
A Second Look at the Crystal Structures of Drosophila melanogaster Acetylcholinesterase in Complex with Tacrine Derivatives Provides Insights Concerning Catalytic Intermediates and the Design of Specific Insecticides



Abstract
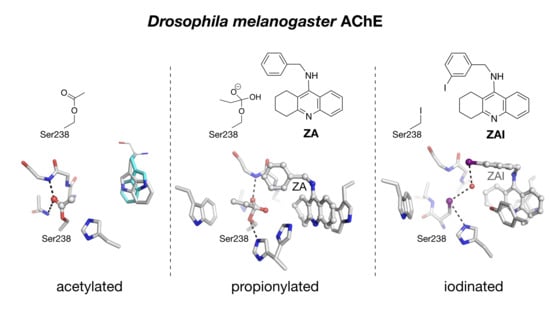
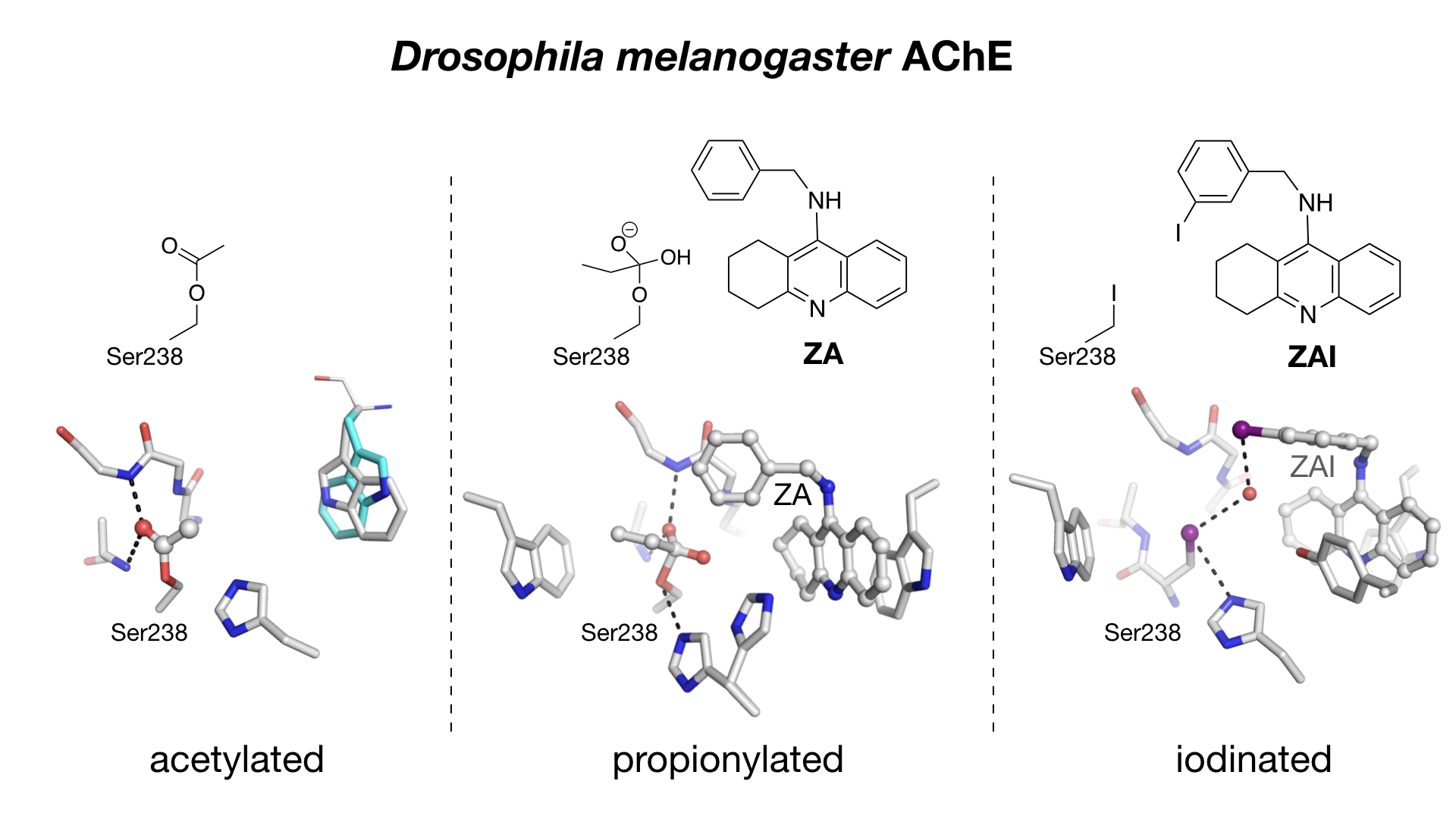
:
1. Introduction
2. Results and Discussion
2.1. Updated Image Processing and Structures Refinement
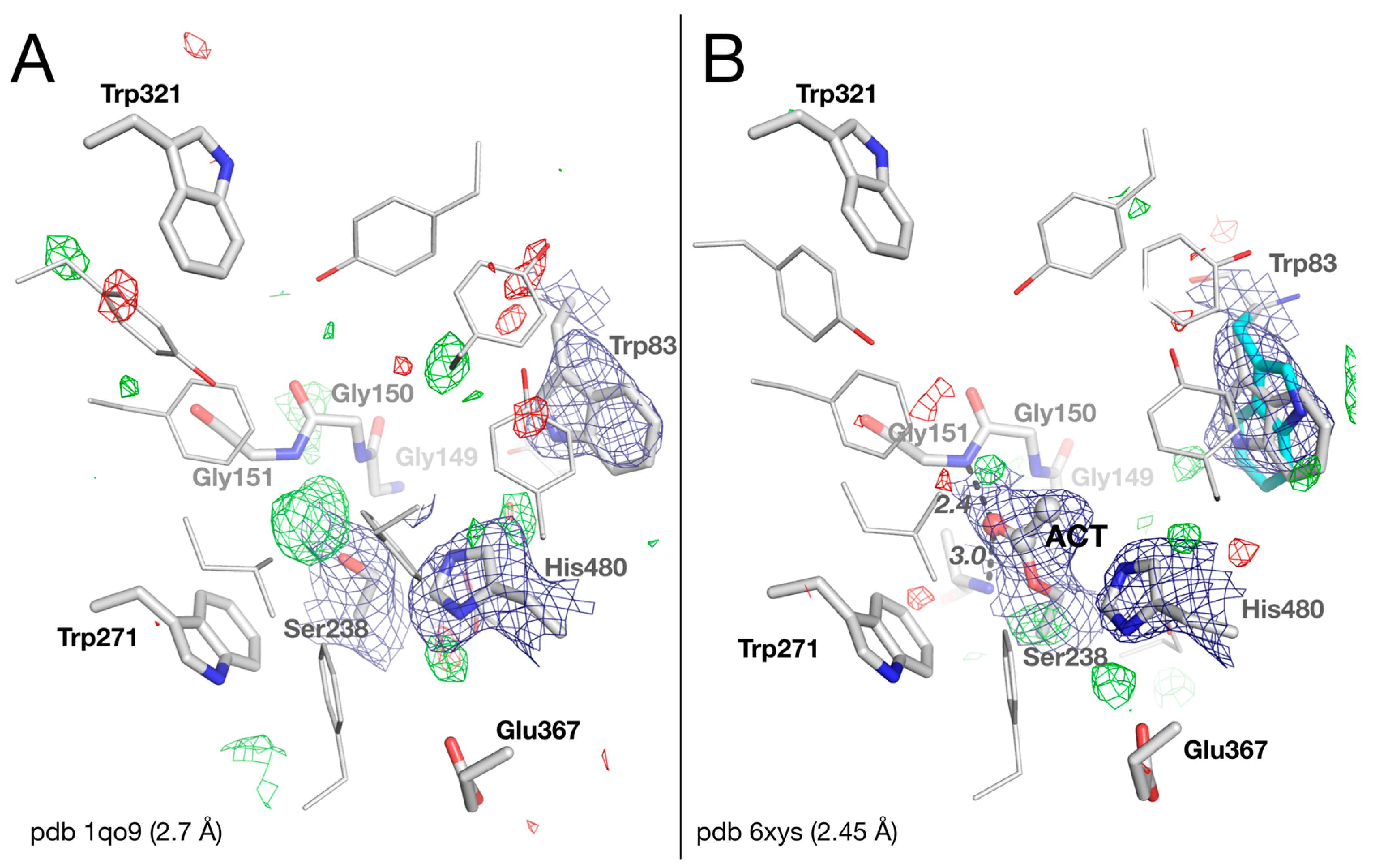
2.2. Crystal Structure of Native Drosophila melanogaster Acetylcholinesterase
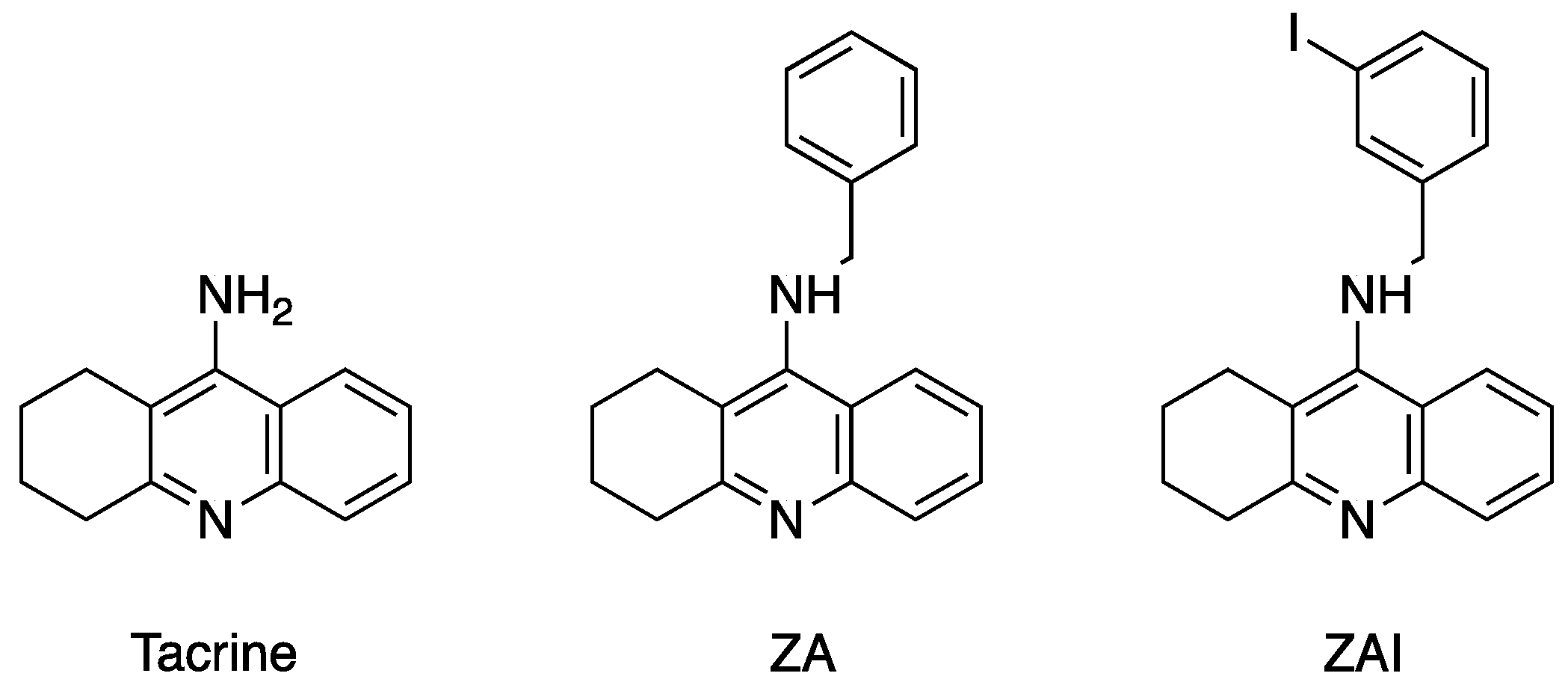
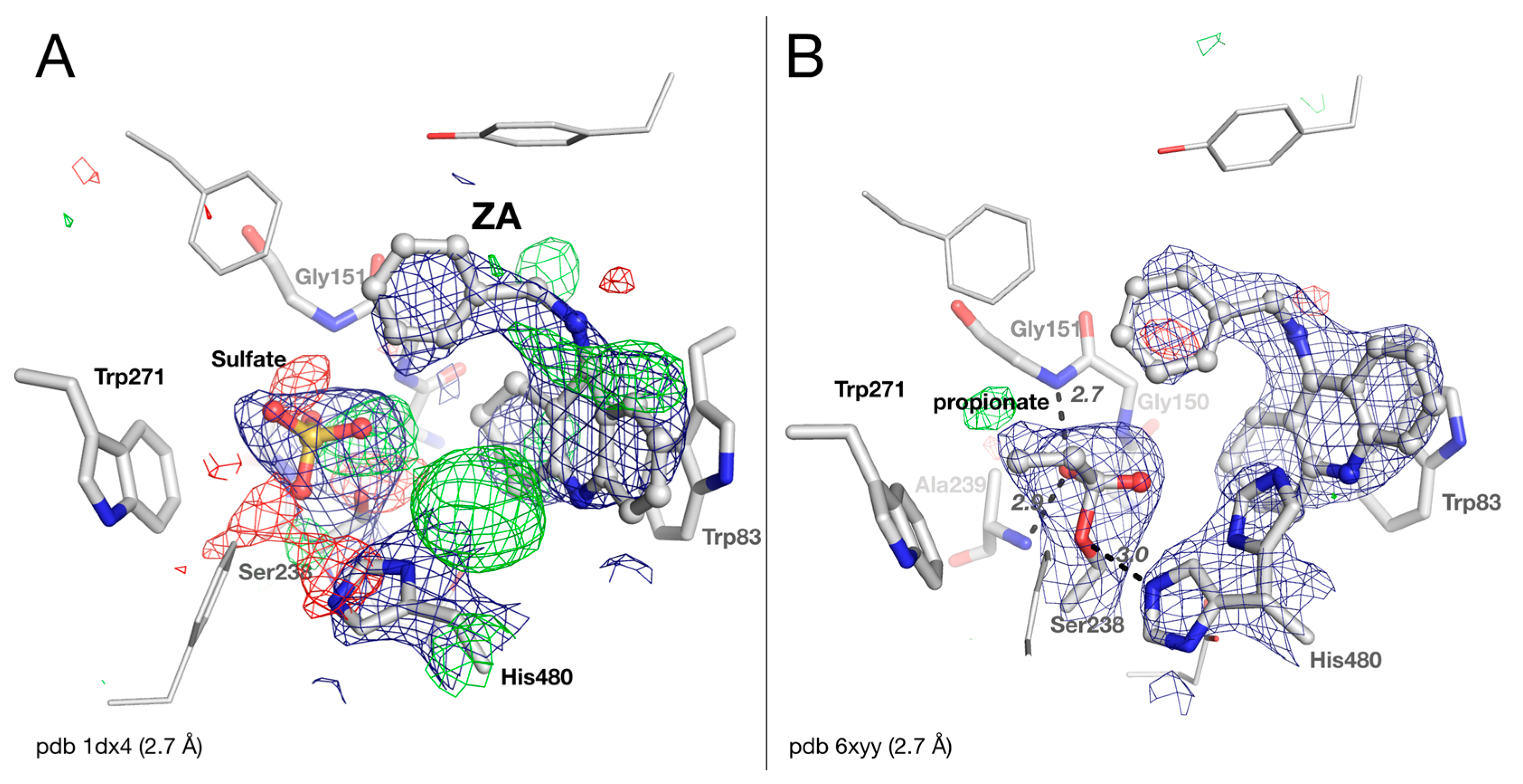
2.3. Crystal structure of Drosophila melanogaster Acetylcholinesterase in Complex with ZA
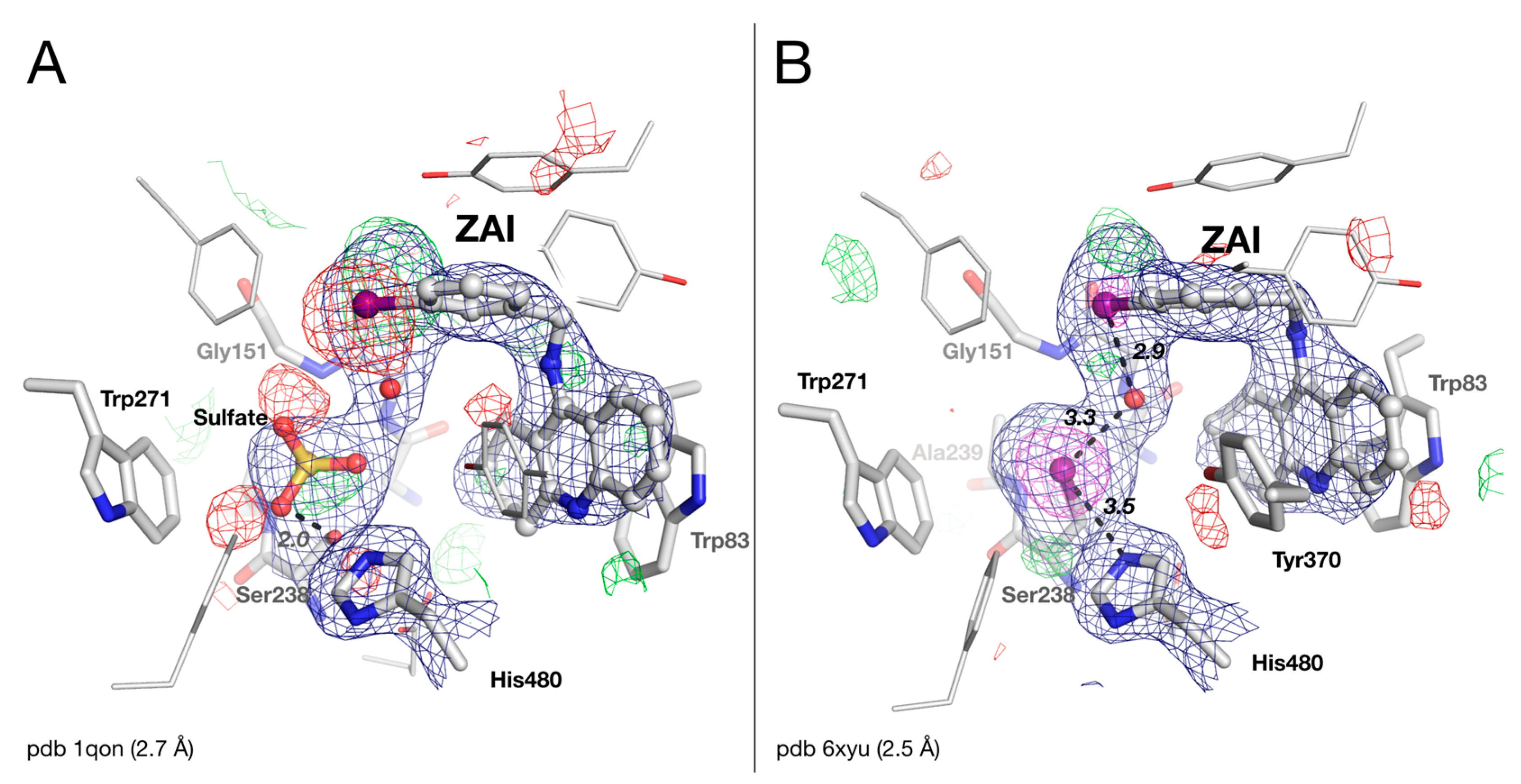
2.4. Crystal Structure of Drosophila melanogaster Acetylcholinesterase in Complex with ZAI
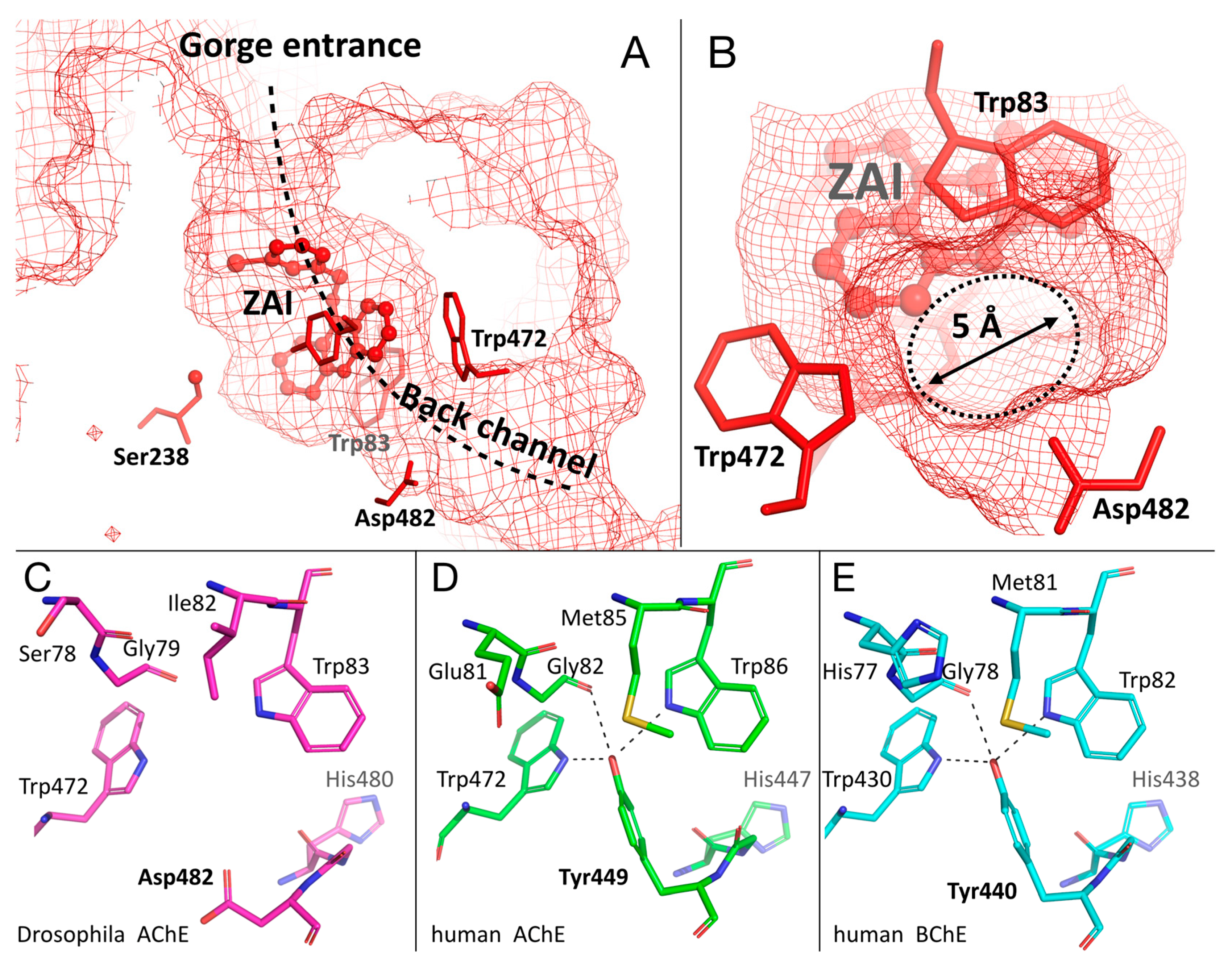
2.5. Specific Channel in the Choline-Binding Pocket of Insect Cholinesterase
2.6. Impact of Preserving Original Date for Potential Reinterpretation
3. Materials and Methods
3.1. X-ray Data Processing and Structure Refinement
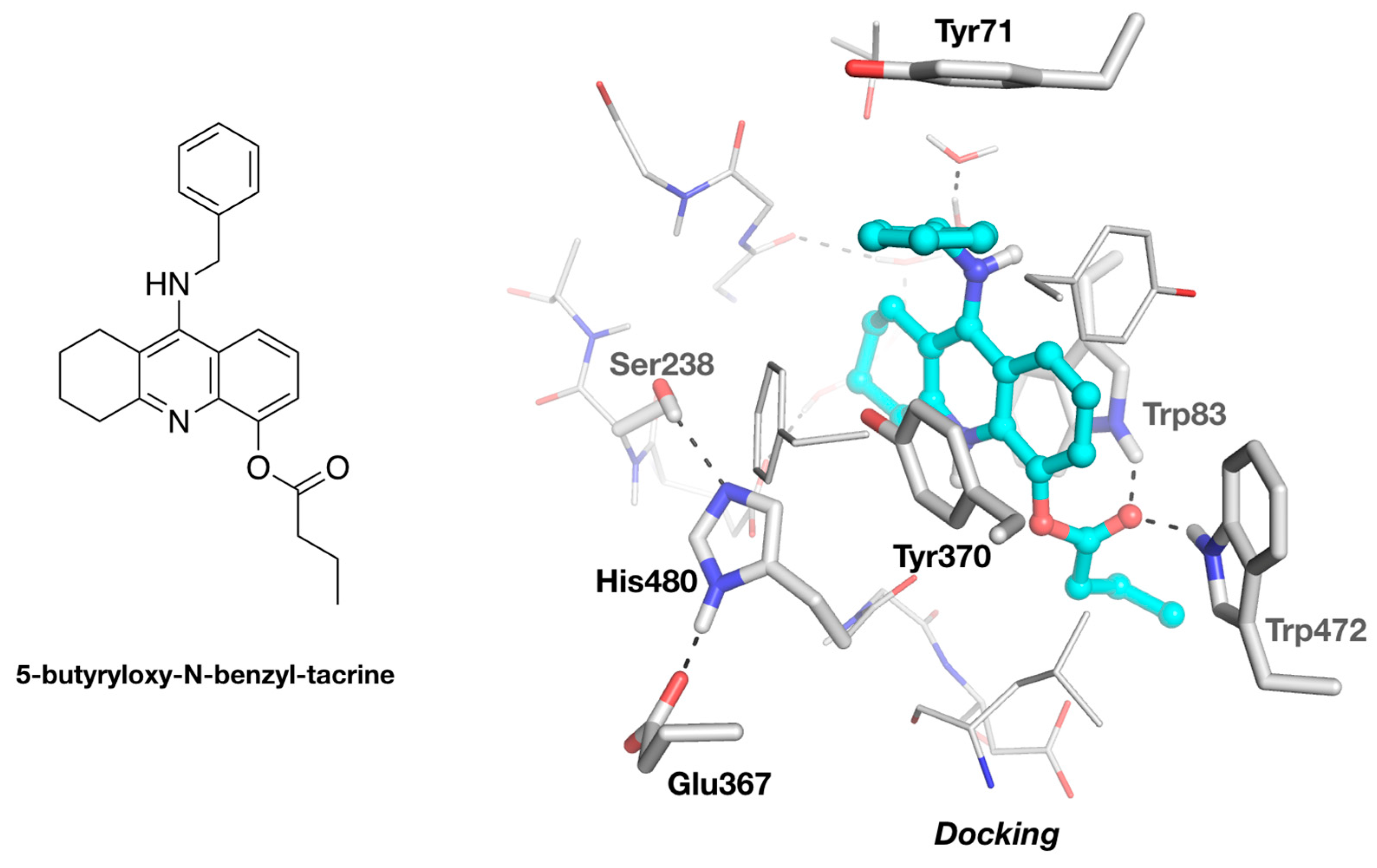
3.2. Molecular Docking of 5-Butyryloxy-9-Benzyltacrine in DmAChE
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Taylor, P.; Radic, Z. The cholinesterases: From genes to proteins. Annu. Rev. Pharmacol. Toxicol. 1994, 34, 281–320. [Google Scholar] [CrossRef]
- Pezzementi, L.; Chatonnet, A. Evolution of cholinesterases in the animal kingdom. Chem. Biol. Interact. 2010, 187, 27–33. [Google Scholar] [CrossRef]
- Casida, J.E.; Durkin, K. Anticholinesterase insecticide retrospective. Chem. Biol. Interact. 2012, 203, 221–225. [Google Scholar] [CrossRef] [Green Version]
- Fournier, D. Mutations of acetylcholinesterase which confer insecticide resistance in insect populations. Chem. Biol. Interact. 2005, 157, 257–261. [Google Scholar] [CrossRef]
- Harel, M.; Kryger, G.; Rosenberry, T.L.; Mallender, W.D.; Lewis, T.; Fletcher, R.J.; Guss, M.; Silman, I.; Sussman, J. Three-dimensional structures of Drosophila melanogaster acetylcholinesterase and of its complexes with two potent inhibitors. Protein Sci. 2000, 9, 1063–1072. [Google Scholar] [CrossRef]
- Nachon, F.; Stojan, J.; Fournier, D. Insights into substrate and product traffic in the Drosophila melanogaster acetylcholinesterase active-site gorge by enlarging a back channel. FEBS J. 2008, 275, 2659–2664. [Google Scholar] [CrossRef]
- Mew, E.J.; Padmanathan, P.; Konradsen, F.; Eddleston, M.; Chang, S.-S.; Phillips, M.R.; Gunnell, D. The global burden of fatal self-poisoning with pesticides 2006-15: Systematic review. J. Affect. Disord. 2017, 219, 93–104. [Google Scholar] [CrossRef] [Green Version]
- Han, Q.; Wong, D.; Robinson, H.; Ding, H.; Lam, P.C.H.; Totrov, M.M.; Carlier, P.R.; Li, J. Crystal structure of acetylcholinesterase catalytic subunits of the malaria vector Anopheles gambiae. Insect Sci. 2017, 25, 721–724. [Google Scholar] [CrossRef]
- Cheung, J.; Mahmood, A.; Kalathur, R.; Liu, L.; Carlier, P.R. Structure of the G119S Mutant Acetylcholinesterase of the Malaria Vector Anopheles gambiae Reveals Basis of Insecticide Resistance. Structure 2017, 26, 130–136. [Google Scholar] [CrossRef]
- Kabsch, W. XDS. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 125–132. [Google Scholar] [CrossRef] [Green Version]
- Afonine, P.V.; Grosse-Kunstleve, R.W.; Echols, N.; Headd, J.J.; Moriarty, N.W.; Mustyakimov, M.; Terwilliger, T.C.; Urzhumtsev, A.; Zwart, P.H.; Adams, P.D. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. Sect. D Biol. Crystallogr. 2012, 68, 352–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K.D. Features and development of Coot. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, P.D.; Afonine, P.V.; Bunkóczi, G.; Chen, V.; Davis, I.W.; Echols, N.; Headd, J.J.; Hung, L.-W.; Kapral, G.J.; Grosse-Kunstleve, R.W.; et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 213–221. [Google Scholar] [CrossRef] [Green Version]
- Cheung, J.; Rudolph, M.J.; Burshteyn, F.; Cassidy, M.S.; Gary, E.N.; Love, J.; Franklin, M.C.; Height, J.J. Structures of Human Acetylcholinesterase in Complex with Pharmacologically Important Ligands. J. Med. Chem. 2012, 55, 10282–10286. [Google Scholar] [CrossRef]
- Brazzolotto, X.; Wandhammer, M.; Ronco, C.; Trovaslet, M.; Jean, L.; Lockridge, O.; Renard, P.-Y.; Nachon, F. Human butyrylcholinesterase produced in insect cells: huprine-based affinity purification and crystal structure. FEBS J. 2012, 279, 2905–2916. [Google Scholar] [CrossRef]
- Nicolet, Y.; Lockridge, O.; Masson, P.; Fontecilla-Camps, J.C.; Nachon, F. Crystal Structure of Human Butyrylcholinesterase and of Its Complexes with Substrate and Products. J. Biol. Chem. 2003, 278, 41141–41147. [Google Scholar] [CrossRef] [Green Version]
- Suarez, D.; Diaz, N.; Fontecilla-Camps, J.; Field, M.J. A Computational Study of the Deacylation Mechanism of Human Butyrylcholinesterase. Biochemistry 2006, 45, 7529–7543. [Google Scholar] [CrossRef]
- Tormos, J.R.; Wiley, K.L.; Seravalli, J.; Nachon, F.; Masson, P.; Nicolet, Y.; Quinn, D.M. The Reactant State for Substrate-Activated Turnover of Acetylthiocholine by Butyrylcholinesterase is a Tetrahedral Intermediate. J. Am. Chem. Soc. 2005, 127, 14538–14539. [Google Scholar] [CrossRef] [Green Version]
- Millard, C.B.; Koellner, G.; Ordentlich, A.; Shafferman, A.; Silman, I.; Sussman, J. Reaction Products of Acetylcholinesterase and VX Reveal a Mobile Histidine in the Catalytic Triad. J. Am. Chem. Soc. 1999, 121, 9883–9884. [Google Scholar] [CrossRef]
- Carletti, E.; Li, H.; Li, B.; Ekström, F.; Nicolet, Y.; Loiodice, M.; Gillon, E.; Froment, M.T.; Lockridge, O.; Schopfer, L.M.; et al. Aging of Cholinesterases Phosphylated by Tabun Proceeds through O-Dealkylation. J. Am. Chem. Soc. 2008, 130, 16011–16020. [Google Scholar] [CrossRef]
- Wandhammer, M.; Carletti, E.; Van Der Schans, M.; Gillon, E.; Nicolet, Y.; Masson, P.; Goeldner, M.; Noort, D.; Nachon, F. Structural Study of the Complex Stereoselectivity of Human Butyrylcholinesterase for the Neurotoxic V-agents. J. Biol. Chem. 2011, 286, 16783–16789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bester, S.; Guelta, M.A.; Cheung, J.; Winemiller, M.D.; Bae, S.Y.; Myslinski, J.; Pegan, S.D.; Height, J.J. Structural Insights of Stereospecific Inhibition of Human Acetylcholinesterase by VX and Subsequent Reactivation by HI-6. Chem. Res. Toxicol. 2018, 31, 1405–1417. [Google Scholar] [CrossRef] [PubMed]
- Garman, E.F.; Weik, M. Radiation Damage in Macromolecular Crystallography. Methods Mol. Biol. 2017, 1607, 467–489. [Google Scholar]
- Weik, M.; Ravelli, R.B.G.; Kryger, G.; McSweeney, S.; Raves, M.L.; Harel, M.; Gros, P.; Silman, I.; Kroon, J.; Sussman, J. Specific chemical and structural damage to proteins produced by synchrotron radiation. Proc. Natl. Acad. Sci. 2000, 97, 623–628. [Google Scholar] [CrossRef] [Green Version]
- Ennifar, E.; Carpentier, P.; Ferrer, J.L.; Walter, P.; Dumas, P. X-ray-induced debromination of nucleic acids at the Br K absorption edge and implications for MAD phasing. Acta Crystallogr. Sect. D Biol. Crystallogr. 2002, 58, 1262–1268. [Google Scholar] [CrossRef]
- Wilcken, R.; Zimmermann, M.O.; Lange, A.; Joerger, A.; Boeckler, F.M. Principles and Applications of Halogen Bonding in Medicinal Chemistry and Chemical Biology. J. Med. Chem. 2013, 56, 1363–1388. [Google Scholar] [CrossRef]
- Nachon, F.; Carletti, E.; Ronco, C.; Trovaslet, M.; Nicolet, Y.; Jean, L.; Renard, P.-Y. Crystal structures of human cholinesterases in complex with huprine W and tacrine: elements of specificity for anti-Alzheimer’s drugs targeting acetyl- and butyryl-cholinesterase. Biochem. J. 2013, 453, 393–399. [Google Scholar] [CrossRef] [Green Version]
- Bourne, Y.; Renault, L.; Marchot, P. Crystal Structure of Snake Venom Acetylcholinesterase in Complex with Inhibitory Antibody Fragment Fab410 Bound at the Peripheral Site. J. Biol. Chem. 2014, 290, 1522–1535. [Google Scholar] [CrossRef]
- Sanson, B.; Colletier, J.-P.; Xu, Y.; Lang, P.T.; Jiang, H.; Silman, I.; Sussman, J.; Weik, M. Backdoor opening mechanism in acetylcholinesterase based on X-ray crystallography and molecular dynamics simulations. Protein Sci. 2011, 20, 1114–1118. [Google Scholar] [CrossRef] [Green Version]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligand | Native | ZAI | ZA | |||
|---|---|---|---|---|---|---|
| pdb code | 1qo9 | 6xys | 1qon | 6xyu | 1dx4 | 6xyy |
| Software | ||||||
| Integration | DENZO | XDS | DENZO | XDS | DENZO | |
| Scaling | SCALEPACK | XSCALE | SCALEPACK | XSCALE | SCALEPACK | |
| Refinement | CNS | Phenix | CNS | Phenix | CNS | Phenix |
| Model building | O | Coot | O | Coot | O | Coot |
| Data Collection | ||||||
| X-ray source—beamline | Elettra, Trieste, XRD | Elettra, Trieste, XRD | NSLS, BNL, X12C | |||
| wavelength (Å) | 1.0 | 1.0 | 0.98 | |||
| Resolution range (Å) | 29.83–2.7 | 30.94– 2.45 | 26.54–2.707 | 40.82–2.51 | 29.78–2.7 | |
| (highest-resolution shell) | (2.796–2.7) | (2.543–2.455) | (2.804–2.707) | (2.6–2.51) | (2.796–2.7) | |
| space group, mol/AU | P43212, 1 | P43212, 1 | P43212, 1 | P43212, 1 | P43212, 1 | |
| unit cell parameters (Å) | 94.3 94.3 159.0 90 90 90 | 94.6 94.6 159.0 90 90 90 | 94.9 94.9 160.0 90 90 90 | 94.9 94.9 160.0 90 90 90 | 95.8 95.8 162.0 90 90 90 | |
| Total reflections | na | 161402 (9870) | na | 70404 (6919) | na | |
| Unique reflections | 20199 (1956) | 26803 (2542) | 19963 (1998) | 24909 (2458) | 20596 (1809) | |
| Multiplicity | na | 6.0 (3.9) | na | 2.8 (2.8) | na | |
| Completeness (%) | 98.95 (98.79) | 98.54 (95.29) | 96.58 (98.71) | 96.64 (98.40) | 95.98 (86.67) | |
| Mean I/σ (I) | 33.5 (5.8) | 20.7 (3.3) | 15.5 (2.4) | 10.97 (1.68) | 24.3 (3.2) | |
| Wilson B-factor | 45.34 | 51.47 | 49.70 | 52.05 | 50.19 | |
| R-merge | na | 0.0487 (0.335) | na | 0.0635 (0.519) | na | |
| R-meas | 0.05 (0.161) | 0.0524 (0.387) | 0.060 (0.254) | 0.0766 (0.638) | 0.038 (0.274) | |
| R-pim | na | 0.0184 (0.1848) | na | 0.0412 (0.358) | na | |
| CC1/2 | na | 0.999 (0.939) | na | 0.997 (0.763) | na | |
| CC* | na | 1 (0.984) | na | 0.999 (0.93) | na | |
| Refinement Statistics | ||||||
| Reflections used | 20196 (1956) | 26739 (2530) | 19930 (1996) | 24876 (2457) | 20578 (1807) | 20577 (1807) |
| Reflections for R-free | 1990 (176) | 1338 (126) | 1704 (152) | 870 (86) | 2030 (193) | 2030 (193) |
| R-work | 0.2728 (0.4117) | 0.2297 (0.3959) | 0.2325 (0.3431) | 0.1857 (0.3252) | 0.2241 (0.3180) | 0.1711 (0.2461) |
| R-free | 0.2701 (0.4481) | 0.2950 (0.5239) | 0.2604 (0.3615) | 0.2522 (0.4266) | 0.2595 (0.3321) | 0.2335 (0.3270) |
| Number of non-H atoms | 4479 | 4398 | 4486 | 4447 | 4413 | 4483 |
| macromolecule | 4273 | 4239 | 4258 | 4230 | 4231 | 4242 |
| ligands | 75 | 65 | 89 | 63 | 91 | 97 |
| solvent | 131 | 94 | 139 | 154 | 97 | 144 |
| Protein residues | 540 | 542 | 686 | 542 | 537 | 537 |
| RMSD (bonds; Å) | 0.010 | 0.008 | 0.011 | 0.008 | 0.008 | 0.008 |
| RMSD (angles; deg) | 1.81 | 1.05 | 1.54 | 0.96 | 1.41 | 0.90 |
| Ramachandran favored (%) | 77.99 | 90.96 | 88.81 | 94.93 | 88.93 | 95.12 |
| Ramachandran allowed (%) | 16.04 | 8.10 | 9.33 | 4.50 | 9.38 | 4.50 |
| Ramachandran outliers (%) | 5.97 | 0.94 | 1.87 | 0.56 | 1.69 | 0.38 |
| Rotamer outliers (%) | 10.34 | 0.22 | 7.16 | 0.00 | 6.28 | 0.22 |
| Clashscore | 64.63 | 18.47 | 35.07 | 10.40 | 28.62 | 7.45 |
| Average B-factor (Å2) | 58.32 | 79.57 | 54.32 | 66.71 | 50.47 | 56.87 |
| macromolecules (Å2) | 58.43 | 79.61 | 54.02 | 66.59 | 50.15 | 56.43 |
| ligands (Å2) | 85.59 | 102.26 | 84.26 | 85.23 | 75.02 | 85.34 |
| solvent (Å2) | 39.15 | 61.86 | 44.27 | 62.47 | 40.67 | 50.57 |
| Number of TLS groups | - | 5 | - | 3 | - | 4 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nachon, F.; Rosenberry, T.L.; Silman, I.; Sussman, J.L. A Second Look at the Crystal Structures of Drosophila melanogaster Acetylcholinesterase in Complex with Tacrine Derivatives Provides Insights Concerning Catalytic Intermediates and the Design of Specific Insecticides. Molecules 2020, 25, 1198. https://doi.org/10.3390/molecules25051198
Nachon F, Rosenberry TL, Silman I, Sussman JL. A Second Look at the Crystal Structures of Drosophila melanogaster Acetylcholinesterase in Complex with Tacrine Derivatives Provides Insights Concerning Catalytic Intermediates and the Design of Specific Insecticides. Molecules. 2020; 25(5):1198. https://doi.org/10.3390/molecules25051198
Chicago/Turabian StyleNachon, Florian, Terrone L. Rosenberry, Israel Silman, and Joel L. Sussman. 2020. "A Second Look at the Crystal Structures of Drosophila melanogaster Acetylcholinesterase in Complex with Tacrine Derivatives Provides Insights Concerning Catalytic Intermediates and the Design of Specific Insecticides" Molecules 25, no. 5: 1198. https://doi.org/10.3390/molecules25051198