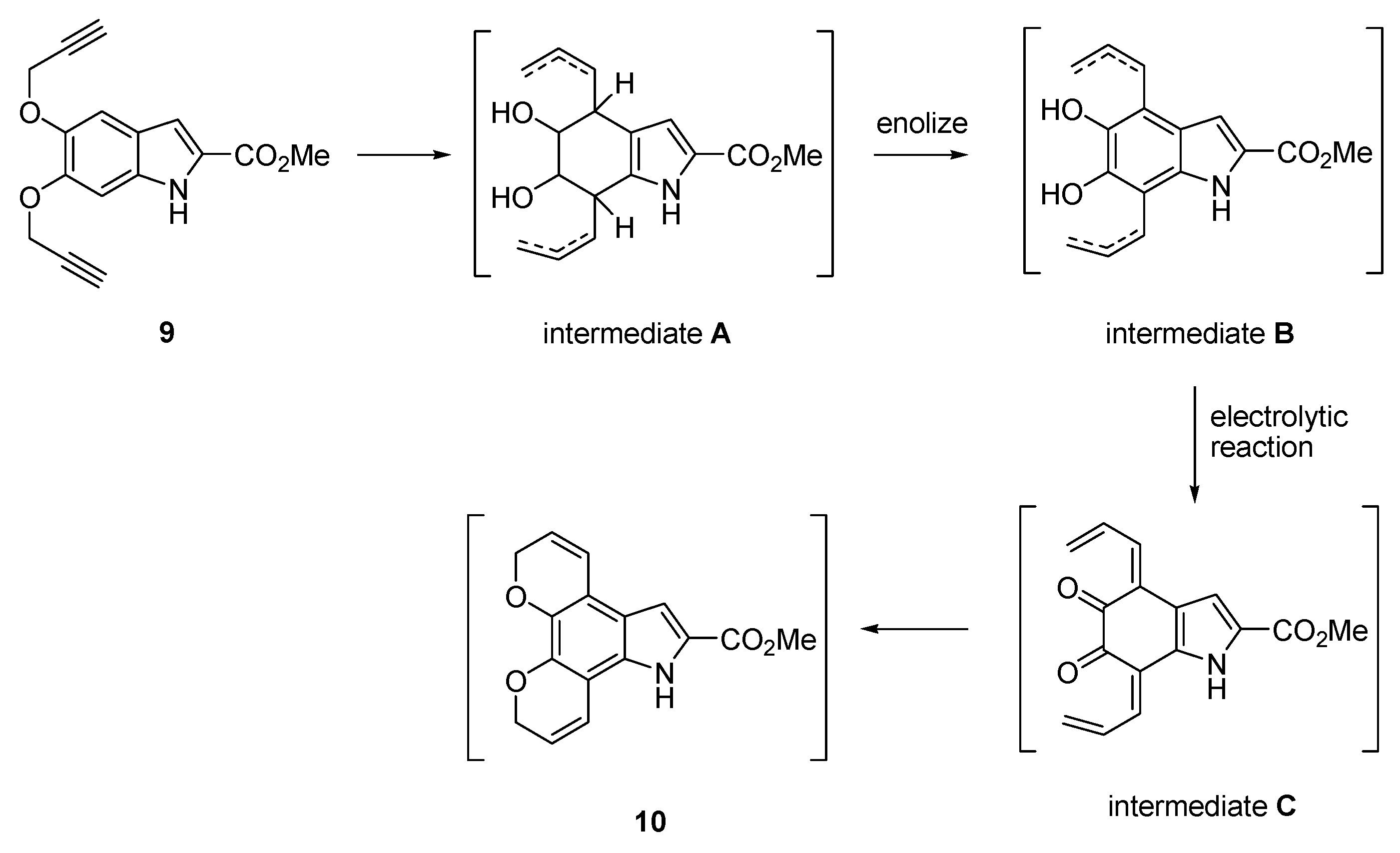
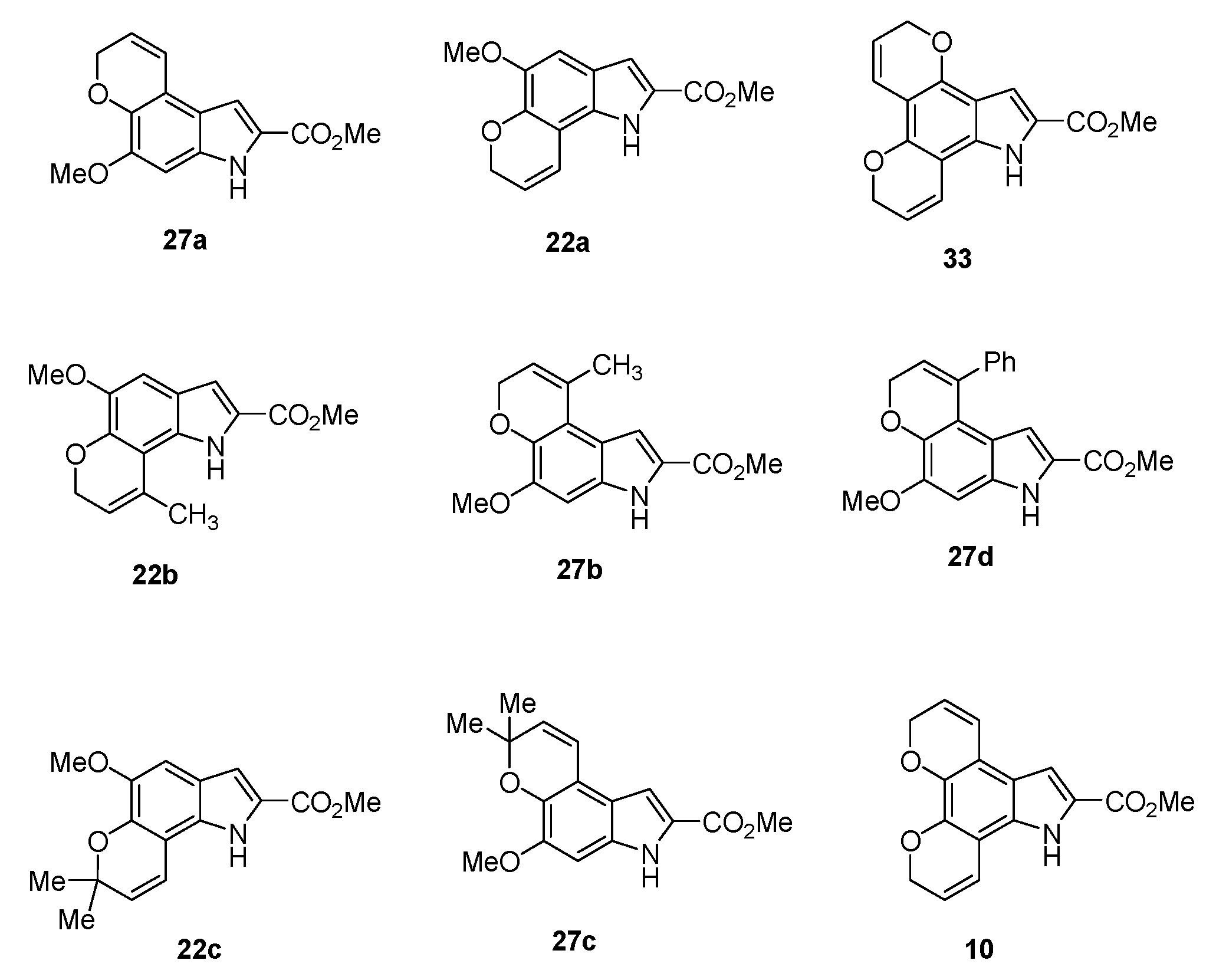
Methyl 5,10-dihydro-7H-dipyrano[3,2-e:2′,3′-g]indole-6-carboxylate (10) Method 2: Route 2 The title compound was prepared as described in GP-3 from methyl (Z)-2-azido-3-(3,4-bis(prop-2-yn-1-yloxy)phenyl) acrylate (9) (646 mg, 2.08 mmol) in xylene (20 mL) to give the product (61.4 mg, 72%) as a yellow solid; m.p. 168–170 °C; IR (KBr): νmax 3415, 3328, 2947, 2831, 2341, 2116, 1674, 1640, 1575, 1524, 1486, 1441, 1400, 1274, 1213, 1158, 1135, 1025, 1000, 920, 813, 755 cm−1; UV-vis (CH3CN): λmax 225 nm (ε 24,100 cm−1 M−1), 269 (13,800), 360 (13,000); 1H-NMR (300 MHz, CDCl3): δ 3.96 (s, 3H, CO2Me), 4.91 (dd, J = 3.8, 1.8 Hz, 2H, O-CH2), 4.94 (dd, J = 3.8, 1.8 Hz, 2H, O-CH2), 5.87–5.97 (m, 2H, H3 and H9), 6.72–6.80 (m, 2H, H4 and H8), 7.19 (d, J = 2.1 Hz, 1H, H5), 8.85 (s, 1H, NH); 13C-NMR (75.6 MHz, CDCl3): δ52.0 (CO2Me), 65.7 (O-CH2), 65.8 (O-CH2), 106.5 (C5), 107.0 (aryl C), 115.3 (aryl C), 118.8 (C4), 119.3 (C8), 120.8 (aryl C), 121.8 (C3), 121.9 (C9), 126.5 (C6), 128.8 (aryl C), 138.1 (aryl C), 141.7 (aryl C), 162.3 (CO2Me); HRMS (+ESI): Found m/z 306.0736 [M + Na]+, C16H13NO4Na requires 306.0737.
This compound was also prepared by the methods described below. Method 1: The title compound was prepared as described in GP-6 from methyl 5,6-bis(prop-2-yn-1-yloxy)-1H-indole-2-carboxylate (16) (118 mg, 0.42 mmol) in chlorobenzene (30 mL) to give the product (43.5 mg, 51%) as a yellow solid.
Method 2: Route 1 The title compound was prepared as described in GP-6 from methyl (Z)-2-azido-3-(2H,10H-pyrano[4,3-h]chromen-5-yl)acrylate (17) (646 mg, 2.08 mmol) in xylene (20 mL) to give the product (52.9 mg, 62%) as a yellow solid.
Methyl 5-methoxy-3,7-dihydropyrano[3,2-e]indole-2-carboxylate (27a) The title compound was prepared as described in GP-6 from methyl 6-methoxy-5-(prop-2-yn-1-yloxy)-1H-indole-2-carboxylate (26a) (108 mg, 0.42 mmol) in chlorobenzene (30 mL) to give the product (80 mg, 74%) as a yellow solid; m.p. 166–168 °C; IR (KBr): νmax 3325, 2948, 2839, 2340, 2110, 1677, 1637, 1515, 1439, 1271, 1193, 1143, 1098, 998, 931, 883, 808, 752 cm−1; UV-vis (CH3CN): λmax 212 nm (ε 37,400 cm−1 M−1), 271 (13,300), 322 (30,600); 1H-NMR (300 MHz, CDCl3): δ 3.92 (s, 3H, OMe), 3.93 (s, 3H, CO2Me), 4.90 (dd, J = 3.7, 1.7 Hz, 2H, O-CH2), 5.89–5.95 (m, 1H, H8), 6.76–6.79 (m, 1H, H9), 6.79 (s, 1H, H4), 7.17 (d, J = 2.1 Hz, 1H, H1), 8.96 (s, 1H, NH); 13C-NMR (75.6 MHz, CDCl3): δ 51.8 (CO2Me), 56.0 (OMe), 65.7 (O-CH2), 93.8 (C4), 106.1 (C1), 114.9 (aryl C), 118.3 (aryl C), 121.6 (C9), 121.8 (C8), 126.0 (C2), 132.2 (aryl C), 139.3 (aryl C), 148.8 (C5), 162.2 (CO2Me); HRMS (+ESI): Found m/z 282.0731 [M + Na]+, C14H13NO4Na requires 282.0737.
Methyl 5-methoxy-1,7-dihydropyrano[2,3-g]indole-2-carboxylate (22a) The title compound was prepared as described in GP-6 from methyl 5-methoxy-6-(prop-2-yn-1-yloxy)-1H-indole-2-carboxylate (21a) (108 mg, 0.42 mmol) in chlorobenzene (30 mL) to give the product (83 mg, 77%) as a yellow solid; m.p. 188–190 °C; IR (KBr): νmax 3327, 2944, 2827, 2366, 2106, 1677, 1528, 1444, 1306, 1221, 1148, 1101, 989, 955, 831, 755 cm−1; UV-vis (CH3CN): λmax 226 nm (ε 28,100 cm−1 M−1), 290 (16,700), 339 (18,400); 1H-NMR (300 MHz, CDCl3): δ 3.94 (s, 3H, OMe), 3.96 (s, 3H, CO2Me), 4.96 (dd, J = 3.8, 1.8 Hz, 2H, O CH2), 5.89–5.95 (m, 1H, H8), 6.75–6.79 (m, 1H, H9), 7.02 (s, 1H, H4), 7.13 (d, J = 2.1 Hz, 1H, H3), 8.94 (s, 1H NH); 13C-NMR (75.6 MHz, CDCl3): δ51.9 (CO2Me), 56.2 (OMe), 65.7 (O-CH2), 103.1 (C4), 107.1 (aryl C), 109.2 (C3), 119.3 (aryl C), 120.8 (C8), 121.0 (C9), 126.0 (C2), 128.7 (aryl C), 137.6 (aryl C), 145.2 (C5), 162.3 (CO2Me); HRMS (+ESI): Found m/z 282.0738 [M + Na]+, C14H13NO4Na requires 282.0737.
Methyl 5-methoxy-9-methyl-3,7-dihydropyrano[3,2-e]indole-2-carboxylate (27b) The title compound was prepared as described in GP-6 from methyl 5-(but-2-yn-1-yloxy)-6-methoxy-1H-indole-2-carboxylate (26b) (114 mg, 0.42 mmol) in chlorobenzene (30 mL) to give the product (62 mg, 54%) as a yellow solid; m.p. 196–198 °C; IR (KBr): νmax 3310, 2948, 2925, 2833, 2106, 1675, 1640, 1517, 1495, 1439, 1366, 1274, 1265, 1194, 1146, 1080, 1004, 960, 880, 815, 765, 672 cm−1; UV-vis (CH3CN): λmax 213 nm (ε 28,900 cm−1 M−1), 267 (9,500), 322 (24,200); 1H-NMR (300 MHz, CDCl3): δ 1.63 (s, 3H, C-CH3), 3.94 (s, 3H, OMe), 3.95 (s, 3H, CO2Me), 4.77 (q, J = 2.4 Hz, 2H, O-CH2), 5.69–5.72 (m, 1H, H8), 6.87 (s, 1H, H1), 7.23 (s, 1H, H4), 8.82 (bs, 1H, NH); 13C-NMR (75.6 MHz, CDCl3): δ 22.6 (C-CH3), 51.8 (CO2Me), 56.1 (OMe), 65.2 (O-CH2), 93.8 (C4), 109.3 (C1), 117.4 (aryl C), 117.9 (aryl C), 118.3 (C8), 126.5 (C2), 132.0 (C-CH3), 132.9 (aryl C), 140.4 (aryl C), 149.0 (C5), 162.2 (CO2Me); HRMS (+ESI): Found m/z 296.0887 [M + Na]+, C15H15NO4Na requires 296.0893.
Methyl 5-methoxy-9-methyl-1,7-dihydropyrano[2,3-g]indole-2-carboxylate (22b) The title compound was prepared as described in GP-6 from methyl 6-(but-2-yn-1-yloxy)-5-methoxy-1H-indole-2-carboxylate (21b) (114 mg, 0.42 mmol) in chlorobenzene (30 mL) to give the product (56 mg, 49%) as a yellow solid; m.p. 176–178 °C; IR (KBr): νmax 3410, 2980, 2930, 2845, 1695, 1645, 1606, 1534, 1445, 1430, 1385, 1375, 1232, 1189, 1139, 1096, 1040, 998, 981, 927, 857, 744, 712cm−1; UV-vis (CH3CN): λmax 224 nm (ε 32,500 cm−1 M−1), 286 (22,200), 337 (23,500); 1H-NMR (300 MHz, CDCl3): δ 1.71 (s, 3H, C-CH3), 3.94 (s, 3H, OMe), 3.96 (s, 3H, CO2Me), 4.77 (q, J = 1.6 Hz, 2H, O-CH2), 5.65–5.68 (m, 1H, H8), 7.05 (s, 1H, H4), 7.14 (d, J = 2.1 Hz, 1H, H3), 8.81 (bs, 1H, NH), 13C-NMR (75.6 MHz, CDCl3): δ 20.8 (C-CH3), 51.8 (CO2Me), 56.2 (OMe), 65.4 (O-CH2), 103.2 (C4), 108.9 (C3), 109.9 (aryl C), 117.7 (C8), 121.9 (aryl C), 126.0 (C2), 128.7 (C-CH3), 129.3 (aryl C), 144.2 (aryl C), 145.4 (C5), 162.1 (CO2Me); HRMS (+ESI): Found m/z 296.0886 [M + Na]+, C15H15NO4Na requires 296.0893.
Methyl 5-methoxy-9-phenyl-3,7-dihydropyrano[3,2-e]indole-2-carboxylate (27d) The title compound was prepared as described in GP-6 from methyl 6-methoxy-5-((3-phenylprop-2-yn-1-yl)oxy)-1H-indole-2-carboxylate (26d) (140 mg, 0.42 mmol) in chlorobenzene (30 mL) to give the product (113 mg, 81%) as a yellow solid; m.p. 188–190 °C; IR (KBr): νmax 3311, 2949, 2816, 2114, 1684, 1639, 1598, 1515, 1500, 1438, 1400, 1360, 1260, 1241, 1193, 1141, 1042, 1011, 995, 920, 823, 759, 703 cm−1; UV-vis (CH3CN): λmax 274 nm (ε 11,600 cm−1 M−1), 326 (26,400); 1H-NMR (300 MHz, CDCl3): δ 3.82 (s, 3H, OMe), 4.00 (s, 3H, CO2Me), 4.85 (d, J = 4.4 Hz, 2H, O-CH2), 5.94–5.97 (m, 1H, H8), 5.98 (s, 1H, H4), 6.88 (s, 1H, H1), 7.30–7.34 (m, 2H, ArH), 7.41–7.44 (m, 3H, ArH), 8.76 (bs, 1H, NH), 13C-NMR (75.6 MHz, CDCl3): δ 51.7 (CO2Me), 58.1 (OMe), 65.1 (O-CH2), 94.2 (C4), 109.4 (C1), 117.0 (aryl C), 117.5 (aryl C), 125.0 (C2), 120.4 (C8), 127.9 (aryl CH), 128.3 (2 × arylCH), 128.5 (2 × aryl CH), 132.8 (aryl C), 139.4 (C-Ph), 139.4 (aryl C), 141.1 (aryl C), 149.0 (C5), 162.1 (CO2Me); HRMS (+ESI): Found m/z 358.1057 [M + Na]+, C20H17NO4Na requires 358.1055.
Methyl 5-methoxy-7,7-dimethyl-3,7-dihydropyrano[3,2-e] indole-2-carboxylate (27c) The title compound was prepared as described in GP-3 from methyl (Z)-2-azido-3-(4-methoxy-3-((2-methylbut-3-yn-2-yl)oxy)phenyl)acrylate (25c) (654 mg, 2.08 mmol) in xylene (20 mL) to give the product (220 mg, 68%) as a yellow solid; m.p. 144–146 °C; IR (KBr): νmax 3316, 2962, 2975, 2111, 1681, 1625, 1600, 1510, 1439, 1400, 1366, 1270, 1230, 1192, 1131, 1075, 999, 934, 887, 818, 754, 728 cm−1; UV-vis (CH3CN): λmax 219 nm (ε 40,000 cm−1 M−1), 271 (13,500), 323 (31,100); 1H-NMR (300 MHz, CDCl3): δ 1.53 (s, 6H, 2 × CH3), 3.92 (s, 3H, OMe), 3.95 (s, 3H, CO2Me), 5.73 (d, J = 9.7 Hz, 1H, H8), 6.68 (d, J = 9.7 Hz, 1H, H9), 6.79 (s, 1H, H4), 7.19 (d, J = 2.1 Hz, 1H, H1), 9.00 (bs, 1H, NH); 13C-NMR (75.6 MHz, CDCl3): δ 27.3 (2 × C-CH3), 51.7 (CO2Me), 58.1 (OMe), 76.2 (C-(CH3)2), 93.1 (C4), 106.1 (C1), 113.4 (aryl C), 118.2 (aryl C), 119.3 (C9), 125.7 (C2), 130.6 (C8), 132.0 (aryl C), 138.1 (aryl C), 149.5 (C5), 162.3 (CO2Me); HRMS (+ESI): Found m/z 310.1044 [M + Na]+, C16H17NO4Na requires 310.1050.
Methyl 5-methoxy-7,7-dimethyl-1,7-dihydropyrano[2,3-g] indole-2-carboxylate (22c) The title compound was prepared as described in GP-3 from methyl (Z)-2-azido-3-(3-methoxy-4-((2-methylbut-3-yn-2-yl)oxy)phenyl)acrylate (20c) (654 mg, 2.08 mmol) in xylene (20 mL) to give the product (203 mg, 74%) as a yellow solid; m.p. 158–160 °C; IR (KBr): νmax 3338, 2955, 2919, 2845, 2111, 1676, 1640, 1595, 1534, 1444, 1370, 1350, 1310, 1265, 1217, 1195, 1134, 1099, 1050, 985, 960, 880, 826, 760, 714 cm−1; UV-vis (CH3CN): λmax 226 nm (ε 29,600 cm−1 M−1), 262 (15,200), 341 (19,000); 1H-NMR (300 MHz, CDCl3): δ 1.55 (s, 6H, 2 × CH3), 3.93 (s, 3H, OMe), 3.96 (s, 3H, CO2Me), 5.71 (d, J = 9.7 Hz, 1H, H8), 6.66 (d, J = 9.7 Hz, 1H, H9), 7.02 (s, 1H, H4), 7.13 (d, J = 2.1 Hz, 1H, H3), 8.95 (bs, 1H, NH); 13C-NMR (75.6 MHz, CDCl3): δ 27.4 (2 × C-CH3), 51.8 (CO2Me), 56.5 (OMe), 76.6 (C-(CH3)2), 103.3 (C4), 105.8 (aryl C), 109.3 (C3), 116.8 (C9), 120.6 (aryl C), 125.7 (C2), 128.9 (aryl C), 129.8 (C8), 142.3 (aryl C), 149.9 (C5), 162.4 (CO2Me); HRMS (+ESI): Found m/z 288.1228 [M + H]+, C16H18NO4 requires 288.1236.


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}








