
Enantioselective Iron/Bisquinolyldiamine Ligand-Catalyzed Oxidative Coupling Reaction of 2-Naphthols


Sauvage Center for Molecular Sciences; Engineering Research Center of Organosilicon Compounds & Materials, Ministry of Education; College of Chemistry and Molecular Sciences; Wuhan University, Wuhan 430072, Hubei, China
*
Author to whom correspondence should be addressed.
Molecules 2020, 25(4), 852; https://doi.org/10.3390/molecules25040852
Submission received: 15 January 2020
/
Revised: 1 February 2020
/
Accepted: 12 February 2020
/
Published: 14 February 2020
(This article belongs to the Special Issue Recent Advances in Iron Catalysis)
Abstract
:An iron-catalyzed asymmetric oxidative homo-coupling of 2-naphthols for the synthesis of 1,1′-Bi-2-naphthol (BINOL) derivatives is reported. The coupling reaction provides enantioenriched BINOLs in good yields (up to 99%) and moderate enantioselectivities (up to 81:19 er) using an iron-complex generated in situ from Fe(ClO4)2 and a bisquinolyldiamine ligand [(1R,2R)-N1,N2-di(quinolin-8-yl)cyclohexane-1,2-diamine, L1]. A number of ligands (L2–L8) and the analogs of L1, with various substituents and chiral backbones, were synthesized and examined in the oxidative coupling reactions.

1. Introduction
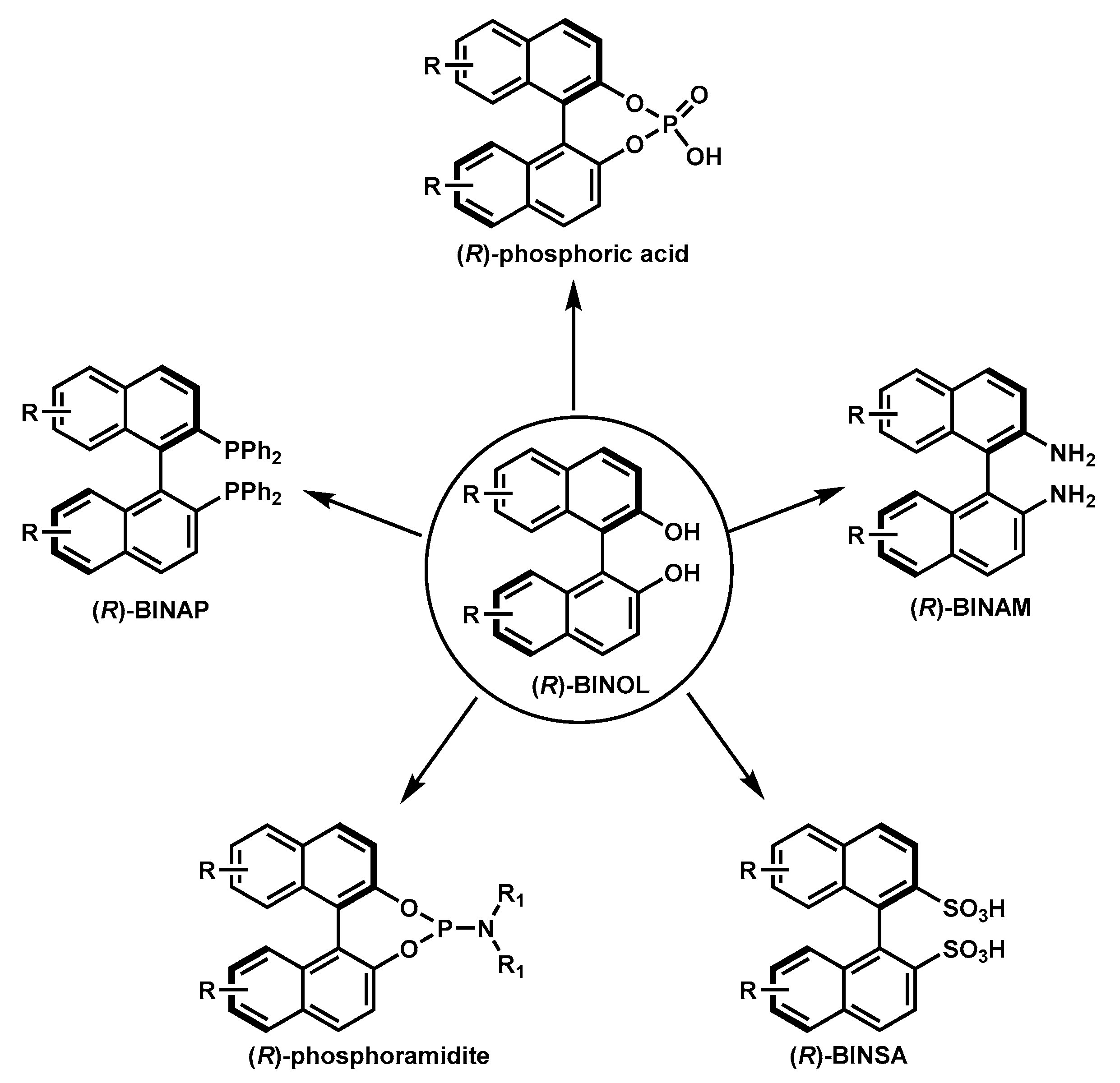
Axially chiral compounds (atropisomers) have aroused much attention from organic chemists due to their prevalence in natural products, bioactive molecules, functional materials, and their wide applications in asymmetric transformations [1]. Many elegant methods have been established for the asymmetric synthesis of axially chiral compounds, both employing transition-metal catalysts [2] and organocatalysts [1]. In particular, 1,1′-Bi-2-naphthol (BINOL) is one of the most useful structural motifs and ligand substructures in asymmetric catalysis [3,4,5,6,7,8,9]. Since the pioneering report by the Noyori group utilizing enantioenriched BINOL as the ligand in asymmetric catalysis [10], numerous BINOL-derived ligands/catalysts (i.e., BINAP [11], BINAM [12], chiral phosphoric acid [13], chiral phosphoramidite [14], and BINSA [15]; Figure 1) have been designed and synthesized. The emergence of such a library of ligands/catalysts has brought marvelous contributions to the synthetic community, with tremendously efficient asymmetric transformations such as reductive coupling [16], allylation [17], ene-type [18], and Aldol [19] reactions and axial chirality assembly [20].
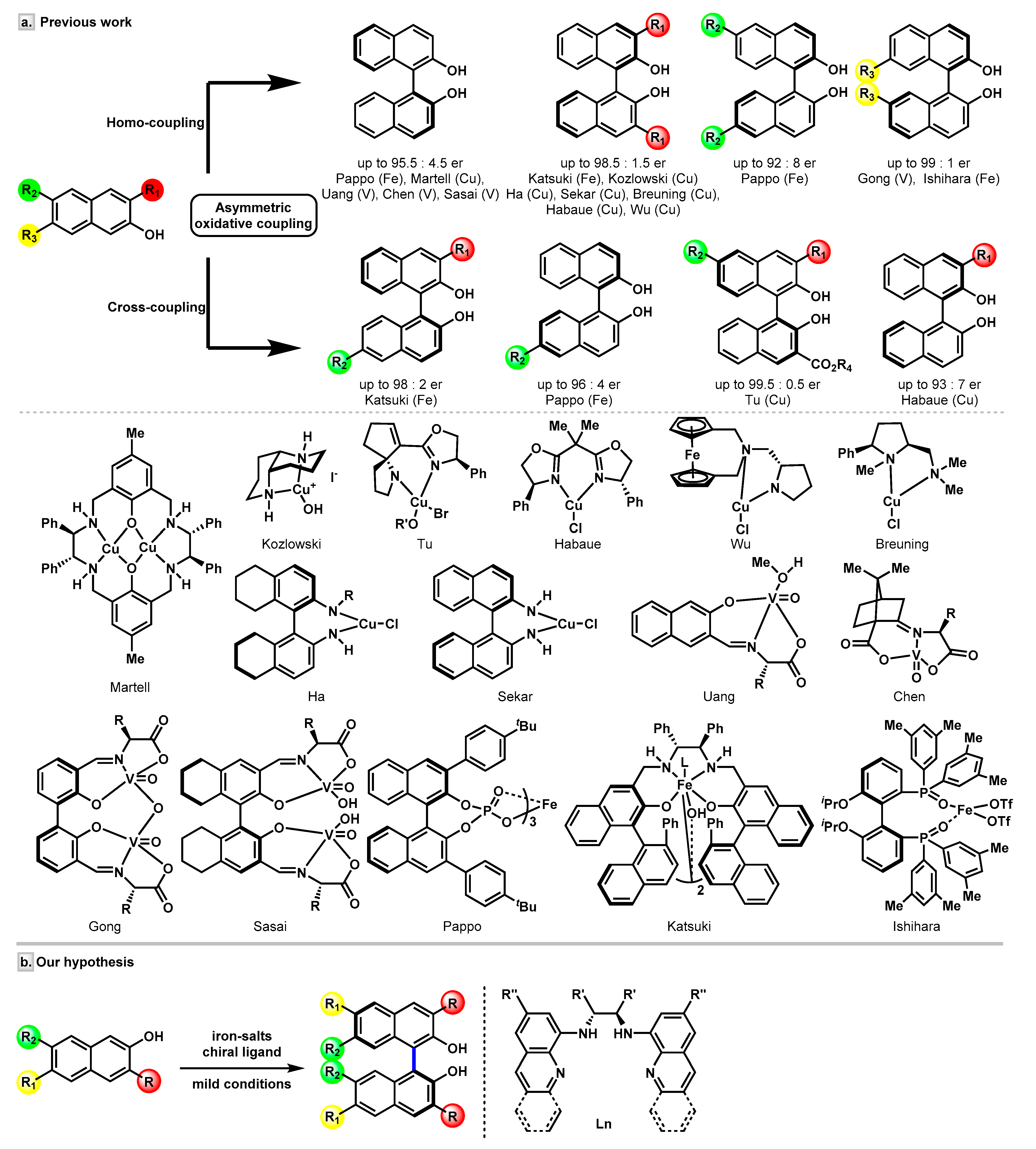
In the past few decades, enormous efforts have been devoted to the enantioselective assembly of BINOL scaffolds. Transition-metal-catalyzed asymmetric oxidative coupling of 2-naphthols have shown its power in the synthesis of BINOLs (Figure 2a). Efficient vanadium-catalyzed [21] protocols were reported by the Uang [22], Chen [23], Gong [24,25,26], and Sasai [27,28,29] groups, independently. Many research groups, including Nakajima, Kozlowski, and others, have successfully developed a series of copper-catalyzed coupling reactions of 2-naphthols [30,31,32,33,34,35,36,37,38,39,40,41,42,43,44]. Recently, a notable work by the Tu group [45] established a Cu/SPDO (spirocyclic pyrrolidine oxazoline) complex-catalyzed cross-coupling reaction to synthesize 3,3′-disubstituted BINOLs. Nevertheless, iron-catalyzed coupling strategies in this area have not been explored so far. Only a handful of remarkable iron catalysts, namely, an iron-salen complex reported by Katsuki and co-workers [46,47], a chiral diphosphine oxide–iron(II) complex developed by the Ishihara group [48], and an iron-chiral phosphoric acid (CPA) catalyst introduced by the Pappo group [49,50], have been disclosed to date (Figure 2a).
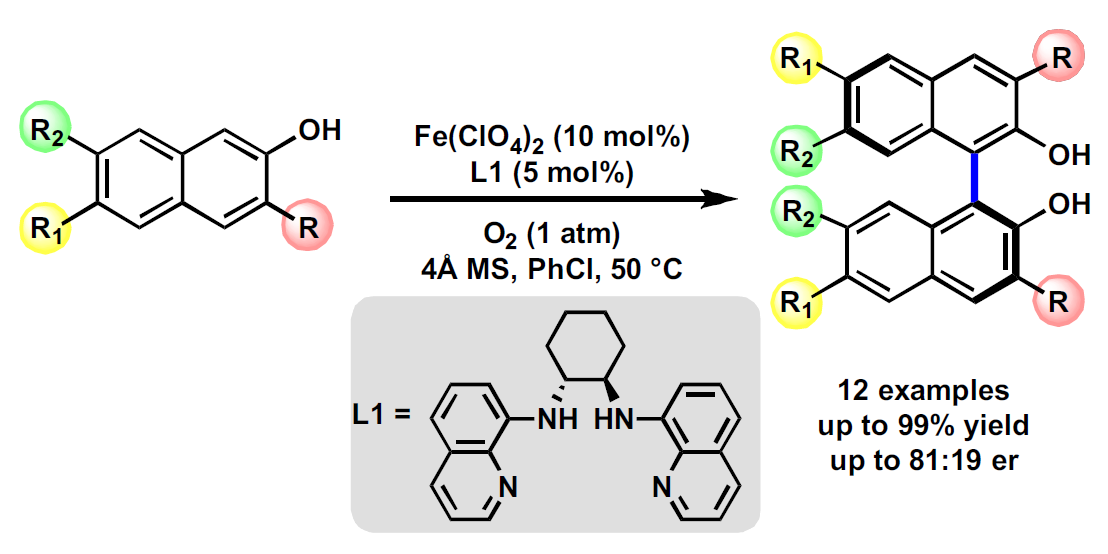
Recently, we have developed an iron-catalyzed direct amination of aliphatic C-H bonds [51,52], and it was interesting to find that the catalysts used were simply generated by in situ mixing of an iron salt and an aminopyridine ligand. Inspired by these results, we envisaged that introducing a chiral aminopyridine-type ligand might impart chirality to the products. Our attention was drawn to N,N′-dimethyl-N,N′-bis(8-quinolyl)-cyclohexanediamine (BQCN), developed by the Che group and successfully applied in iron-catalyzed asymmetric cis-dihydroxylation of alkenes [53] and, most recently, in Friedel-Crafts reactions [54]. Since our previous studies revealed that free secondary amine ligand presented a good reactivity [51], we were wondering whether the N-unprotected bis(8-quinolyl)-cyclohexanediamine ligand (bisquinolyldiamine, L1), which is synthesized straightforwardly from 8-haloquinoline and diamine, is capable of controlling the selectivities in the iron-catalyzed oxidative coupling of 2-naphthols. Herein, we report the studies toward the synthesis of the amino ligands and their applications in the synthesis of optically active BINOL derivatives via an oxidative homo-coupling reaction of 2-naphthols under mild conditions (Figure 2b).
2. Results and Discussion
2.1. Synthesis of Bisquinolyldiamine Ligands
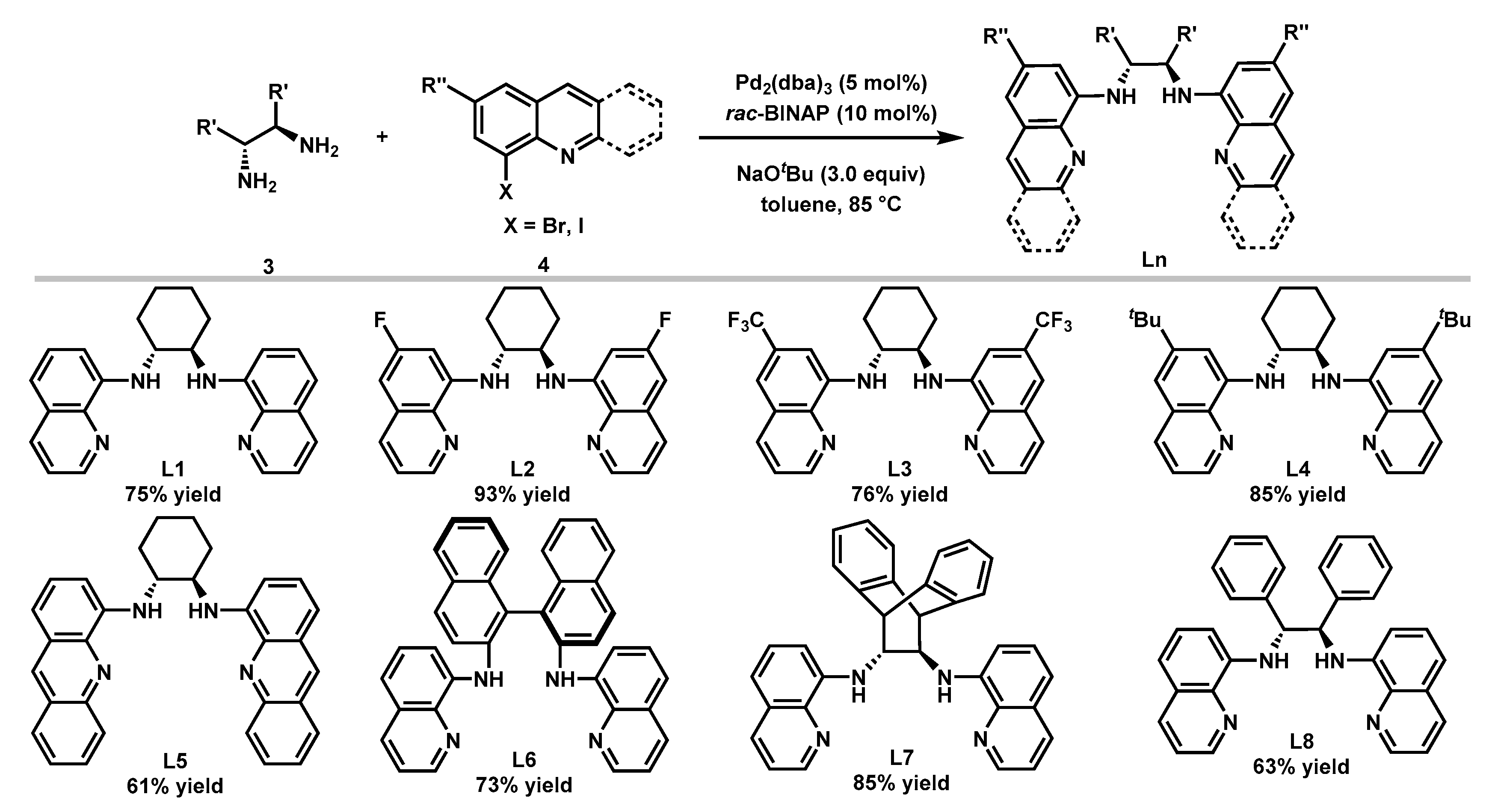
The Buchwald-Hartwig C–N coupling reaction [55,56] was used for the synthesis of a variety of bisquinolyldiamine ligands following the literature procedure [53]. As shown in Figure 3, with the catalyst derived from 5 mol% Pd2(dba)3 and 10 mol% rac-BINAP, eight ligands were synthesized in moderate to good yields. The chiral backbones of these ligands include (1R,2R)-cyclohexane-1,2-diamine (L1), (R)-[1,1′-binaphthalene]-2,2′-diamine (L6), (12R)-9,10-dihydro-9,10-ethanoanthracene-11,12-diamine (L7), and (1R,2R)-1,2-diphenylethane-1,2-diamine (L8). Ligands with a variety of electronically differentiated substituents at the C6 position of the quinoline moiety (L2–L4) and an acridine-derived ligand (L5) were also prepared to probe the electronic and steric effects of the ligands on the reaction.
2.2. Reaction Investigation
To test our hypothesis, we selected 2-naphthol (1a) as the model substrate for optimization of reaction conditions (Table 1). All reactions were carried out with the catalyst generated in situ by stirring Fe(ClO4)2 and L1 for 30 min before the addition of the substrate, and under atmospheric dioxygen. Various solvents (methanol, dichloroethane, chloroform, toluene, and chlorobenzene) were screened first (entries 1–5) and resulted in moderate conversions and enantioselectivities, except toluene and methanol. The reaction in chlorobenzene gave a better balance between reactivity and selectivity (53% conv. and 79:21 er, entry 5). The addition of 4Å MS led to a slightly higher enantioselectivity with partial conversion in a much shorter reaction time (entries 5 vs. 6). We were delighted to find that increasing the temperature from 30 °C to 50 °C delivered 39% conversion with comparable enantioselectivity (entry 7). Further increase in temperature (i.e., 70 °C and 90 °C) resulted in lower enantioselectivities (entries 8 and 9). Then, the reaction with different iron salts were investigated, including Fe(ClO4)3, Fe(OAc)2, Fe(OTf)2, Fe(acac)2, and FeCl2, but failed to provide better results (entries 10–14). The ratio of iron precursor versus L1 was also examined (entries 14–19). Surprisingly, increasing Fe(ClO4)2-loading from 5 mol% to 10 mol% improves the efficiency without affecting the enantioselectivity and delivered the coupling product 2a in 84% isolated yield with 80:20 er (entries 15 and 16). Although further increasing Fe(ClO4)2 to 12.5 mol% improved the yield, a slightly diminished er was also observed (entry 17). Finally, reactions with the catalysts derived from the diamine ligand (L2–L8) were inspected. Electron-withdrawing groups-substituted ligands (L2 and L3) showed excellent reactivities but with low enantioselectivities (entries 20 and 21). In contrast, ligand L4 with an electron-donating substituent delivered the product in reduced yield, albeit with good er (entry 22). Upon further screening, the ligands (L5–L8) bearing different chiral backbones, lower yield, and er were obtained with L5, while ligands L6–L8 failed to give any product (entries 23–26).
2.3. Substrates Scope
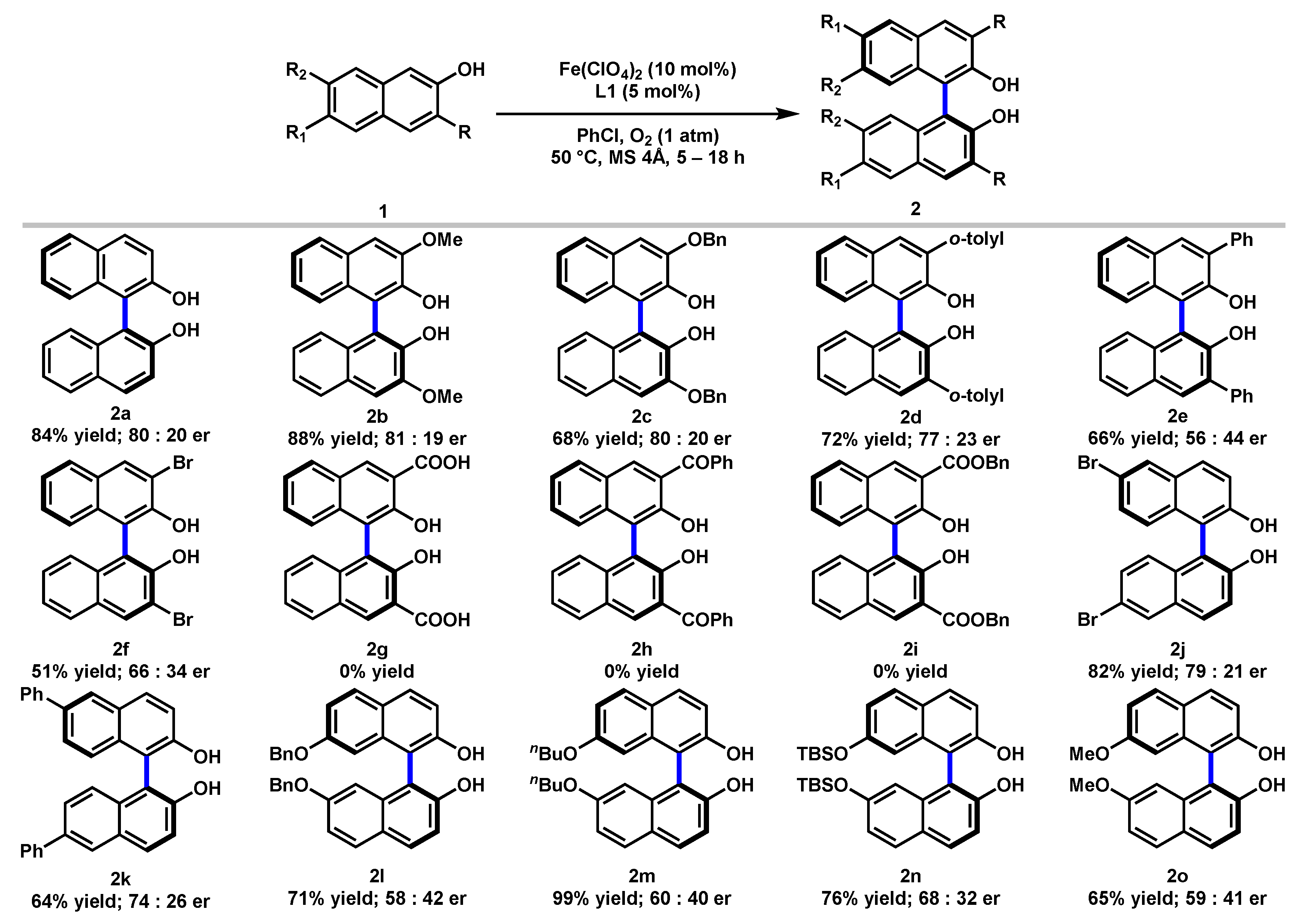
Next, we investigated the scope of the iron-catalyzed asymmetric oxidative coupling reaction, and the results were summarized in Scheme 1. A variety of substituted 2-naphthols with electronic and steric properties were examined. Substrate 1 containing functionalities at the C3-position, including OMe, OBn, o-tolyl, and Ph were successfully converted into the coupling products (2b–2e) in 56–88% yields with 56:44 to 81:19 er. The substrate bearing an electron-withdrawing Br substitution at the C3-position was also converted into the corresponding product (2f) in 51% yield with 79:21 er. The electronic effects of different functionalities are clearly demonstrated by the observation that electron-donating groups delivered higher yield (e.g., C3-OMe 88%, 2b vs. C3-Br 51%, 2f). However, C3-substituted substrates with carbonyl functionalities like CO2H, COPh, and CO2Bn failed to give any products (2g–2i). When C6-Br-substituted 2-naphthol was applied, the desired product (2j) was obtained in 82% yield with 79:21 er. A C6-phenyl-substituted 2-naphthol also resulted in 64% yield and 74:26 er (2k). Moreover, C7-substituted (BnO, nBuO, TBSO, and MeO) substrates were also effectively coupled and delivered the corresponding products (2l–2o) in 65–99% yields, albeit with dramatically diminished er.
3. Materials and Methods
3.1. General Information
Unless otherwise noted, all reagents were purchased commercially and used without further purification. Petroleum ether (PE) (60–90 °C), ethyl acetate (EA), and dichloromethane (DCM) were used for silica gel chromatography. MeCN, toluene, DMF, and THF were purchased commercially or were dried by passage through an activated alumina column under argon [57]. PhCl, CHCl3, MeOH, and acetone were freshly distilled after drying over CaH2. 1H-NMR spectra were recorded at room temperature on a Bruker ADVANCE III 400 MHz spectrometer and were reported relative to residual Chloroform-d (δ 7.26 ppm) or DMSO-d6 (δ 2.50 ppm). 13C-NMR spectra were recorded on a Bruker ADVANCE III 400 MHz spectrometer (100 MHz) and were reported relative to Chloroform-d (δ 77.16 ppm). 19F-NMR spectra were recorded on a Bruker ADVANCE III 400 MHz spectrometer (376 MHz). Data for 1H-NMR were reported as chemical shift (δ ppm) (multiplicity, coupling constant (Hz), and integration) using standard abbreviations for multiplicities: s = singlet, d = doublet, t = triplet, q = quartet, and m = multiplet. Data for 13C-NMR and 19F-NMR were reported in terms of chemical shifts (δ ppm). High-resolution mass spectra (HRMS) were obtained by using a Bruker Compact TOF mass spectrometer in electrospray ionization mode (ESI). Enantiomeric ratio (er) was determined by an Agilent 1260 Series HPLC utilizing DAICEL Chiralpak (AD-H, AS-H, or IC) or Chiralcel (OD-H) columns (4.6 mm × 250 mmL). Optical rotations were measured with a Perkin Elmer 343 polarimeter and were reported as: [α]DT (concentration in g/100 mL, solvent). The NMR spectra of all new compounds and HPLC spectra of oxidative coupling products were provided in the Supplementary Materials.
3.2. Preparation of Ligands
General Procedure (Scheme 2): To an oven-dried Schlenk flask were added diamine 3 (1.0 equiv), Pd2(dba)3 (5 mol%), rac-BINAP (10 mol%), NaOtBu (3.0 equiv), and toluene under Ar atmosphere. Then 8-haloqunoline 4 (2.2 equiv) was added directly. The flask was sealed, and the reaction was stirred at 85 °C until the complete consumption of the starting material 3. The mixture was cooled to room temperature, filtered through a silica plug, and the plug was washed with EA. The combined filtrates were concentrated under reduced pressure, and the residue was purified by silica gel chromatography to give the desired product Ln.
(1R,2R)-N1,N2-Di(quinolin-8-yl)cyclohexane-1,2-diamine (L1) [53]: Following the general procedure, the reaction was carried out with (1R,2R)-cyclohexane-1,2-diamine 3a (0.36 g, 3.2 mmol, 1.0 equiv); Pd2(dba)3 (0.15 g, 0.16 mmol, 5 mol%); rac-BINAP (0.20 g, 0.32 mmol, 10 mol%); NaOtBu (0.92 g, 9.6 mmol, 3.0 equiv); and 8-bromoqunoline 4a (1.46 g, 7.0 mmol, 2.2 equiv) in 30 mL of toluene. The desired product was obtained (0.88 g, 75% yield) as a pale yellow solid after purification by silica gel chromatography (PE/EA = 30/1 to 10/1). 1H-NMR (400 MHz, Chloroform-d) δ 8.57 (dd, J = 4.2, 1.7 Hz, 2H), 7.98 (dd, J = 8.3, 1.7 Hz, 2H), 7.36 (td, J = 8.0, 0.9 Hz, 2H), 7.30–7.25 (m, 2H), 6.98 (dd, J = 8.2, 1.3 Hz, 2H), 6.84 (dd, J = 7.7, 1.1 Hz, 2H), 6.43 (brs, 2H), 3.86–3.67 (m, 2H), 2.49–2.31 (m, 2H), 1.86 (td, J = 4.6, 4.1, 2.2 Hz, 2H), 1.67–1.48 (m, 4H).
(1R,2R)-N1,N2-Bis(6-fluoroquinolin-8-yl)cyclohexane-1,2-diamine (L2): Following the general procedure, the reaction was carried out with (1R,2R)-cyclohexane-1,2-diamine 3a (81.6 mg, 0.7 mmol, 1.0 equiv); Pd2(dba)3 (32.8 mg, 0.04 mmol, 5 mol%); rac-BINAP (45.1 mg, 0.07 mmol, 10 mol%); NaOtBu (206.9 mg, 2.2 mmol, 3.0 equiv); and 8-bromo-6-fluoroquinoline 4b (356.7 mg, 1.6 mmol, 2.2 equiv) in 2 mL of toluene. The desired product was obtained (262.3 mg, 93% yield) as a pale yellow solid after purification by silica gel chromatography (PE/EA = 5/1). 1H-NMR (400 MHz, Chloroform-d) δ 8.49 (dd, J = 4.2, 1.5 Hz, 2H), 7.87 (dd, J = 8.3, 1.2 Hz, 2H), 7.34–7.21 (m, 2H), 6.68–6.49 (m, 4H), 6.46 (dd, J = 9.3, 2.5 Hz, 2H), 3.76–3.56 (m, 2H), 2.45–2.28 (m, 2H), 1.97–1.79 (m, 2H), 1.66–1.47 (m, 4H) (Figure S2); 13C-NMR (100 MHz, Chloroform-d) δ 162.4 (d, JC–F = 243.3 Hz), 146.3 (d, JC–F = 13.5 Hz), 145.6 (d, JC–F = 2.4 Hz), 135.6, 135.4 (d, JC–F = 5.8 Hz), 129.3 (d, JC–F = 12.8 Hz), 122.2, 96.1 (d, JC–F = 22.8 Hz), 95.1 (d, JC–F = 30.6 Hz), 56.6, 31.7, 24.5 (Figure S3); 19F NMR (376 MHz, Chloroform-d) δ –110.9 (Figure S4); HRMS (ESI+) calcd for C24H23F2N4 [M + H]+: 405.1885, found 405.1880; = −315.6 (c = 0.2, CHCl3); M. p. 162–166 °C.
(1R,2R)-N1,N2-Bis(6-(trifluoromethyl)quinolin-8-yl)cyclohexane-1,2-diamine (L3): Following the general procedure, the reaction was carried out with (1R,2R)-cyclohexane-1,2-diamine 3a (0.31 g, 2.7 mmol, 1.0 equiv); Pd2(dba)3 (0.13 g, 0.14 mmol, 5 mol%); rac-BINAP (0.17 g, 0.28 mmol, 10 mol%); NaOtBu (0.79 g, 8.2 mmol, 3.0 equiv); and 8-bromo-6-trifluoromethylquinoline 4c (1.57 g, 5.7 mmol, 2.1 equiv) in 25 mL of toluene. The desired product was obtained (1.04 g, 76% yield) as a yellow green solid after purification by silica gel chromatography (PE/DCM = 10/1 to 5/1 to 1/1). 1H-NMR (400 MHz, Chloroform-d) δ 8.61 (dd, J = 4.2, 1.5 Hz, 2H), 7.95 (d, J = 8.1 Hz, 2H), 7.32 (dd, J = 8.2, 4.2 Hz, 2H), 7.14–7.03 (m, 2H), 6.95–6.83 (m, 2H), 6.55 (s, 2H), 3.87–3.62 (m, 2H), 2.45–2.24 (m, 2H), 2.03–1.85 (m, 2H), 1.70–1.48 (m, 4H) (Figure S5); 13C-NMR (100 MHz, Chloroform-d) δ 148.4, 144.9, 138.8, 136.9, 129.4 (q, JC–F = 31.7 Hz), 127.5, 124.5 (q, JC–F = 272.7 Hz), 122.3, 110.6 (q, JC–F = 4.7 Hz), 100.0, 57.3, 32.3, 24.8 (Figure S6); 19F NMR (376 MHz, Chloroform-d) δ –62.8 (Figure S7); HRMS (ESI+) calcd for C26H23F6N4 [M + H]+: 505.1821, found 505.1817; = −329.1 (c = 1.0, CHCl3); M. p. 120–124 °C.
(1R,2R)-N1,N2-Bis(6-(tert-butyl)quinolin-8-yl)cyclohexane-1,2-diamine (L4): Following the general procedure, the reaction was carried out with (1R,2R)-cyclohexane-1,2-diamine 3a (0.68 g, 6.0 mmol, 1.0 equiv); Pd2(dba)3 (0.28 g, 0.3 mmol, 5 mol%); rac-BINAP (0.37 g, 0.6 mmol, 10 mol%); NaOtBu (1.73 g, 18 mmol, 3.0 equiv); and 8-bromo-6-(tert-butyl)quinoline 4d (3.46 g, 13.1 mmol, 2.2 equiv) in 35 mL of toluene. The desired product was obtained (2.44 g, 85% yield) as a yellow solid after purification by silica gel chromatography (PE/DCM = 10/1 to PE/EA = 5/1). 1H-NMR (400 MHz, Chloroform-d) δ 8.55 (dd, J = 4.3, 1.7 Hz, 2H), 7.99 (d, J = 7.5 Hz, 2H), 7.31–7.25 (m, 2H), 7.02–6.89 (m, 4H), 3.87–3.81 (m, 2H), 2.38 (d, J = 12.2 Hz, 2H), 1.91–1.83 (m, 2H), 1.71–1.50 (m, 4H), 1.36 (s, 18H) (Figure S8); 13C-NMR (100 MHz, Chloroform-d) δ 150.6, 146.2, 143.5, 137.4, 136.0, 128.5, 121.3, 109.4, 104.3, 55.5, 35.2, 31.4, 30.7, 24.0 (Figure S9); HRMS (ESI+) calcd for C32H41N4 [M + H]+: 481.3326, found 481.3323; = −39.2 (c = 1.0, CHCl3); M. p. 172–174 °C.
(1R,2R)-N1,N2-Di(acridin-4-yl)cyclohexane-1,2-diamine (L5): Following the general procedure, the reaction was carried out with (1R,2R)-cyclohexane-1,2-diamine 3a (0.19 g, 1.6 mmol, 1.0 equiv); Pd2(dba)3 (0.08 g, 0.08 mmol, 5 mol%); rac-BINAP (0.10 g, 0.16 mmol, 10 mol%); NaOtBu (0.47 g, 4.9 mmol, 3.0 equiv); and 4-iodoacridine 4e (1.07 g, 3.5 mmol, 2.2 equiv) in 30 mL of toluene. The desired product was obtained (0.46 g, 61% yield) as a yellow solid after purification by silica gel chromatography (PE/DCM = 2/1 to PE/DCM = 1/1 to DCM). 1H-NMR (400 MHz, DMSO-d6) δ 8.78 (s, 2H), 8.01 (d, J = 8.3 Hz, 2H), 7.91 (d, J = 8.7 Hz, 2H), 7.67 (t, J = 8.0 Hz, 2H), 7.49 (t, J = 8.0 Hz, 2H), 7.43 (t, J = 7.9 Hz, 2H), 7.18 (d, J = 8.4 Hz, 2H), 6.97 (d, J = 7.5 Hz, 2H), 6.75 (d, J = 7.0 Hz, 2H), 3.98–3.90 (m, 2H), 2.37–2.30 (m, 2H), 1.86–1.80 (m, 2H), 1.62–1.55 (m, 4H) (Figure S10); 13C-NMR (100 MHz, Chloroform-d) δ 146.5, 144.2, 140.7, 135.1, 129.7, 128.9, 127.8, 127.3, 127.2, 127.0, 125.5, 113.8, 103.1, 56.6, 31.6, 24.4 (Figure S11); HRMS (ESI+) calcd for C32H29N4 [M + H]+: 469.2387, found 469.2371; = −678.0 (c = 0.5, CHCl3); M. p. 198–202 °C.
(R)-N2,N2′-Di(quinolin-8-yl)-[1,1′-binaphthalene]-2,2′-diamine (L6) [53]: Following the general procedure, the reaction was carried out with (R)-[1,1′-binaphthalene]-2,2′-diamine 3b (141.9 mg, 0.5 mmol, 1.0 equiv); Pd2(dba)3 (23.2 mg, 0.025 mmol, 5 mol%); rac-BINAP (31.4 mg, 0.05 mmol, 10 mol%); NaOtBu (148.2 mg, 1.5 mmol, 3.0 equiv); and 8-bromoqunoline 4a (224.0 mg, 1.1 mmol, 2.2 equiv) in 10 mL of toluene. The desired product was obtained (196.6 mg, 73% yield) as a yellow solid after purification by recrystallization from EA. 1H-NMR (400 MHz, Chloroform-d) δ 8.42 (d, J = 3.0 Hz, 2H), 7.99–7.95 (m, 4H), 7.94–7.86 (m, 6H), 7.37 (dt, J = 8.0, 4.0 Hz, 2H), 7.30–7.19 (m, 8H), 6.94–6.89 (m, 4H).
(12R)-N11,N12-Di(quinolin-8-yl)-9,10-dihydro-9,10-ethanoanthracene-11,12-diamine (L7) [53]: Following the general procedure, the reaction carried out with (12R)-9,10-dihydro-9,10-ethanoanthracene-11,12-diamine 3c (20 mg, 0.08 mmol, 1.0 equiv); Pd2(dba)3 (5.3 mg, 5 mol%); rac-BINAP (6.2 mg, 10 mol%); NaOtBu (25.5 mg, 0.26 mmol, 3.0 equiv); and 8-bromoqunoline 4a (41.8 mg, 0.2 mmol, 2.2 equiv) in 1 mL of toluene. The desired product was obtained (33.4 mg, 85% yield) as a white solid after purification by silica gel chromatography (PE/EA = 5/1). 1H-NMR (400 MHz, Chloroform-d) δ 8.61 (dd, J = 4.1, 1.4 Hz, 2H), 8.06 (d, J = 4.0 Hz, 2H), 7.44 (d, J = 7.1 Hz, 2H), 7.40–7.13 (m, 10H), 7.06 (d, J = 8.1 Hz, 2H), 6.91 (d, J = 8.0 Hz, 2H), 6.18 (s, 2H), 4.63 (s, 2H), 3.97 (s, 2H).
(1R,2R)-1,2-Diphenyl-N1,N2-di(quinolin-8-yl)ethane-1,2-diamine (L8) [58]: Following the general procedure, the reaction was carried out with (1R,2R)-1,2-diphenylethane-1,2-diamine 3d (1.06 g, 5.0 mmol, 1.0 equiv); Pd2(dba)3 (0.23 g, 0.25 mmol, 5 mol%); rac-BINAP (0.33 g, 0.5 mmol, 10 mol%); NaOtBu (1.47 g, 15 mmol, 3.0 equiv); and 8-bromoqunoline 4a (2.51 g, 12 mmol, 2.4 equiv) in 90 mL of toluene. The desired product was obtained (1.47 g, 63% yield) as a white solid after purification by silica gel chromatography (PE/DCM = 30/1 to PE/EA = 5/1). 1H-NMR (400 MHz, Chloroform-d) δ 8.67 (dd, J = 4.3, 1.6 Hz, 2H), 8.05 (d, J = 8.4 Hz, 2H), 7.36 (m, 4H), 7.28–7.09 (m, 12H), 7.03 (d, J = 8.0 Hz, 2H), 6.50 (d, J = 7.6 Hz, 2H), 5.01 (s, 2H).
3.3. Preparation of Substituted Quinolines
General procedure for synthesis of substituted 8-bromoquinoline (Scheme 3): to a 50 mL round bottom flask was added 4-substituted 2-bromoaniline, glycerol (17.0 equiv), m-nitrobenzenesulfonate sodium (1.2 equiv), FeSO4•7H2O (0.05 equiv), and MsOH. The reaction mixture was heated at 125 °C for 24 h. After cooling to room temperature, aqueous NaOH solution (2.5 M) was added to the reaction mixture to adjust pH to 12. Then EtOH was added to form a black solution, which was extracted with EA or DCM (3 × 100 mL). The combined organic phase was washed with H2O (100 mL), brine (100 mL), and dried with anhydrous Na2SO4. After removing the solvents, the residue was purified by silica gel chromatography.
8-Bromo-6-fluoroquinoline (4b) [59]: Following the general procedure, the reaction was carried out with 2-bromo-4-fluoroaniline (1.57 g, 8.3 mmol, 1.0 equiv); glycerol (11 mL, 149.0 mmol, 18.0 equiv); m-nitrobenzenesulfonate sodium (2.24 g, 10.0 mmol, 1.2 equiv); FeSO4•7H2O (0.12 g, 0.4 mmol, 0.05 equiv), and MsOH (11 mL). The desired product was obtained (0.86 g, 43% yield) as a pale yellow solid after purification by silica gel chromatography (PE/EA = 10/1 to 5/1). 1H-NMR (400 MHz, Chloroform-d) δ 9.02 (dd, J = 4.2, 1.2 Hz, 1H), 8.14 (dd, J = 8.3, 1.6 Hz, 1H), 7.90 (dd, J = 8.1, 2.7 Hz, 1H), 7.50 (ddd, J = 8.3, 4.2, 0.6 Hz, 1H), 7.46 (dd, J = 8.3, 2.7 Hz, 1H).
8-Bromo-6-(trifluoromethyl)quinoline (4c): Following the general procedure, the reaction was carried out with 2-bromo-4-(trifluoromethyl)aniline (6.38 g, 26.6 mmol, 1.0 equiv); glycerol (20 mL, 271.5 mmol, 10.0 equiv); m-nitrobenzenesulfonate sodium (7.19 g, 32.0 mmol, 1.2 equiv); FeSO4•7H2O (0.37 g, 1.3 mmol, 0.05 equiv), and MsOH (35 mL). The desired product was obtained (1.57 g, 21% yield) as a pale orange solid after purification by silica gel chromatography (PE/EA = 10/1 to 5/1). 1H-NMR (400 MHz, Chloroform-d) δ 9.16 (dd, J = 4.2, 1.7 Hz, 1H), 8.28 (dd, J = 8.3, 1.7 Hz, 1H), 8.24 (d, J = 1.9 Hz, 1H), 8.16–8.10 (m, 1H), 7.60 (dd, J = 8.3, 4.2 Hz, 1H) (Figure S12); 13C-NMR (100 MHz, Chloroform-d) δ 153.4, 146.5, 137.7, 129.1(q, JC–F = 33.4 Hz), 129.0(q, JC–F = 3.1 Hz), 128.4, 126.3, 125.7 (q, JC–F = 4.3 Hz), 123.2, 123.2(q, JC–F = 272.8 Hz) (Figure S13); 19F NMR (376 MHz, Chloroform-d) δ –62.5 (Figure S14); HRMS (ESI+) calcd for C10H6BrF3N [M + H]+: 275.9630, found 275.9620; M. p. 58–62 °C.
8-Bromo-6-(tert-butyl)quinoline (4d) [60]: Following the general procedure, the reaction was carried out with 2-bromo-4-(tert-butyl)aniline (1.78 g, 7.8 mmol, 1.0 equiv); glycerol (10 mL, 135.7 mmol, 17.0 equiv); m-nitrobenzenesulfonate sodium (2.11 g, 9.4 mmol, 1.2 equiv); FeSO4•7H2O (0.11 g, 0.41 mmol, 0.05 equiv); and MsOH (10 mL). The desired product was obtained (1.73 g, 84% yield) as a yellow solid after purification by silica gel chromatography (PE/EA = 20/1). 1H-NMR (400 MHz, Chloroform-d) δ 9.00 (dd, J = 4.2, 1.5 Hz, 1H), 8.18–8.12 (m, 2H), 7.70 (d, J = 2.0 Hz, 1H), 7.45 (dd, J = 8.2, 4.2 Hz, 1H), 1.42 (s, 9H).
4-Iodoacridine [4e] (Scheme 4)
To a 100 mL Schlenk flask were added TMP (1.14 g, 8.1 mmol, 1.5 equiv) and 20 mL of THF. The solution was cooled to 0 °C, and nBuLi (2.4 M, 4 mL, 9.6 mmol, 1.7 equiv) was added dropwise by syringe. Upon the completion of the addition, the mixture was stirred at 0 °C for another 30 min. Then ZnCl2•TMEDA (0.68 g, 2.7 mmol, 0.5 equiv) was added at 0 °C, and the resultant mixture was stirred for 20 min before acridine (0.98 g, 5.5 mmol, 1.0 equiv) was added. After the reaction was warmed up to 25 °C, I2 (2.17 g, 8.5 mmol, 1.5 equiv) in THF (20 mL) was added dropwise. The reaction mixture was stirred for 2 h and then quenched with saturated Na2S2O3 solution and extracted with EA (3 × 30 mL). The combined organic phase was washed with brine and dried over anhydrous Na2SO4. The desired product was obtained (1.08 g, 64% yield) as a yellow solid after purification by silica gel chromatography (PE/DCM = 20/1 to 5/1). 1H-NMR (400 MHz, Chloroform-d) δ 8.72 (s, 1H), 8.46 (dd, J = 7.1, 1.1 Hz, 1H), 8.39 (d, J = 8.8 Hz, 1H), 8.09–7.93 (m, 2H), 7.83 (ddd, J = 8.5, 6.6, 1.3 Hz, 1H), 7.63–7.52 (m, 1H), 7.31–7.19 (m, 1H) (Figure S15); 13C-NMR (100 MHz, Chloroform-d) δ 149.8, 147.1, 141.0, 137.2, 130.9, 130.1, 129.4, 127.9, 127.2, 126.7, 126.63, 126.58, 104.0 (Figure S16); HRMS (ESI+) calcd for C13H9IN [M + H]+: 305.9774, found 305.9763; M.p. 100–104 °C.
2-Methoxynaphthalene [61]: Following the reported procedure, the reaction was carried out with 2-naphthol (1.14 g, 10 mmol, 1.0 equiv); NaH (60% wt, 0.41 g, 17 mmol, 1.7 equiv); and MeI (1.76 g, 12 mmol, 1.2 equiv) in 10 mL of DMF. The desired product was obtained (1.28 g, 81% yield) as a white solid after purification by silica gel chromatography (DCM). 1H-NMR (400 MHz, Chloroform-d) δ 7.78 (dd, J = 11.5, 8.4 Hz, 3H), 7.47 (ddd, J = 8.1, 6.8, 1.3 Hz, 1H), 7.36 (ddd, J = 8.1, 6.8, 1.2 Hz, 1H), 7.22–7.13 (m, 2H), 3.94 (s, 3H).
2-Bromo-3-methoxynaphthalene [62]: Following the reported procedure, the reaction was carried out with 2-methoxynaphthalene (0.79 g, 5.0 mmol, 1.0 equiv); nBuLi solution (1.67 M in hexane, 3 mL, 5.3 mmol, 1.1 equiv); and 1,2-dibromoethane (1.30 g, 6.9 mmol, 1.3 equiv) in 10 mL of THF. The desired product was obtained (0.87 g, 73% yield) as a white solid after recrystallization from hot hexane for 3 times. 1H-NMR (400 MHz, Chloroform-d) δ 8.06 (s, 1H), 7.71 (dd, J = 13.0, 8.2 Hz, 2H), 7.51–7.42 (m, 1H), 7.40–7.32 (m, 1H), 7.16 (s, 1H), 4.01 (s, 3H).
3-Methoxynaphthalen-2-ol (1b) [63]: Following the reported procedure, the reaction was carried out with naphthalene-2, 3-diol (1.60 g, 10 mmol, 1.0 equiv); K2CO3 (1.81 g, 13 mmol, 1.3 equiv); and MeI (1.73 g, 12 mmol, 1.2 equiv) in 10 mL of acetone. The desired product was obtained (0.57 g, 33% yield) as a white solid after purification by silica gel chromatography (PE/EA = 50/1 to PE/EA = 20/1 to PE/EA = 10/1). 1H-NMR (400 MHz, Chloroform-d) δ 8.06 (s, 1H), 7.71 (dd, J = 13.0, 8.2 Hz, 2H), 7.50–7.42 (m, 1H), 7.40–7.33 (m, 1H), 7.16 (s, 1H), 4.01 (s, 3H).
3-(Benzyloxy)naphthalen-2-ol (1c) [64]: Following the reported procedure, the reaction was carried out with naphthalene-2, 3-diol (1.61 g, 10 mmol, 1.0 equiv); K2CO3 (1.82 g, 13 mmol, 1.3 equiv); and BnBr (2.58 g, 15 mmol, 1.5 equiv) in 20 mL of DMF. The desired product was obtained (0.88 g, 35% yield) as a yellow solid after purification by silica gel chromatography (PE/EA = 50/1 to 20/1 to 10/1). 1H-NMR (400 MHz, Chloroform-d) δ 7.71–7.64 (m, 2H), 7.52–7.31 (m, 7H), 7.29 (s, 1H), 7.22 (s, 1H), 5.97 (s, 1H), 5.24 (s, 2H).
General procedure (Scheme 5): To a Schlenk flask were added 2-bromo-3-methoxynaphthalene (1.0 equiv), aryl boronic acid (2.2 equiv), K2CO3 (3.0 equiv), Pd(PPh3)4 (2.5 mol%), and degassed EtOH/toluene/water (1/1/1) under Ar atmosphere. The mixture was heated at 90 °C until the completion of the reaction. Then the mixture was cooled to room temperature, and DCM was added. The mixture was washed with NaOH solution (20% wt), and the aqueous phase was extracted with DCM (2 × 20 mL). The combined organic phase was washed with brine (20 mL) and dried over anhydrous MgSO4. After removing the solvent, the residue was dissolved in anhydrous DCM. The solution was cooled to −78 °C, and BBr3 (1 M in DCM, 5.0 equiv) was added slowly by syringe. Then the mixture was warmed up to room temperature and stirred until the complete consumption of the starting material. The mixture was poured into the ice water (50 mL) and extracted with DCM (3 × 50 mL). The combined organic phase was washed with brine (100 mL) and dried over anhydrous Na2SO4. After removing the solvent, the residue was purified by silica gel chromatography to give the desired product.
3-(o-Tolyl)naphthalen-2-ol (1d) [65]: Following the general procedure, the reaction was carried out with 2-bromo-3-methoxynaphthalene (236.2 mg, 1.0 mmol, 1.0 equiv); o-tolylboronic acid (304.8 mg, 2.2 mmol, 2.2 equiv); K2CO3 (417.7 mg, 3.0 mmol, 3.0 equiv); and Pd(PPh3)4 (29.9 mg, 2.5 mol%) in 6 mL of degassed solvents. Then BBr3 (1 M in DCM, 5 mL, 5 mmol, 5.0 equiv) was used to remove the methyl group. The desired product was obtained (185.1 mg, 79% yield overall) as a brown sticky liquid after purification by silica gel chromatography (PE/DCM = 10/1). 1H-NMR (400 MHz, Chloroform-d) δ 7.80–7.72 (m, 2H), 7.63 (s, 1H), 7.45 (ddd, J = 8.3, 6.9, 1.2 Hz, 1H), 7.44–7.28 (m, 6H), 4.92 (s, 1H), 2.20 (s, 3H).
3-Phenylnaphthalen-2-ol (1e) [66]: Following the general procedure, the reaction was carried out with 2-bromo-3-methoxynaphthalene (0.71 g, 3.0 mmol, 1.0 equiv); phenylboronic acid (0.55 g, 4.5 mmol, 1.5 equiv); K2CO3 (1.90 g, 13.8 mmol, 4.5 equiv); and Pd(PPh3)4 (0.09 g, 2.5 mol%) in 30 mL of degassed solvent. Then BBr3 (1 M in DCM, 15 mL, 15 mmol, 5.0 equiv) was used to remove the methyl group. The desired product was obtained (0.63 g, 95% yield overall) as a pale brown solid after purification by silica gel chromatography (DCM). 1H-NMR (400 MHz, Chloroform-d) δ 7.81–7.76 (m, 1H), 7.74 (d, J = 7.2 Hz, 2H), 7.60–7.51 (m, 4H), 7.49–7.41 (m, 2H), 7.39–7.32 (m, 2H), 5.30 (s, 1H).
3-Bromonaphthalen-2-ol (1f) [67]: Following the general procedure, the reaction was carried out with 2-bromo-3-methoxynaphthalene (240.6 mg, 1.0 mmol, 1.0 equiv) and BBr3 (1 M in DCM, 5 mL, 5.0 mmol, 5.0 equiv). The desired product was obtained (226.0 mg, quantitative yield) as a white solid after purification by silica gel chromatography (DCM). 1H-NMR (400 MHz, Chloroform-d) δ 8.03 (s, 1H), 7.69 (ddt, J = 7.4, 2.2, 1.2 Hz, 2H), 7.45 (ddd, J = 8.2, 6.8, 1.3 Hz, 1H), 7.39 (s, 1H), 7.35 (ddd, J = 8.2, 6.8, 1.2 Hz, 1H), 5.64 (s, 1H).
6-Phenylnaphthalen-2-ol (1k) [5]: Following the general procedure, the reaction was carried out with 6-bromonaphthalen-2-ol (1.12 g, 5.0 mmol, 1.0 equiv); phenylboronic acid (0.73 g, 6.0 mmol, 1.2 equiv); K2CO3 (3.00 g, 21.8 mmol, 4.4 equiv); and Pd(PPh3)4 (0.15 g, 2.5 mol%) in 30 mL of degassed solvent. The desired product was obtained (0.77 g, 70% yield) as a white solid after purification by silica gel chromatography (DCM). 1H-NMR (400 MHz, Chloroform-d) δ 7.98 (d, J = 1.7 Hz, 1H), 7.82 (d, J = 8.8 Hz, 1H), 7.76 (d, J = 8.5 Hz, 1H), 7.71 (ddd, J = 8.2, 2.8, 1.5 Hz, 3H), 7.48 (dd, J = 8.5, 6.9 Hz, 2H), 7.41–7.33 (m, 1H), 7.18 (d, J = 2.5 Hz, 1H), 7.14 (dd, J = 8.8, 2.5 Hz, 1H).
7-(Benzyloxy)naphthalen-2-ol (1l) [68]: Following the reported procedure, the reaction was carried out with naphthalene-2,7-diol (1.60 g, 10 mmol, 1.0 equiv); K2CO3 (1.80 g, 13 mmol, 1.3 equiv), and BnBr (2.65 g, 15 mmol, 1.5 equiv) in 20 mL of DMF. The desired product was obtained (0.75 g, 30% yield) as a white solid after purification by silica gel chromatography (PE/EA = 5/1). 1H-NMR (400 MHz, Chloroform-d) δ 7.71–7.63 (m, 2H), 7.52–7.45 (m, 2H), 7.45–7.38 (m, 2H), 7.38–7.31 (m, 1H), 7.15–7.06 (m, 2H), 7.04 (d, J = 2.5 Hz, 1H), 6.94 (dd, J = 8.8, 2.4 Hz, 1H), 5.16 (s, 2H).
7-Butoxynaphthalen-2-ol (1m) [69]: Following the reported procedure, the reaction was carried out with naphthalene-2,7-diol (1.60 g, 10 mmol, 1.0 equiv); K2CO3 (1.80 g, 13 mmol, 1.3 equiv); and nBuI (2.26 g, 12 mmol, 1.2 equiv) in 20 mL of acetone. The desired product was obtained (0.32 g, 15% yield) as a white solid after purification by silica gel chromatography (PE/DCM = 1/1 to PE/EA = 5/1). 1H-NMR (400 MHz, Chloroform-d) δ 7.65 (dd, J = 8.7, 2.5 Hz, 2H), 7.17–6.83 (m, 4H), 5.04 (s, 1H), 4.06 (t, J = 6.5 Hz, 2H), 1.83 (dq, J = 8.7, 6.6 Hz, 2H), 1.62–1.43 (m, 2H), 1.00 (t, J = 7.4 Hz, 3H);
7-((tert-Butyldimethylsilyl)oxy)naphthalen-2-ol (1n) [70]: Following the reported procedure, the reaction was carried out with naphthalene-2,7-diol (1.60 g, 10 mmol, 1.0 equiv); imidazole (0.68 g, 10 mmol, 1.0 equiv); and TBSCl (1.35 g, 9 mmol, 0.9 equiv) in 15 mL of DMF. The desired product was obtained (0.75 g, 33% yield) as a yellow solid after purification by silica gel chromatography (PE/EA = 5/1). 1H-NMR (400 MHz, Chloroform-d) δ 7.65 (t, J = 9.3 Hz, 1H), 7.03 (d, J = 2.2 Hz, 1H), 7.00 (d, J = 2.4 Hz, 1H), 6.93 (ddd, J = 11.0, 8.8, 2.4 Hz, 1H), 1.01 (s, 4H), 0.24 (s, 3H).
7-Methoxynaphthalen-2-ol (1o) [69]: Following the reported procedure, the reaction was carried out with naphthalene-2,7-diol (1.61 g, 10 mmol, 1.0 equiv); K2CO3 (1.80 g, 13 mmol, 1.3 equiv); and MeI (1.75 g, 12 mmol, 1.2 equiv) in 20 mL of acetone. The desired product was obtained (0.53 g, 30% yield) as a white solid after purification by silica gel chromatography (PE/DCM = 1/1 to PE/EA = 5/1). 1H-NMR (400 MHz, Chloroform-d) δ 7.66 (dd, J = 9.2, 3.6 Hz, 2H), 7.06 (d, J = 2.3 Hz, 1H), 7.01–6.97 (m, 2H), 6.94 (dd, J = 8.7, 2.4 Hz, 1H), 3.90 (s, 3H).
3.4. Iron-Catalyzed Asymmetric Oxidative Coupling Reaction of 2-Naphthols
(S)-[1,1′-Binaphthalene]-2,2′-diol (2a) [47]: Fe(ClO4)2 (12.7 mg, 10 mol%; NOTE: perchlorate salt is a potential explosive [71] and should be handled with extreme caution) and L1 (9.2 mg, 5 mol%) were dissolved in anhydrous PhCl (5 mL) in a 25 mL Schlenk tube, and the mixture was stirred at room temperature for 30 min. Then, 2-naphthol (72.3 mg, 0.5 mmol, 1.0 equiv) and MS 4Å (152.7 mg) were added. The reaction mixture was quickly evacuated and refilled with oxygen (1 atm), and this operation was repeated for three cycles. Then the mixture was stirred at 50 °C under oxygen, as monitored by TLC. The desired product was obtained (60.6 mg, 84% yield) as a pale yellow solid after purification by silica gel chromatography (PE to PE/EA = 10/1 to 5/1). 80:20 er (HPLC: Chiralpak AS-H, hexane/propan-2-ol = 90/10, 0.5 mL/min, λ = 230 nm, tR (min): major = 24.9, minor = 38.9). 1H-NMR (400 MHz, Chloroform-d) δ 7.99 (d, J = 8.9 Hz, 2H), 7.90 (d, J = 7.9 Hz, 2H), 7.38 (td, J = 7.7, 1.6 Hz, 4H), 7.31 (ddd, J = 8.2, 6.9, 1.4 Hz, 2H), 7.19–7.12 (m, 2H), 5.04 (s, 2H).
(S)-3,3′-Dimethoxy-[1,1′-binaphthalene]-2,2′-diol (2b) [30]: The reaction was conducted with Fe(ClO4)2 (12.7 mg, 10 mol%) and L1 (9.2 mg, 5 mol%); 3-methoxynaphthalen-2-ol (87.5 mg, 0.5 mmol, 1.0 equiv); and MS 4Å (152.5 mg). The desired product was obtained (76.2 mg, 88% yield) as a white solid after purification by silica gel chromatography (PE to PE/EA = 5/1). 81:19 er (HPLC: Chiralpak AS-H, hexane/propan-2-ol = 50/50, 1.0 mL/min, λ = 230 nm, tR (min): major = 14.3, minor = 24.4). 1H-NMR (400 MHz, Chloroform-d) δ 7.83–7.74 (m, 2H), 7.38–7.28 (m, 4H), 7.23–7.08 (m, 4H), 5.90 (s, 2H), 4.10 (s, 6H).
(S)-3,3′-Bis(benzyloxy)-[1,1′-binaphthalene]-2,2′-diol (2c) [72]: The reaction was conducted with Fe(ClO4)2 (12.7 mg, 10 mol%) and L1 (9.2 mg, 5 mol%); 3-(benzyloxy)naphthalen-2-ol (125.2 mg, 0.5 mmol, 1.0 equiv); and MS 4Å (150.0 mg). The desired product was obtained (84.9 mg, 68% yield) as a white solid after purification by silica gel chromatography (PE to PE/EA = 5/1). 80:20 er (HPLC: Chiralpak AS-H, hexane/propan-2-ol = 85/15, 1.0 mL/min, λ = 254 nm, tR (min): major = 35.1, minor = 42.1). 1H-NMR (400 MHz, Chloroform-d) δ 7.77 (d, J = 8.1 Hz, 2H), 7.57–7.48 (m, 4H), 7.47–7.36 (m, 8H), 7.32 (dt, J = 8.1, 4.0 Hz, 2H), 7.17 (d, J = 3.9 Hz, 4H), 6.01 (s, 2H), 5.33 (s, 4H).
(S)-3,3′-Di-o-tolyl-[1,1′-binaphthalene]-2,2′-diol (2d) [73]: The reaction was conducted with Fe(ClO4)2 (13.4 mg, 10 mol%) and L1 (9.4 mg, 5 mol%); 3-(o-tolyl)naphthalen-2-ol (116.0 mg, 0.5 mmol, 1.0 equiv); and MS 4Å (155.6 mg). The desired product was obtained (84.2 mg, 72% yield) as a brown solid after purification by silica gel chromatography (PE/DCM = 10/1 to 1/1 to PE/EA = 5/1). 77:23 er (HPLC: Chiralpak AD-H, hexane/propan-2-ol = 70/30, 0.8 mL/min, λ = 254 nm, tR (min): major = 19.8, minor = 6.4). 1H-NMR (400 MHz, Chloroform-d) δ 7.94–7.86 (m, 2H), 7.86 (s, 2H), 7.46–7.25 (m, 14H), 5.15 (s, 2H), 2.27 (s, 6H).
(S)-3,3′-Diphenyl-[1,1′-binaphthalene]-2,2′-diol (2e) [74]: The reaction was conducted with Fe(ClO4)2 (13.1 mg, 10 mol%) and L1 (9.0 mg, 5 mol%); 3-phenylnaphthalen-2-ol (111.6 mg, 0.5 mmol, 1.0 equiv); and MS 4Å (158.0 mg). The desired product was obtained (72.2 mg, 66% yield) as a pale yellow solid after purification by silica gel chromatography (PE/DCM = 1/1). 56:44 er (HPLC: Chiralpak IC, hexane/propan-2-ol = 90/10, 0.8 mL/min, λ = 230 nm, tR (min): major = 6.6, minor = 10.1). 1H-NMR (400 MHz, Chloroform-d) δ 8.03 (s, 2H), 7.97–7.89 (m, 2H), 7.78–7.70 (m, 4H), 7.53–7.47 (m, 4H), 7.44–7.36 (m, 4H), 7.36–7.30 (m, 2H), 7.23 (dd, J = 8.3, 1.1 Hz, 2H), 5.38 (s, 2H).
(S)-3,3′-Dibromo-[1,1′-binaphthalene]-2,2′-diol (2f) [75]: The reaction was conducted with Fe(ClO4)2 (12.7 mg, 10 mol%) and L1 (9.2 mg, 5 mol%); 3-bromonaphthalen-2-ol (111.5 mg, 0.5 mmol, 1.0 equiv); and MS 4Å (150.4 mg). The desired product was obtained (56.2 mg, 51% yield) as a pale yellow solid after purification by silica gel chromatography (PE to PE/EA = 10/1). 66:34 er (HPLC: Chiralpak IC, hexane/propan-2-ol = 97/3, 1.0 mL/min, λ = 230 nm, tR (min): major = 12.2, minor = 14.3). 1H-NMR (400 MHz, Chloroform-d) δ 8.25 (d, J = 0.7 Hz, 2H), 7.86–7.77 (m, 2H), 7.39 (ddd, J = 8.2, 6.8, 1.3 Hz, 2H), 7.31 (ddd, J = 8.3, 6.8, 1.4 Hz, 2H), 7.10 (dq, J = 7.7, 0.7 Hz, 2H), 5.55 (s, 2H).
(S)-6,6′-Dibromo-[1,1′-binaphthalene]-2,2′-diol (2j) [76]: The reaction was conducted with Fe(ClO4)2 (12.7 mg, 10 mol%) and L1 (9.2 mg, 5 mol%); 6-bromonaphthalen-2-ol (112.7 mg, 0.5 mmol, 1.0 equiv); and MS 4Å (156.0 mg). The desired product was obtained (90.6 mg, 82% yield) as a pale yellow solid after purification by silica gel chromatography (PE to PE/EA = 10/1 to 5/1). 79:21 er (HPLC: Chiralpak AS-H, hexane/propan-2-ol = 90/10, 0.5 mL/min, λ = 254 nm, tR (min): major = 27.8, minor = 38.9). 1H-NMR (400 MHz, Chloroform-d) δ 8.25 (d, J = 0.7 Hz, 2H), 7.85–7.78 (m, 2H), 7.39 (ddd, J = 8.2, 6.8, 1.3 Hz, 2H), 7.31 (ddd, J = 8.3, 6.8, 1.4 Hz, 2H), 7.10 (dq, J = 7.6, 0.7 Hz, 2H), 5.55 (s, 2H).
(S)-6,6′-Diphenyl-[1,1′-binaphthalene]-2,2′-diol (2k) [23]: The reaction was conducted with Fe(ClO4)2 (12.7 mg, 10 mol%) and L1 (9.2 mg, 5 mol%); 6-phenylnaphthalen-2-ol (109.5 mg, 0.5 mmol, 1.0 equiv); and MS 4Å (160.6 mg). The desired product was obtained (70.2 mg, 64% yield) as pale yellow solid after purification by silica gel chromatography (PE to PE/EA = 10/1 to 5/1). 74:26 er (HPLC: Chiralpak AS-H, hexane/propan-2-ol = 90/10, 0.8 mL/min, λ = 254 nm, tR (min): major = 13.1, minor = 10.2). 1H-NMR (400 MHz, Chloroform-d) δ 8.03 (s, 2H), 7.96–7.90 (m, 2H), 7.77–7.71 (m, 4H), 7.53–7.46 (m, 4H), 7.45–7.36 (m, 4H), 7.35–7.29 (m, 2H), 7.23 (dd, J = 8.3, 1.1 Hz, 2H), 5.38 (s, 2H).
(S)-7,7′-Bis(benzyloxy)-[1,1′-binaphthalene]-2,2′-diol (2l) [77]: The reaction was conducted with Fe(ClO4)2 (12.9 mg, 10 mol%) and L1 (9.0 mg, 5 mol%); 7-(benzyloxy)naphthalen-2-ol (125.4 mg, 0.5 mmol, 1.0 equiv); and MS 4Å (160.6 mg). The desired product was obtained (88.5 mg, 71% yield) as a pale yellow solid after purification by silica gel chromatography (PE to PE/EA = 5/1). 58:42 er (HPLC: Chiralcel OD-H, hexane/propan-2-ol = 85/15, 1.0 mL/min, λ = 230 nm, tR (min): major = 14.8, minor = 26.7). 1H-NMR (400 MHz, Chloroform-d) δ 7.89 (d, J = 8.8 Hz, 2H), 7.80 (d, J = 8.9 Hz, 2H), 7.22 (dt, J = 6.1, 2.2 Hz, 8H), 7.16 (dd, J = 6.6, 3.1 Hz, 4H), 7.11 (dd, J = 8.9, 2.5 Hz, 2H), 6.49 (d, J = 2.4 Hz, 2H), 4.99 (s, 2H), 4.80 (d, J = 11.7 Hz, 2H), 4.74 (d, J = 11.7 Hz, 2H).
(S)-7,7′-Dibutoxy-[1,1′-binaphthalene]-2,2′-diol (2m) [29]: The reaction was conducted with Fe(ClO4)2 (12.3 mg, 10 mol%) and L1 (9.0 mg, 5 mol%); 7-butoxynaphthalen-2-ol (108.5 mg, 0.5 mmol, 1.0 equiv); and MS 4Å (150.0 mg). The desired product was obtained (106.5 mg, 99% yield) as a white solid after purification by silica gel chromatography (PE/EA = 10/1 to 5/1). 60:40 er (HPLC: Chiralpak AD-H, hexane/propan-2-ol = 90/10, 1.0 mL/min, λ = 254 nm, tR (min): major = 8.3, minor = 19.3). 1H-NMR (400 MHz, Chloroform-d) δ 7.84 (d, J = 8.8 Hz, 2H), 7.76 (d, J = 8.9 Hz, 2H), 7.20 (d, J = 8.8 Hz, 2H), 7.03 (dd, J = 8.9, 2.4 Hz, 2H), 6.48 (d, J = 1.9 Hz, 2H), 5.08 (s, 2H), 3.71 (ddt, J = 27.6, 9.3, 6.5 Hz, 4H), 1.67–1.53 (m, 4H), 1.34 (tt, J = 16.4, 8.3 Hz, 4H), 0.86 (t, J = 7.4 Hz, 6H).
(S)-7,7′-Bis((tert-butyldimethylsilyl)oxy)-[1,1′-binaphthalene]-2,2′-diol (2n): The reaction was conducted with Fe(ClO4)2 (12.6 mg, 10 mol%) and L1 (9.8 mg, 5 mol%); 7-((tert-butyldimethylsilyl)oxy)naphthalen-2-ol (138.2 mg, 0.5 mmol, 1.0 equiv); and MS 4Å (153.7 mg). The desired product was obtained (103.8 mg, 76% yield) as a brown solid after purification by silica gel chromatography (PE to PE/EA = 5/1). 68:32 er (HPLC: Chiralpak IC, hexane/propan-2-ol = 97/3, 1.0 mL/min, λ = 254 nm, tR (min): major = 6.0, minor = 8.5). 1H-NMR (400 MHz, Chloroform-d) δ 7.86 (dd, J = 9.0, 0.7 Hz, 2H), 7.75 (d, J = 8.8 Hz, 2H), 7.21 (d, J = 8.9 Hz, 2H), 6.95 (dd, J = 8.8, 2.4 Hz, 2H), 6.46 (d, J = 2.4 Hz, 2H), 5.07 (s, 2H), 0.83 (s, 18H), −0.03 (s, 6H), −0.06 (s, 6H) (Figure S17); 13C-NMR (100 MHz, Chloroform-d) δ 155.3, 153.2, 134.9, 131.1, 129.9, 125.1, 119.9, 115.3, 111.7, 109.8, 25.8, 18.3, −4.5 (Figure S18); HRMS (ESI−), m/z calc′d for C32H41O4Si2 [M − H]−: 545.2549, found 545.2578; = 56.6 (c = 1.0, CHCl3); M.p. 118–122 °C.
(S)-7,7′-Dimethoxy-[1,1′-binaphthalene]-2,2′-diol (2o) [78]: The reaction was conducted with Fe(ClO4)2 (12.4 mg, 10 mol%) and L1 (9.4 mg, 5 mol%); 7-methoxynaphthalen-2-ol (87.3 mg, 0.5 mmol, 1.0 equiv); and MS 4Å (156.3 mg). The desired product was obtained (56.3 mg, 65% yield) as a white solid after purification by silica gel chromatography (PE to PE/EA = 5/1). 59:41 er (HPLC: Chiralcel OD-H, hexane/propan-2-ol = 85/15, 1.0 mL/min, λ = 230 nm, tR (min): major = 11.1, minor = 18.8). 1H-NMR (400 MHz, Chloroform-d) δ 7.88 (dd, J = 9.0, 0.7 Hz, 2H), 7.78 (d, J = 8.9 Hz, 2H), 7.22 (d, J = 8.8 Hz, 2H), 7.03 (dd, J = 8.9, 2.5 Hz, 2H), 6.48 (d, J = 2.5 Hz, 2H), 3.57 (s, 6H).
4. Conclusions
In summary, we have developed an iron/bisquinolyldiamine-catalyzed asymmetric oxidative coupling of 2-naphthols. This method employs in situ-formed iron complexes from Fe(ClO4)2 and readily available ligand L1 and uses 1 atm oxygen as the oxidant. The atom economy of this transformation, the easily available catalyst, and operationally simple procedure provide new applications of asymmetric iron catalysis. Further studies on synthesizing a library of nitrogen ligands and extending their applications are underway in our laboratory.
Supplementary Materials
The following are available online. NMR and HPLC spectra.
Author Contributions
L.-Y.W. performed the experiments; L.-Y.W., M.U., and W.-B.L. wrote the paper; and W.-B.L. conceived and designed the experiments. All authors have read and agreed to the published version of the manuscript.
Funding
We thank the National Natural Science Foundation of China (21602160 and 21772148) and Wuhan University for financial support.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Wang, Y.-B.; Tan, B. Construction of Axially Chiral Compounds via Asymmetric Organocatalysis. Acc. Chem. Res. 2018, 51, 534–547. [Google Scholar] [CrossRef] [PubMed]
- Wencel-Delord, J.; Panossian, A.; Leroux, F.R.; Colobert, F. Recent Advances and New Concepts for the Synthesis of Axially Stereoenriched Biaryls. Chem. Soc. Rev. 2015, 44, 3418–3430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moliterno, M.; Cari, R.; Puglisi, A.; Antenucci, A.; Sperandio, C.; Moretti, E.; Sabato, A.D.; Salvio, R.; Bella, M. Quinine-Catalyzed Asymmetric Synthesis of 2,2′-Binaphthol-type Biaryls under Mild Reaction Conditions. Angew. Chem. Int. Ed. 2016, 128, 6635–6639. [Google Scholar] [CrossRef]
- Wang, J.-Z.; Zhou, J.; Xu, C.; Sun, H.; Kürti, L.; Xu, Q.-L. Symmetry in Cascade Chirality-Transfer Processes: A Catalytic Atroposelective Direct Arylation Approach to BINOL Derivatives. J. Am. Chem. Soc. 2016, 138, 5202–5205. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.-H.; Cheng, D.-J.; Zhang, J.; Wang, Y.; Liu, X.-Y.; Tan, B. Atroposelective Synthesis of Axially Chiral Biaryldiols via Organocatalytic Arylation of 2-Naphthols. J. Am. Chem. Soc. 2015, 137, 15062–15065. [Google Scholar] [CrossRef]
- Wang, H. Recent Advances in Asymmetric Oxidative Coupling of 2-Naphthol and Its Derivatives. Chirality 2010, 22, 827–837. [Google Scholar] [CrossRef]
- Brunel, J.M. BINOL: A Versatile Chiral Reagent. Chem. Rev. 2005, 105, 857–898. [Google Scholar] [CrossRef]
- Chen, Y.; Yekta, S.; Yudin, A.K. Modified BINOL Ligands in Asymmetric Catalysis. Chem. Rev. 2003, 103, 3155–3212. [Google Scholar] [CrossRef]
- Pu, L. 1,1′-Binaphthyl Dimers, Oligomers, and Polymers: Molecular Recognition, Asymmetric Catalysis, and New Materials. Chem. Rev. 1998, 98, 2405–2494. [Google Scholar] [CrossRef]
- Tomino, I.; Tanimoto, Y.; Noyori, R. Virtually Complete Enantioface Differentiation in Carbonyl Group Reduction by a Complex Aluminum Hydride Reagent. J. Am. Chem. Soc. 1979, 101, 3129–3131. [Google Scholar]
- Berthod, M.; Mignani, G.; Woodward, G.; Lemaire, M. Modified BINAP: The How and the Why. Chem. Rev. 2005, 105, 1801–1836. [Google Scholar] [CrossRef] [PubMed]
- Kočovský, P.; Vyskočil, Š.; Smrčina, M. Non-Symmetrically Substituted 1,1′-Binaphthyls in Enantioselective Catalysis. Chem. Rev. 2003, 103, 3213–3245. [Google Scholar] [CrossRef] [PubMed]
- Parmar, D.; Sugiono, E.; Raja, S.; Rueping, M. Complete Field Guide to Asymmetric BINOL-Phosphate Derived Brønsted Acid and Metal Catalysis: History and Classification by Mode of Activation; Brønsted Acidity, Hydrogen Bonding, Ion Pairing, and Metal Phosphates. Chem. Rev. 2014, 114, 9047–9153. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.-F.; Xie, F.; Yang, B.; Yu, H.; Zhang, W.-B. Chiral Phosphoramidite Ligand and Its Application in Asymmetric Catalysis. Chin. J. Org. Chem. 2011, 31, 429–442. [Google Scholar]
- Hatano, M.; Ishihara, K. Chiral 1,1′-Binaphthyl-2,2′-Disulfonic Acid (BINSA) and Its Derivatives for Asymmetric Catalysis. Asian J. Org. Chem. 2014, 3, 352–365. [Google Scholar] [CrossRef]
- Komanduri, V.; Krische, M.J. Enantioselective Reductive Coupling of 1,3-Enynes to Heterocyclic Aromatic Aldehydes and Ketones via Rhodium-Catalyzed Asymmetric Hydrogenation: Mechanistic Insight into the Role of Brønsted Acid Additives. J. Am. Chem. Soc. 2006, 128, 16448–16449. [Google Scholar] [CrossRef]
- Costa, A.L.; Piazza, M.G.; Tagliavini, E.; Trombini, C.; Ronchi, A.U. Catalytic Asymmetric Synthesis of Homoallylic Alcohols. J. Am. Chem. Soc. 1993, 115, 7001–7002. [Google Scholar] [CrossRef]
- Sakane, S.; Fujiwara, J.; Maruoka, K.; Yamamoto, H. Chiral Leaving Group. Biogenetic-Type Asymmetric Synthesis of Limonene and Bisabolenes. J. Am. Chem. Soc. 1983, 105, 6154–6155. [Google Scholar] [CrossRef]
- Mukaiyama, T.; Inubushi, A.; Suda, S.; Hara, R.; Kobayashi, S. [1,1′-Bi-2-naphthalenediolato(2-)-O,O′]oxotitanium. An Efficient Chiral Catalyst for the Asymmetric Aldol Reaction of Silyl Enol Ethers with Aldehydes. Chem. Lett. 1990, 19, 1015–1018. [Google Scholar] [CrossRef]
- Qi, L.-W.; Mao, J.-H.; Zhang, J.; Tan, B. Organocatalytic Asymmetric Arylation of Indoles Enabled by Azo Groups. Nat. Chem. 2018, 10, 58–64. [Google Scholar] [CrossRef]
- Takizawa, S. Development of Dinuclear Vanadium Catalysts for Enantioselective Coupling of 2-Naphthols via a Dual Activation Mechanism. Chem. Pharm. Bull. 2009, 57, 1179–1188. [Google Scholar] [CrossRef] [Green Version]
- Chu, C.-Y.; Hwang, D.R.; Wang, S.-K.; Uang, B.J. Chiral Oxovanadium Complex Catalyzed Enantioselective Oxidative Coupling of 2-Naphthols. Chem. Commun. 2001, 37, 980–981. [Google Scholar] [CrossRef] [Green Version]
- Barhate, N.B.; Chen, C.-T. Catalytic Asymmetric Oxidative Couplings of 2-Naphthols by Tridentate N-Ketopinidene-Based Vanadyl Dicarboxylates. Org. Lett. 2002, 4, 2529–2532. [Google Scholar] [CrossRef]
- Guo, Q.-X.; Wu, Z.-J.; Luo, Z.-B.; Liu, Q.-Z.; Ye, J.-L.; Luo, S.-W.; Cun, L.-F.; Gong, L.-Z. Highly Enantioselective Oxidative Couplings of 2-Naphthols Catalyzed by Chiral Bimetallic Oxovanadium Complexes with either Oxygen or Air as Oxidant. J. Am. Chem. Soc. 2007, 129, 13927–13938. [Google Scholar] [CrossRef]
- Luo, Z.; Liu, Q.; Gong, L.-Z.; Cui, X.; Mi, A.; Jiang, Y. Novel Achiral Biphenol-Derived Diastereomeric Oxovanadium(IV) Complexes for Highly Enantioselective Oxidative Coupling of 2-Naphthols. Angew. Chem. Int. Ed. 2002, 41, 4532–4535. [Google Scholar] [CrossRef]
- Luo, Z.-B.; Liu, Q.-Z.; Gong, L.-Z. The Rational Design of Novel Chiral Oxovanadium(IV) Complexes for Highly Enantioselective Oxidative Coupling of 2-Naphthols. Chem. Commun. 2002, 38, 914–915. [Google Scholar] [CrossRef] [PubMed]
- Sako, M.; Takizawa, S.; Yoshida, Y.; Sasai, H. Enantioselective and Aerobic Oxidative Coupling of 2-Naphthol Derivatives Using Chiral Dinuclear Vanadium(V) Complex in Water. Tetrahedron Asymmetry 2015, 26, 613–616. [Google Scholar] [CrossRef] [Green Version]
- Takizawa, S.; Katayama, T.; Sasai, H. Dinuclear Chiral Vanadium Catalysts for Oxidative Coupling of 2-Naphtholsvia a Dual Activation Mechanism. Chem. Commun. 2008, 44, 4113–4122. [Google Scholar] [CrossRef]
- Takizawa, S.; Katayama, T.; Somei, H.; Asano, Y.; Yoshida, T.; Kameyama, C.; Rajesh, D.; Onitsuka, K.; Suzuki, T.; Mikami, M.; et al. Dual Activation in Oxidative Coupling of 2-Naphthols Catalyzed by Chiral Dinuclear Vanadium Complexes. Tetrahedron 2008, 64, 3361–3371. [Google Scholar] [CrossRef]
- Nakajima, M.; Miyoshi, I.; Kanayama, K.; Hashimoto, S.; Noji, M.; Koga, K. Enantioselective Synthesis of Binaphthol Derivatives by Oxidative Coupling of Naphthol Derivatives Catalyzed by Chiral Diamine•Copper Complexes. J. Org. Chem. 1999, 64, 2264–2271. [Google Scholar] [CrossRef]
- Gao, J.; Reibenspies, J.H.; Martell, A.E. Structurally Defined Catalysts for Enantioselective Oxidative Coupling Reactions. Angew. Chem. Int. Ed. 2003, 42, 6008–6012. [Google Scholar] [CrossRef] [PubMed]
- Kozlowski, M.C.; Morgan, B.J.; Linton, E.C. Total Synthesis of Chiral Biaryl Natural Products by Asymmetric Biaryl Coupling. Chem. Soc. Rev. 2009, 38, 3193–3207. [Google Scholar] [CrossRef] [PubMed]
- Hewgley, J.B.; Stahl, S.S.; Kozlowski, M.C. Mechanistic Study of Asymmetric Oxidative Biaryl Coupling: Evidence for Self-Processing of the Copper Catalyst to Achieve Control of Oxidase vs. Oxygenase Activity. J. Am. Chem. Soc. 2008, 130, 12232–12233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Podlesny, E.E.; Kozlowski, M.C. Enantioselective Total Synthesis of (S)-Bisoranjidiol, an Axially Chiral Bisanthraquinone. Org. Lett. 2012, 14, 1408–1411. [Google Scholar] [CrossRef]
- O’Brien, E.M.; Morgan, B.J.; Mulrooney, C.A.; Carroll, P.J.; Kozlowski, M.C. Perylenequinone Natural Products: Total Synthesis of Hypocrellin A. J. Org. Chem. 2010, 75, 57–68. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Hewgley, J.B.; Mulrooney, C.A.; Yang, J.; Kozlowski, M.C. Enantioselective Oxidative Biaryl Coupling Reactions Catalyzed by 1,5-Diazadecalin Metal Complexes: Efficient Formation of Chiral Functionalized BINOL Derivatives. J. Org. Chem. 2003, 68, 5500–5511. [Google Scholar] [CrossRef]
- Mulrooney, C.A.; Li, X.; DiVirgilio, E.S.; Kozlowski, M.C. General Approach for the Synthesis of Chiral Perylenequinones via Catalytic Enantioselective Oxidative Biaryl Coupling. J. Am. Chem. Soc. 2003, 125, 6856–6857. [Google Scholar] [CrossRef]
- Li, X.; Yang, J.; Kozlowski, M.C. Enantioselective Oxidative Biaryl Coupling Reactions Catalyzed by1,5-Diazadecalin Metal Complexes. Org. Lett. 2001, 3, 1137–1140. [Google Scholar] [CrossRef]
- Temma, T.; Hatano, B.; Habaue, S. Cu(I)-Catalyzed Asymmetric Oxidative Cross-Coupling of 2-Naphthol Derivatives. Tetrahedron 2006, 62, 8559–8563. [Google Scholar] [CrossRef]
- Temma, T.; Habaue, S. Highly Selective Oxidative Cross-Coupling of 2-Naphthol Derivatives with Chiral Copper(I)–Bisoxazoline Catalysts. Tetrahedron Lett. 2005, 46, 5655–5657. [Google Scholar] [CrossRef]
- Prause, F.; Arensmeyer, B.; Fröhlich, B.; Breuning, M. In-Depth Structure–Selectivity Investigations on Asymmetric, Copper-Catalyzed Oxidative Biaryl Coupling in the Presence of 5-cis-Substituted Prolinamines. Catal. Sci. Technol. 2015, 5, 2215–2226. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.-H.; Lee, D.-W.; Lee, Y.-S.; Ko, D.-H.; Ha, D.-C. Enantioselective Oxidative Coupling of Methyl 3-Hydroxy-2-naphthoate Using Mono-N-alkylated Octahydrobinaphthyl-2,2′-Diamine Ligand. Tetrahedron 2004, 60, 9037–9042. [Google Scholar] [CrossRef]
- Alamsetti, S.K.; Poonguzhali, E.; Ganapathy, D.; Sekar, G. Enantioselective Oxidative Coupling of 2-Naphthol Derivatives by Copper-(R)-1,1′-Binaphthyl-2,2′-Diamine-TEMPO Catalyst. Adv. Synth. Catal. 2013, 355, 2803–2808. [Google Scholar] [CrossRef]
- Zhang, Q.; Cui, X.; Chen, L.; Liu, H.; Wu, Y. Syntheses of Chiral Ferrocenophanes and Their Application to Asymmetric Catalysis. Eur. J. Org. Chem. 2014, 2014, 7823–7829. [Google Scholar] [CrossRef]
- Tian, J.-M.; Wang, A.-F.; Yang, J.-S.; Zhao, X.-J.; Tu, Y.-Q.; Zhang, S.-Y.; Chen, Z.-M. Copper-Complex-Catalyzed Asymmetric Aerobic Oxidative Cross-Coupling of 2-Naphthols: Enantioselective Synthesis of 3,3′-Substituted C1-Symmetric BINOLs. Angew. Chem. Int. Ed. 2019, 58, 11023–11027. [Google Scholar] [CrossRef] [PubMed]
- Egami, H.; Katsuki, T. Iron-Catalyzed Asymmetric Aerobic Oxidation: Oxidative Coupling of 2-Naphthols. J. Am. Chem. Soc. 2009, 131, 6082–6083. [Google Scholar] [CrossRef] [PubMed]
- Egami, H.; Matsumoto, K.; Oguma, T.; Kunisu, T.; Katsuki, T. Enantioenriched Synthesis of C1-Symmetric BINOLs: Iron-Catalyzed Cross-Coupling of 2-Naphthols and Some Mechanistic Insight. J. Am. Chem. Soc. 2010, 132, 13633–13635. [Google Scholar] [CrossRef]
- Horibe, T.; Nakagawa, K.; Hazeyama, T.; Takeda, K.; Ishihara, K. An Enantioselective Oxidative Coupling Reaction of 2-Naphthol Derivatives Catalyzed by Chiral Diphosphine Oxide–iron(II) Complexes. Chem. Commun. 2019, 55, 13677–13680. [Google Scholar] [CrossRef]
- Narute, S.; Parnes, R.; Toste, F.D.; Pappo, D. Enantioselective Oxidative Homocoupling and Cross-Coupling of 2-Naphthols Catalyzed by Chiral Iron Phosphate Complexes. J. Am. Chem. Soc. 2016, 138, 16553–16560. [Google Scholar] [CrossRef] [Green Version]
- Shalit, H.; Dyadyuk, A.; Pappo, D. Selective Oxidative Phenol Coupling by Iron Catalysis. J. Org. Chem. 2019, 84, 1677–1686. [Google Scholar] [CrossRef]
- Liu, W.; Zhong, D.-Y.; Yu, C.-L.; Zhang, Y.; Wu, D.; Feng, Y.-L.; Cong, H.-J.; Lu, X.-Q.; Liu, W.-B. Iron-Catalyzed Intramolecular Amination of Aliphatic C–H Bonds of Sulfamate Esters with High Reactivity and Chemoselectivity. Org. Lett. 2019, 21, 2673–2678. [Google Scholar] [CrossRef] [PubMed]
- Zhong, D.-Y.; Wu, D.; Zhang, Y.; Lu, Z.-W.; Usman, M.; Liu, W.; Lu, X.-Q.; Liu, W.-B. Synthesis of Sultams and Cyclic N-Sulfonyl Ketimines via Iron-Catalyzed Intramolecular Aliphatic C-H Amidation. Org. Lett. 2019, 21, 5808–5812. [Google Scholar] [CrossRef] [PubMed]
- Zang, C.; Liu, Y.-G.; Xu, Z.-J.; Tse, C.-T.; Guan, X.-G.; Wei, J.-H.; Huang, J.-S.; Che, C.-M. Highly Enantioselective Iron-Catalyzed cis-Dihydroxylation of Alkenes with Hydrogen Peroxide Oxidant via an Fe(III)-OOH Reactive Intermediate. Angew. Chem. Int. Ed. 2016, 55, 10253–10257. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.-H.; Cao, B.; Tse, C.-W.; Chang, X.-Y.; Zhou, C.-Y.; Che, C.-M. Chiral cis-Iron(II) Complexes with Metal- and Ligand-Centered Chirality for Highly Regio- and Enantioselective Alkylation of N-heteroaromatics. Chem. Sci. 2020, 11, 684–693. [Google Scholar] [CrossRef] [Green Version]
- Ruiz-Castillo, P.; Buchwald, S.L. Applications of Palladium-Catalyzed C–N Cross-Coupling Reactions. Chem. Rev. 2016, 116, 12564–12649. [Google Scholar] [CrossRef]
- Baumgartner, L.M.; Dennis, J.M.; White, N.A.; Buchwald, S.L.; Jensen, K.F. Use of a Droplet Platform to Optimize Pd-Catalyzed C–N Coupling Reactions Promoted by Organic Bases. Org. Process Res. Dev. 2019, 23, 1594–1601. [Google Scholar] [CrossRef]
- Pangborn, A.B.; Giardello, M.A.; Grubbs, R.H.; Rosen, R.K.; Timmers, F.J. Safe and Convenient Procedure for Solvent Purification. Organometallics 1996, 15, 1518–1520. [Google Scholar] [CrossRef]
- Michon, C.; Ellern, A.; Angelici, R.J. Chiral Tetradentate Amine and Tridentate Aminocarbene Ligands: Synthesis, Reactivity and X-Ray Structural Characterizations. Inorganica Chimica Acta 2006, 39, 4549–4556. [Google Scholar] [CrossRef]
- Ramann, G.A.; Cowen, B.J. Quinoline Synthesis by Improved Skraup–Doebner–Von Miller Reactions Utilizing Acrolein Diethyl Acetal. Tetrahedron Lett. 2015, 56, 6436–6439. [Google Scholar] [CrossRef]
- Chou, C.-C.; Hu, F.-C.; Yeh, H.-H.; Wu, H.-P.; Chi, Y.; Clifford, J.N.; Palomares, E.; Liu, S.-H.; Chou, P.-T.; Lee, G.-H. Highly Efficient Dye-Sensitized Solar Cells Based on Panchromatic Ruthenium Sensitizers with Quinolinyl Bipyridine Anchors. Angew. Chem. Int. Ed. 2014, 53, 178–183. [Google Scholar] [CrossRef]
- Patra, T.; Agasti, S.; Akanksha; Maiti, D. Nickel-Catalyzed Decyanation of Inert Carbon–Cyano Bonds. Chem. Commun. 2013, 49, 69–71. [Google Scholar] [CrossRef] [PubMed]
- Niimi, K.; Mori, H.; Miyazaki, E.; Osaka, I.; Kakizoe, H.; Takimiya, K.; Adachi, C. [2,2ʹ] Bi[Naphtho [2,3-B] Furanyl]: A Versatile Organic Semiconductor with a Furan–Furan Junction. Chem. Commun. 2012, 48, 5892–5894. [Google Scholar] [CrossRef] [PubMed]
- Bolchi, C.; Catalano, P.; Fumagalli, L.; Gobbi, M.; Pallavicini, M.; Pedretti, A.; Villa, L.; Vistoli, G.; Valoti, E. Structure–Affinity Studies for a Novel Series of Homochiral Naphthol and Tetrahydronaphthol Analogues of A1 Antagonist WB-4101. Bioorganic Med. Chem. 2004, 12, 4937–4951. [Google Scholar] [CrossRef]
- Liu, Y.-C.; Trzoss, M.; Brimble, M.A. A Facile Synthesis of Aryl Spirodioxines Based on a 3H,3ʹH-2,2ʹ-Spirobi (Benzo [b][1,4] Dioxine) Skeleton. Synthesis 2007, 9, 1392–1402. [Google Scholar]
- Forkosh, H.; Vershinin, V.; Reiss, H.; Pappo, D. Stereoselective Synthesis of Optically Pure 2-Amino-2′-Hydroxy-1,1′-Binaphthyls. Org. Lett. 2018, 20, 2459–2463. [Google Scholar] [CrossRef] [PubMed]
- Xiao, B.; Fu, Y.; Xu, J.; Gong, G.-T.; Dai, J.-J.; Yi, J.; Liu, L. Pd(II)-Catalyzed C–H Activation/Aryl-Aryl Coupling of Phenol Esters. J. Am. Chem. Soc. 2010, 132, 468–469. [Google Scholar] [CrossRef]
- Ueta, Y.; Mikami, K.; Ito, S. Accessto Air-STable 1,3-Diphosphacyclobutane-2,4-Diyls by an Arylation Reaction with Arynes. Angew. Chem. Int. Ed. 2016, 55, 7525–7529. [Google Scholar] [CrossRef]
- Buzard, D.J.; Olsson, C.; Noson, K.; Lipshutz, B.H. A Modular Route to Nonracemic Cyclo-Nobins. Preparation of the Parent Ligand for Homo- and Heterogeneous Catalysis. Tetrahedron 2004, 60, 4443–4449. [Google Scholar]
- Schreiner, J.L.; Pirkle, W.H. Chiral High-Pressure Liquid Chromatographic Stationary Phases. Separation of the Enantiomers of Bi-β-naphthols and Analogues. J. Org. Chem. 1981, 46, 4988–4991. [Google Scholar]
- Kingsbury, W.D. Synthesis of Structural Analogs of Leukotriene B4 and Their Receptor Binding Activity. J. Med. Chem. 1993, 36, 3308–3320. [Google Scholar] [CrossRef]
- Wolsey, W.C. Perchlorate Salts, Their Uses and Alternatives. J. Chem. Educ. 1973, 50, A335–A337. [Google Scholar] [CrossRef]
- Theveau, L.; Bellini, R.; Dydio, P.; Szabo, Z.; Werf, A.; Sander, R.A.; Reek, J.N.K.; Moberg, C. Cofactor-Controlled Chirality of Tropoisomeric Ligand. Organometallics 2016, 35, 1956–1963. [Google Scholar] [CrossRef]
- Ahmed, I.; Clark, D.-A. Rapid Synthesis of 3,3′ Bis-Arylated BINOL Derivatives Using a C−H Borylation in Situ Suzuki−Miyaura Coupling Sequence. Org. Lett. 2014, 16, 4332–4335. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.-J.; Clarkson, G.C.; Docherty, G.; North, C.L.; Woodward, G.; Wills, M. Ruthenium (II) Complexes of Monodonor Ligands: Efficient Reagents for Asymmetric Ketone Hydrogenation. J. Org. Chem. 2005, 70, 8079–8087. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Sun, X.; Li, X.-W.; Yuan, Y.-Z.; Jiao, N. Efficient and Practical Oxidative Bromination and Iodination of Arenes and Heteroarenes with DMSO and Hydrogen Halide: A Mild Protocol for Late-Stage Functionalization. Org. Lett. 2015, 17, 2886–2889. [Google Scholar] [CrossRef]
- Meesala, Y.; Wu, H.-L.; Koteswararao, B.; Kuo, T.-S.; Lee, W.-Z. Aerobic Oxidative Coupling of 2-Naphthol Derivatives Catalyzed by a Hexanuclear Bis(μ-hydroxo) Copper(II) Catalyst. Organometallics 2014, 33, 4385–4393. [Google Scholar] [CrossRef]
- Simonsen, K.B.; Gothelf, K.V.; Jørgensen, K.A. A Simple Synthetic Approach to 3,3′-Diaryl BINOLs. J. Org. Chem. 1998, 63, 7536–7538. [Google Scholar] [CrossRef]
- Qu, B.; Haddad, N. Ligand-Accelerated Stereoretentive Suzuki−Miyaura Coupling of Unprotected 3,3′-Dibromo-BINOL. J. Org. Chem. 2016, 81, 745–750. [Google Scholar] [CrossRef]
Sample Availability: Not available. |
Figure 1.
Representative 1,1′-Bi-2-naphthol (BINOL)-derived ligands/catalysts.

Figure 2.
Transition metal-catalyzed coupling for the enantioselective synthesis of BINOLs. (a) The previous examples of homo- or cross-coupling of 2-naphthols. (b) Our proposed asymmetric oxidative homo-coupling of 2-naphthols using iron/bisquinolyldiamine catalyst.
Figure 2.
Transition metal-catalyzed coupling for the enantioselective synthesis of BINOLs. (a) The previous examples of homo- or cross-coupling of 2-naphthols. (b) Our proposed asymmetric oxidative homo-coupling of 2-naphthols using iron/bisquinolyldiamine catalyst.

Figure 3.
Ligands synthesis.

Scheme 1.
Substrates Scope a. a Reactions conducted with Fe(ClO4)2 (10 mol%), L1 (5 mol%), substrate 1 (0.5 mmol), and 4Å MS (150 mg) in PhCl (5 mL) under oxygen atmosphere (1 atm) at 50 °C. Percentage represented isolated yields. Er determined by HPLC.
Scheme 1.
Substrates Scope a. a Reactions conducted with Fe(ClO4)2 (10 mol%), L1 (5 mol%), substrate 1 (0.5 mmol), and 4Å MS (150 mg) in PhCl (5 mL) under oxygen atmosphere (1 atm) at 50 °C. Percentage represented isolated yields. Er determined by HPLC.

Scheme 2.
Synthesis of Bisquinolyldiamine Ligands.

Scheme 3.
Synthesis of Substituted Quinolines (Skraup Reaction).

Scheme 4.
Synthesis of 4-Iodoacridine.

Scheme 5.
Synthesis of Substrates 1d–1e.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
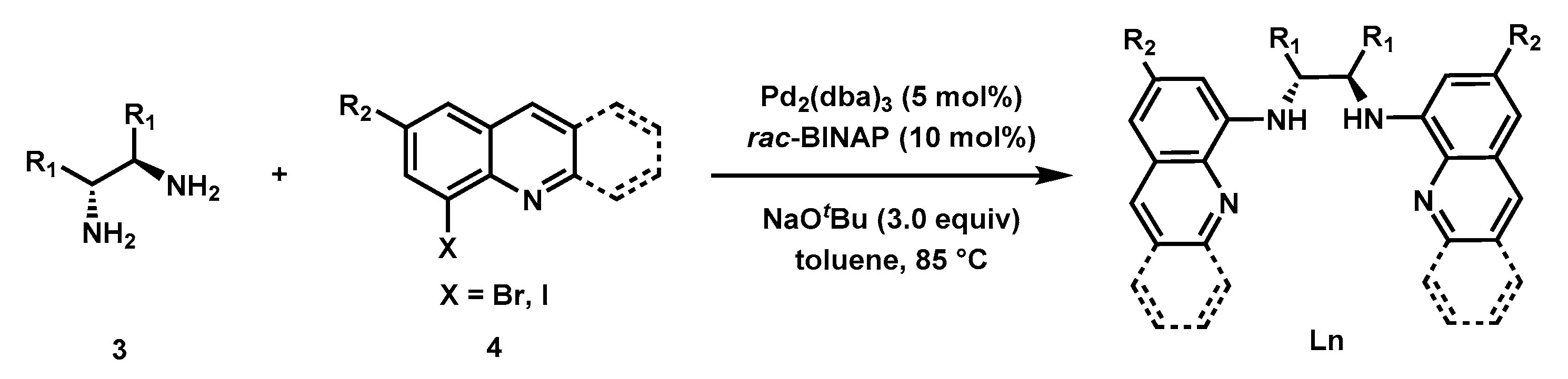{kind=link}
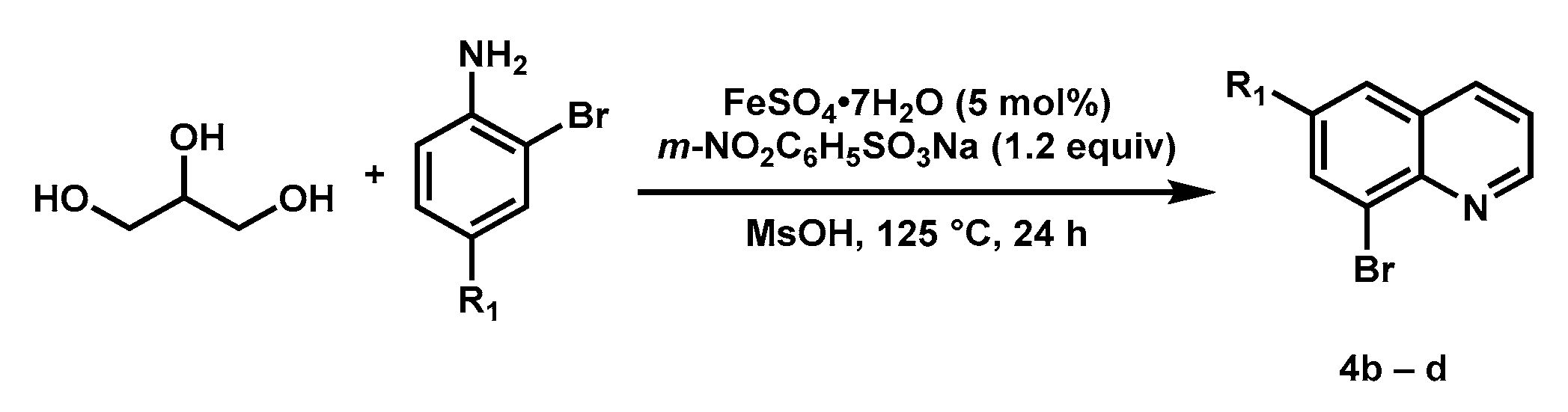{kind=link}
{kind=link}
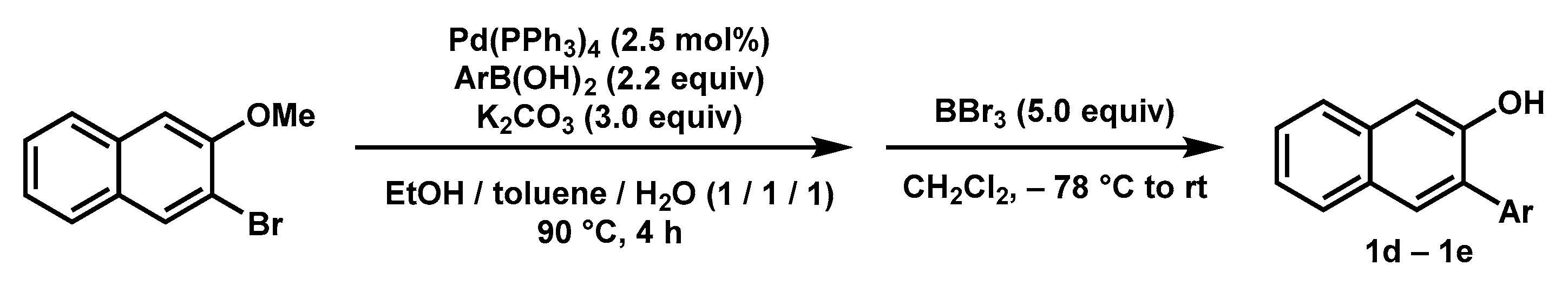{kind=link}
Table 1.
Conditions Screening a.

| Entry | Fe (x mol%) | Ln (y mol%) | Additives | Solvent | T °C | t/h | conv. (%) b | er (%) c |
|---|---|---|---|---|---|---|---|---|
| 1 | Fe(ClO4)2 (5.0) | L1(5.0) | - | MeOH | 30 | 28.5 | - | n.d. f |
| 2 | Fe(ClO4)2 (5.0) | L1 (5.0) | - | DCE | 30 | 28.5 | 57 d | 73:27 |
| 3 | Fe(ClO4)2 (5.0) | L1 (5.0) | - | CHCl3 | 30 | 28.5 | 68 d | 77:23 |
| 4 | Fe(ClO4)2 (5.0) | L1 (5.0) | - | toluene | 30 | 28.5 | 22 d | 80:20 |
| 5 | Fe(ClO4)2 (5.0) | L1 (5.0) | - | PhCl | 30 | 28.5 | 53 d | 79:21 |
| 6 | Fe(ClO4)2 (5.0) | L1 (5.0) | MS 4Å | PhCl | 30 | 5.0 | 12 d | 80:20 |
| 7 | Fe(ClO4)2 (5.0) | L1 (5.0) | MS 4Å | PhCl | 50 | 5.0 | 39 d | 78:22 |
| 8 | Fe(ClO4)2 (5.0) | L1 (5.0) | MS 4Å | PhCl | 70 | 5.0 | 38 d | 73:27 |
| 9 | Fe(ClO4)2 (5.0) | L1 (5.0) | MS 4Å | PhCl | 90 | 5.0 | 28 d | 70:30 |
| 10 | Fe(ClO4)3 (5.0) | L1 (5.0) | MS 4Å | PhCl | 50 | 5.0 | 76 | 76:24 |
| 11 | Fe(OAc)2 (5.0) | L1 (5.0) | MS 4Å | PhCl | 50 | 5.0 | 16 | n.d. f |
| 12 | Fe(OTf)2 (5.0) | L1 (5.0) | MS 4Å | PhCl | 50 | 5.0 | 19 | n.d. f |
| 13 | Fe(acac)2 (5.0) | L1 (5.0) | MS 4Å | PhCl | 50 | 5.0 | 23 | n.d. f |
| 14 | FeCl2 (5.0) | L1 (5.0) | MS 4Å | PhCl | 50 | 5.0 | 20 | n.d. f |
| 15 | Fe(ClO4)2 (7.5) | L1 (5.0) | MS 4Å | PhCl | 50 | 5.0 | 78 | 77:23 |
| 16 | Fe(ClO4)2 (10.0) | L1 (5.0) | MS 4Å | PhCl | 50 | 5.0 | 88(84 e) | 80:20 |
| 17 | Fe(ClO4)2 (12.5) | L1 (5.0) | MS 4Å | PhCl | 50 | 5.0 | 95 | 72:28 |
| 18 | Fe(ClO4)2 (5.0) | L1 (10.0) | MS 4Å | PhCl | 50 | 5.0 | 39 | 77:23 |
| 19 | Fe(ClO4)2 (10.0) | L1 (10.0) | MS 4Å | PhCl | 50 | 5.0 | 86 | 77:23 |
| 20 | Fe(ClO4)2 (10.0) | L2 (5.0) | MS 4Å | PhCl | 50 | 5.0 | 90 | 70:30 |
| 21 | Fe(ClO4)2 (10.0) | L3 (5.0) | MS 4Å | PhCl | 50 | 5.0 | 85 | 60:40 |
| 22 | Fe(ClO4)2 (10.0) | L4 (5.0) | MS 4Å | PhCl | 50 | 5.0 | 53 e | 78:22 |
| 23 | Fe(ClO4)2 (10.0) | L5 (5.0) | MS 4Å | PhCl | 50 | 5.0 | 67 e | 55:45 |
| 24 | Fe(ClO4)2 (10.0) | L6 (5.0) | MS 4Å | PhCl | 50 | 5.0 | - | n.d. f |
| 25 | Fe(ClO4)2 (10.0) | L7 (5.0) | MS 4Å | PhCl | 50 | 5.0 | - | n.d. f |
| 26 | Fe(ClO4)2 (10.0) | L8 (5.0) | MS 4Å | PhCl | 50 | 5.0 | - | n.d. f |
a All reactions carried out with 1a (0.5 mmol), MS 4Å (150 mg) in 5 mL solvent under O2 atmosphere (1 atm). b Conversions determined by GC using dodecane as an internal standard. c Determined by HPLC (Chiralpak AS-H). d Determined by 1H-NMR analysis. e Isolated yield. f n.d.: not detected.
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Wu, L.-Y.; Usman, M.; Liu, W.-B. Enantioselective Iron/Bisquinolyldiamine Ligand-Catalyzed Oxidative Coupling Reaction of 2-Naphthols. Molecules 2020, 25, 852. https://doi.org/10.3390/molecules25040852
AMA Style
Wu L-Y, Usman M, Liu W-B. Enantioselective Iron/Bisquinolyldiamine Ligand-Catalyzed Oxidative Coupling Reaction of 2-Naphthols. Molecules. 2020; 25(4):852. https://doi.org/10.3390/molecules25040852
Chicago/Turabian StyleWu, Lin-Yang, Muhammad Usman, and Wen-Bo Liu. 2020. "Enantioselective Iron/Bisquinolyldiamine Ligand-Catalyzed Oxidative Coupling Reaction of 2-Naphthols" Molecules 25, no. 4: 852. https://doi.org/10.3390/molecules25040852