
Synthesis, Anticancer Activity, and Preliminary Pharmacokinetic Evaluation of 4,4-Disubstituted Curcuminoid 2,2-bis(Hydroxymethyl)Propionate Derivatives

 ,
,
Abstract
:
1. Introduction
2. Results and Discussion
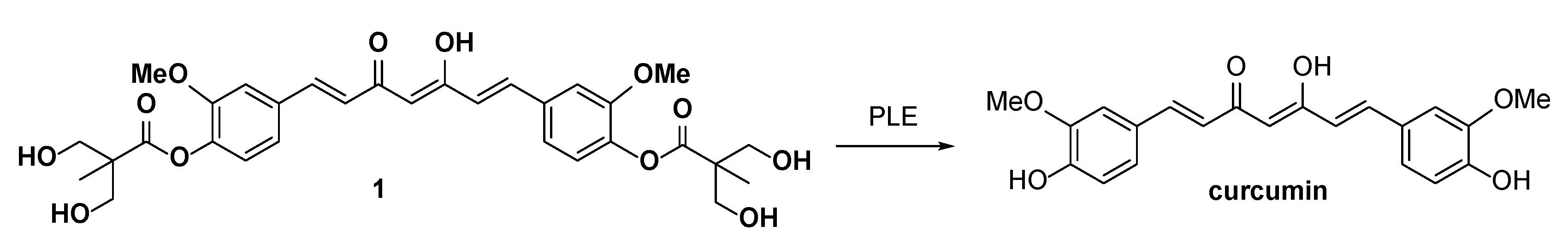
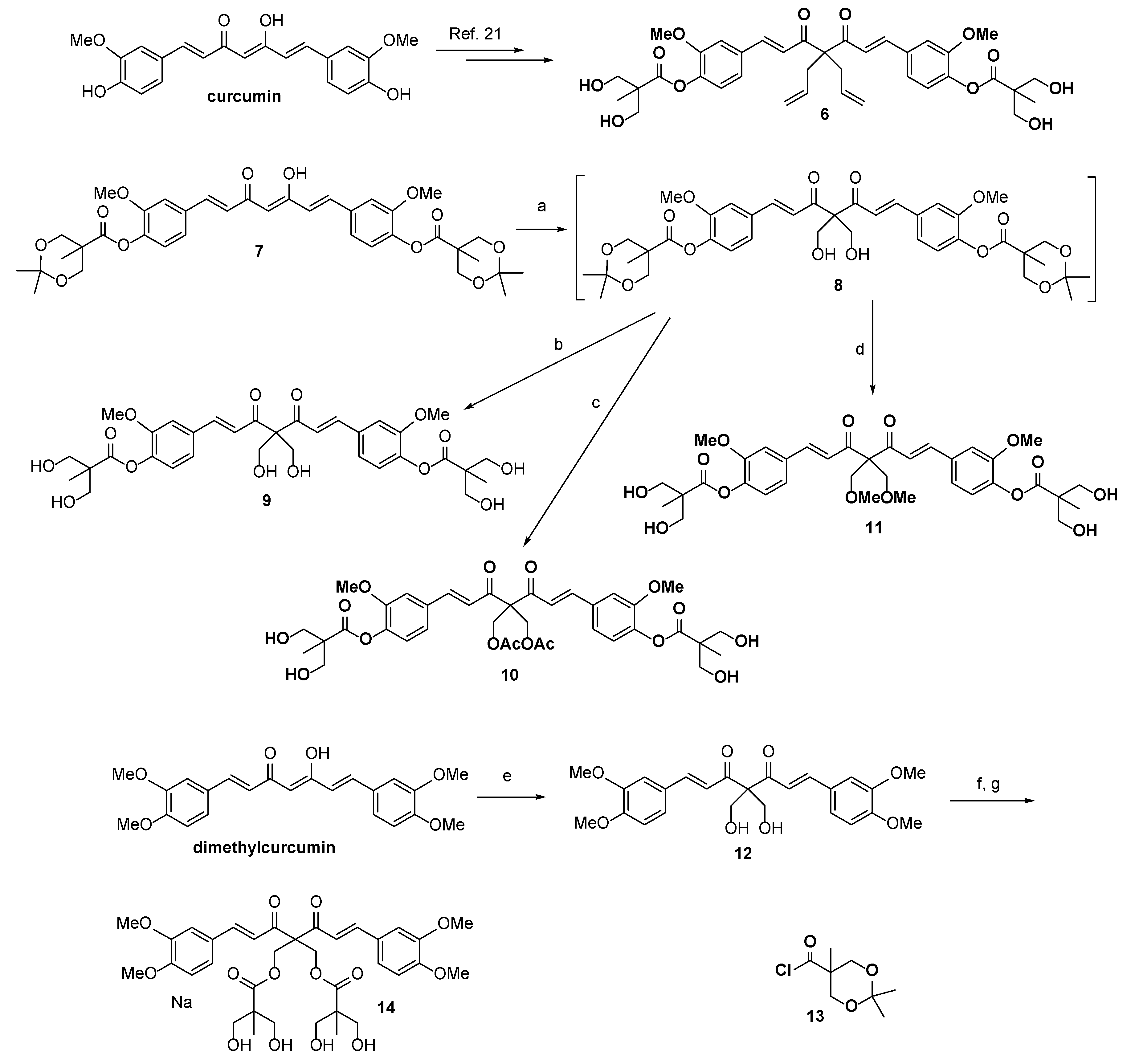
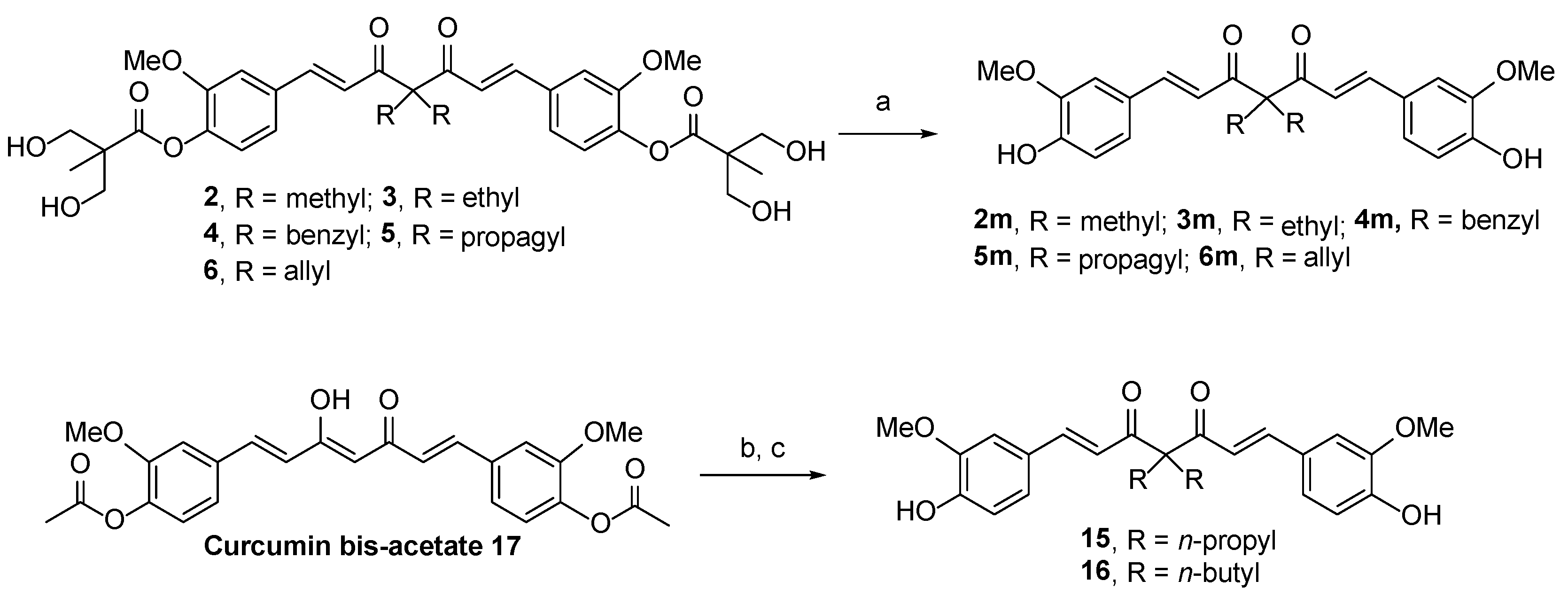
2.1. Chemistry
2.2. Structure–Activity Relationship
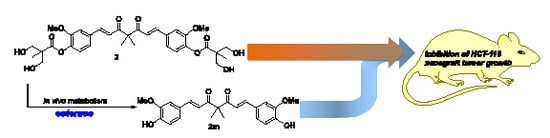
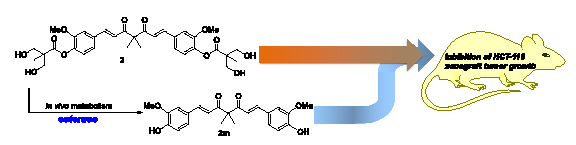
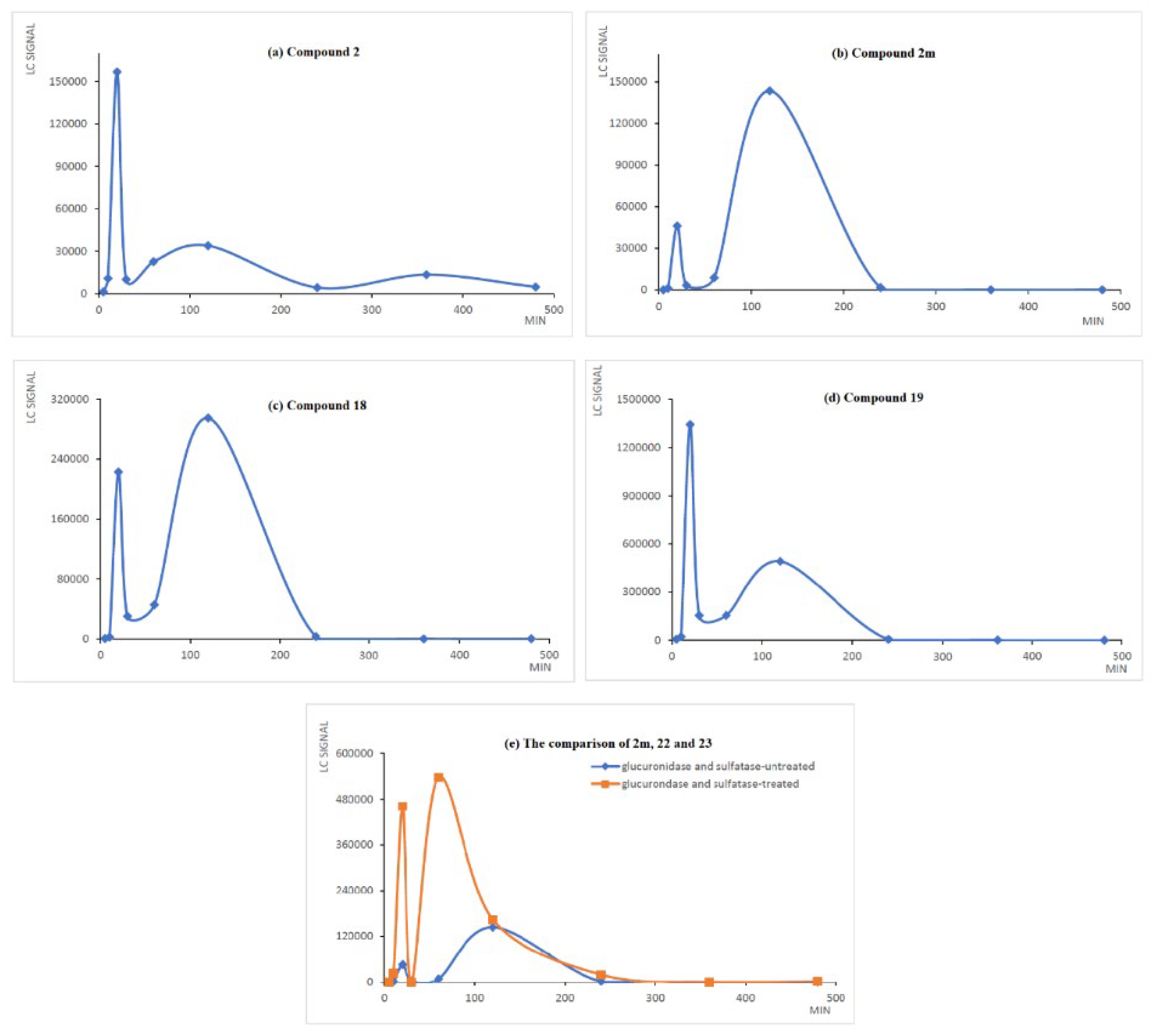
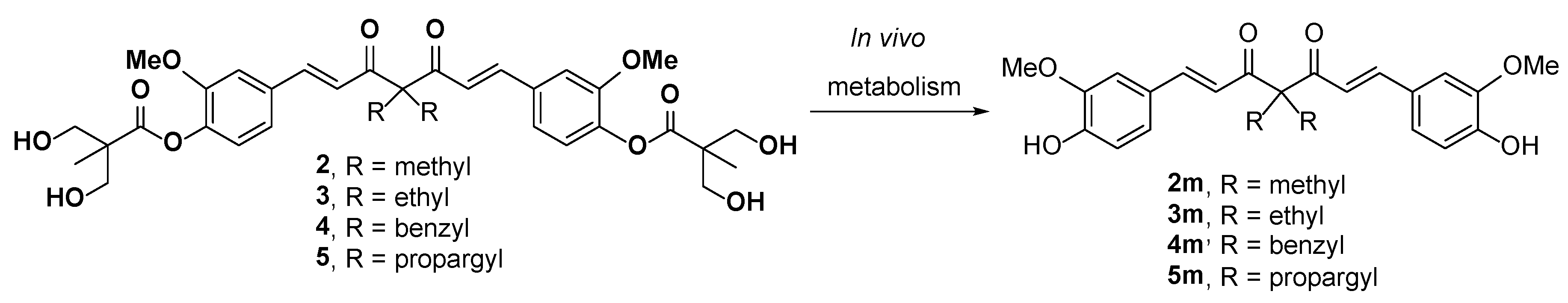
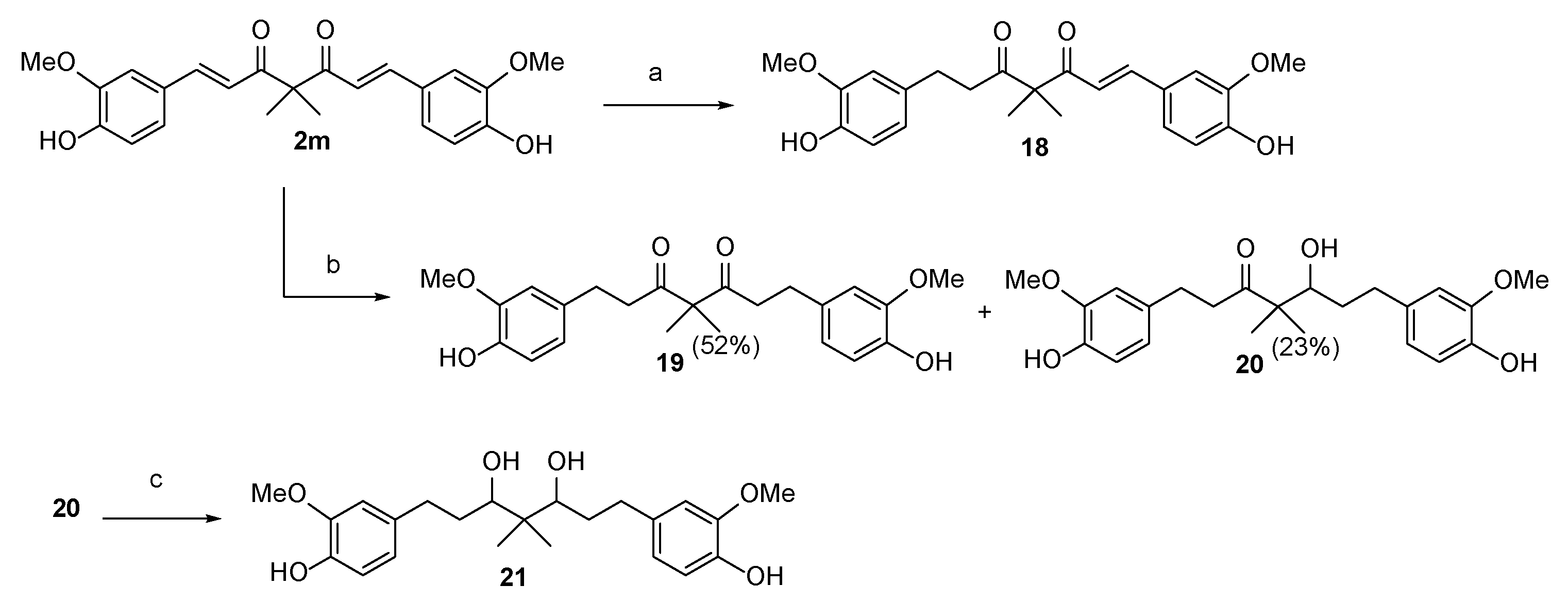
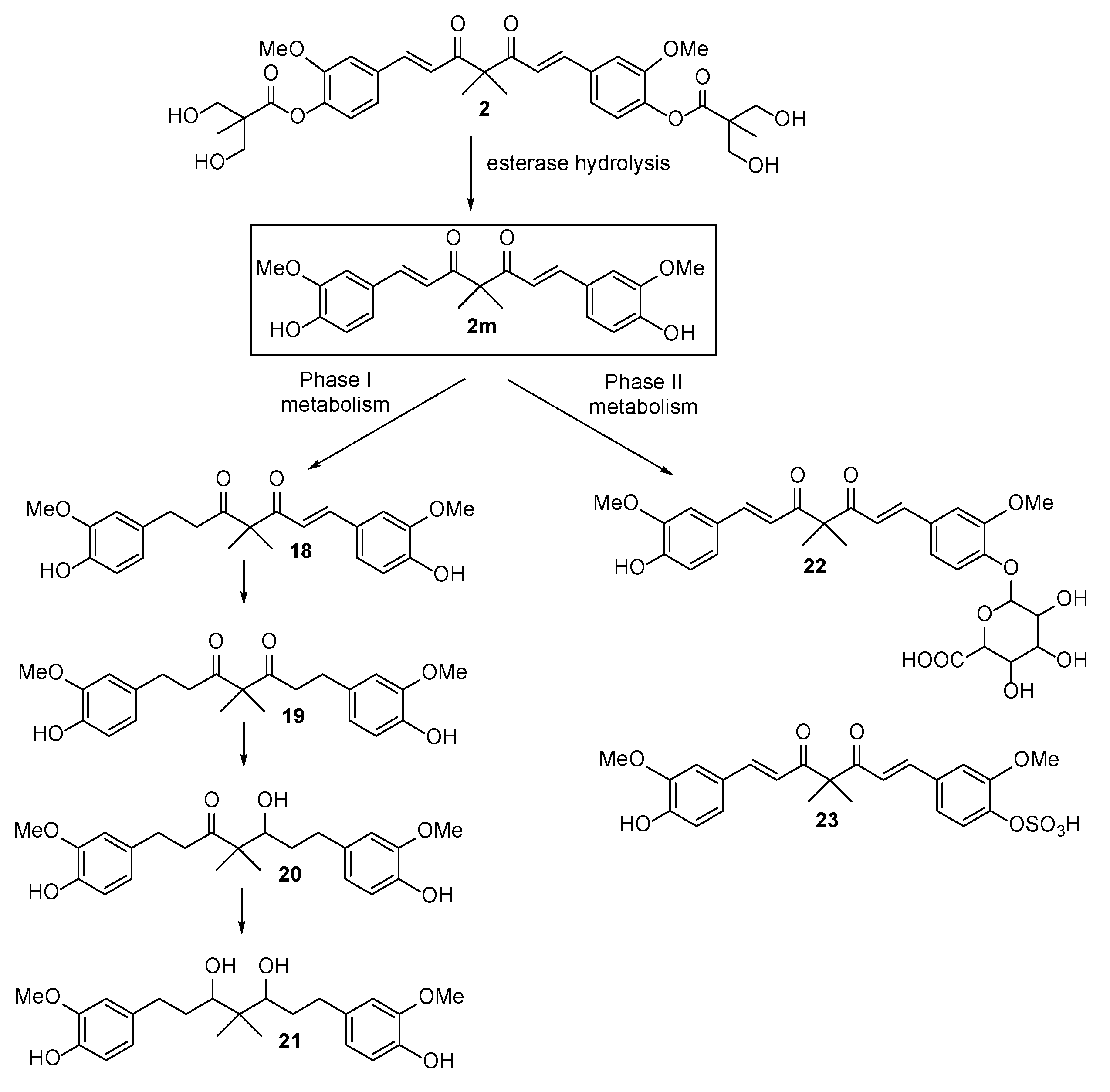
2.3. The Preliminary Pharmacokinetic Evaluation of 2
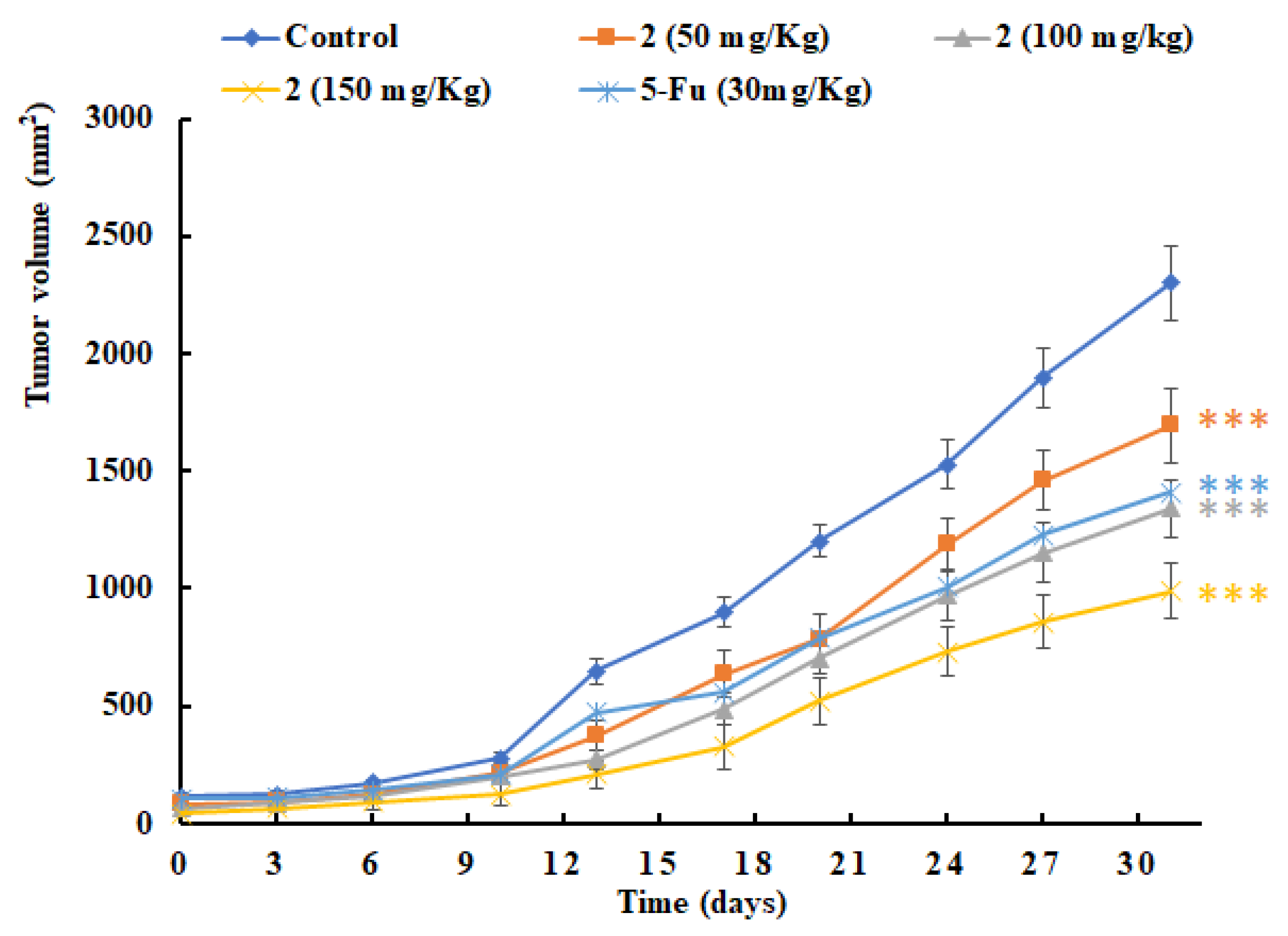
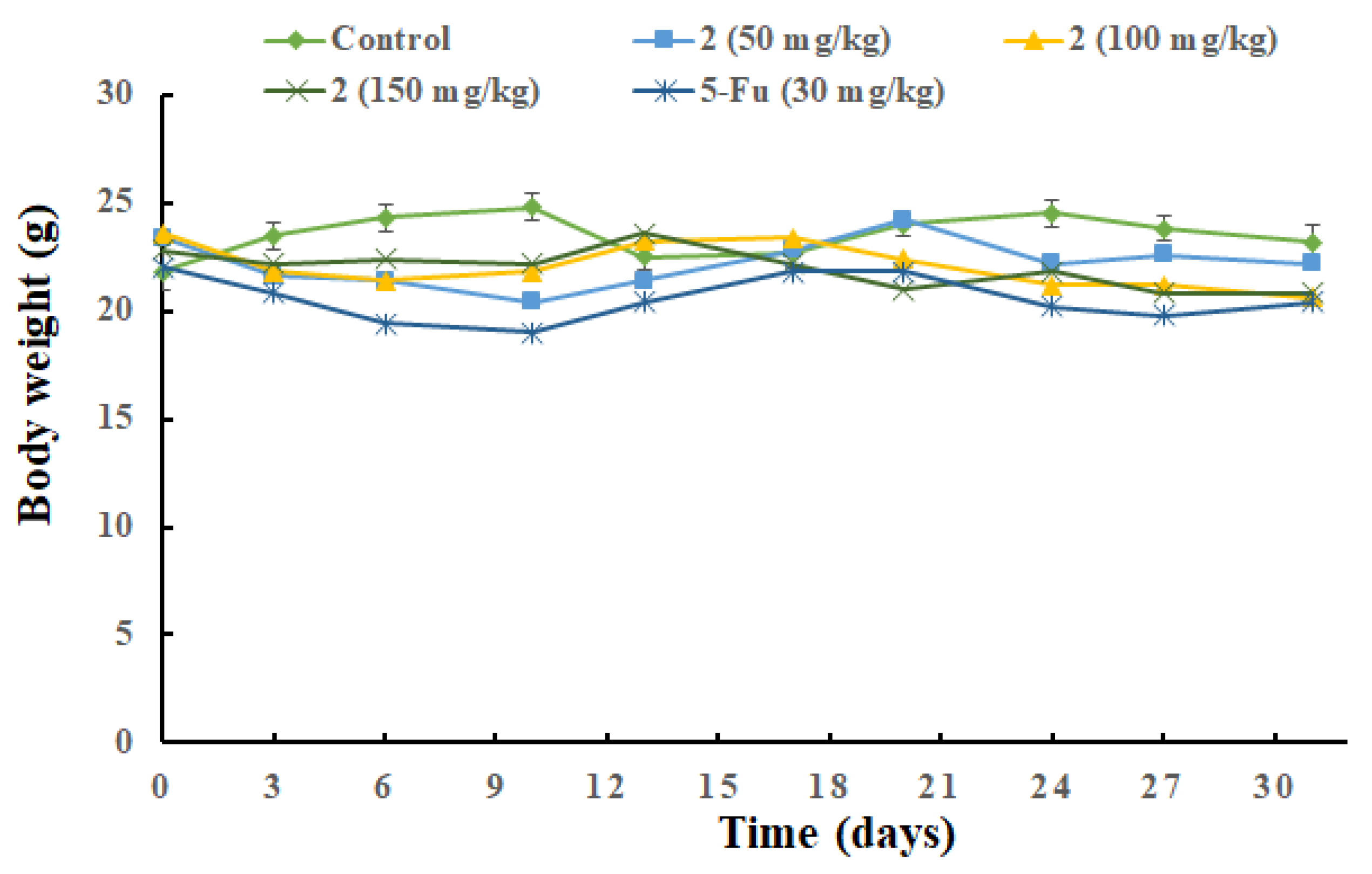
2.4. In Vivo Antitumor Efficacy of 2
3. Materials and Methods
3.1. General Information
3.1.1. [(1E,6E)-4,4-Diallyl-3,5-dioxohepta-1,6-diene-1,7-diyl]bis(2-methoxy-4,1-phenylene) bis[3-hydroxy-2-(hydroxymethyl)-2-methylpropanoate] (6)
3.1.2. [(1E,6E)-4,4-bis(Hydroxymethyl)-3,5-dioxohepta-1,6-diene-1,7-diyl]bis(2-methoxy-4,1-phenylene) bis(2,2,5-trimethyl-1,3-dioxane-5-carboxylate) (8)
3.1.3. [(1E,6E)-4,4-bis(Hydroxymethyl)-3,5-dioxohepta-1,6-diene-1,7-diyl]bis(2-methoxy-4,1-phenylene) bis[3-hydroxy-2-(hydroxymethyl)-2-methylpropanoate] (9)
3.1.4. [(1E,6E)-4,4-bis(Acetoxymethyl)-3,5-dioxohepta-1,6-diene-1,7-diyl]bis(2-methoxy-4,1-phenylene) bis[3-hydroxy-2-(hydroxymethyl)-2-methylpropanoate] (10)
3.1.5. [(1E,6E)-4,4-bis(Methoxymethyl)-3,5-dioxohepta-1,6-diene-1,7-diyl]bis(2-methoxy-4,1-phenylene) bis[3-hydroxy-2-(hydroxymethyl)-2-methylpropanoate] (11)
3.1.6. (1E,6E)-1,7-bis(3,4-Dimethoxyphenyl)-4,4-bis(hydroxymethyl)hepta-1,6-diene-3,5-dione (12)
3.1.7. 2,2-bis[(E)-3-(3,4-Dimethoxyphenyl)acryloyl]propane-1,3-diylbis[3-hydroxy-2-(hydroxymethyl)-2-methylpropanoate] (14)
3.1.8. General Procedure for the Synthesis of Compounds 2m–6m
(1E,6E)-1,7-bis(4-Hydroxy-3-methoxyphenyl)-4,4-dimethylhepta-1,6-diene-3,5-dione (2m)
(1E,6E)-4,4-Diethyl-1,7-bis(4-hydroxy-3-methoxyphenyl)hepta-1,6-diene-3,5-dione (3m)
(1E,6E)-4,4-Dibenzyl-1,7-bis(4-hydroxy-3-methoxyphenyl)hepta-1,6-diene-3,5-dione (4m)
(1E,6E)-1,7-bis(4-Hydroxy-3-methoxyphenyl)-4,4-di(prop-2-yn-1-yl)hepta-1,6-diene-3,5-dione (5m)
(1E,6E)-4,4-Diallyl-1,7-bis(4-hydroxy-3-methoxyphenyl)hepta-1,6-diene-3,5-dione (6m)
3.1.9. General Procedure for the Synthesis of Compounds 15 and 16
(1E,6E)-1,7-bis(4-Hydroxy-3-methoxyphenyl)-4,4-dipropylhepta-1,6-diene-3,5-dione (15)
(1E,6E)-4,4-Dibutyl-1,7-bis(4-hydroxy-3-methoxyphenyl)hepta-1,6-diene-3,5-dione (16)
3.1.10. (E)-1,7-bis(4-Hydroxy-3-methoxyphenyl)-4,4-dimethylhept-1-ene-3,5-dione (18)
3.1.11. 1,7-bis(4-Hydroxy-3-methoxyphenyl)-4,4-dimethylheptane-3,5-dione (19) and 5-hydroxy-1,7-bis(4-hydroxy-3-methoxyphenyl)-4,4-dimethylheptan-3-one (20)
3.1.12. 1,7-bis(4-Hydroxy-3-methoxyphenyl)-4,4-dimethylheptane-3,5-diol (21)
3.2. Biological Assays
3.2.1. In Vitro MTT [3-(4,5-Dimethylthiazaol-2-yl)-2,4-diphenyltetrazolium bromide] Assay
3.2.2. In Vivo Antitumor Activity Assay
3.2.3. The Preliminary Pharmacokinetic Evaluation of 2 in Rat
Drug administration and blood collection
3.2.4. Analysis of 2, 2m, 18, 19, 20, 21, 22 and 23 in Serum
3.2.5. LC-MS Analysis Method
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Strimpakos, A.S.; Sharma, R.A. Curcumin: Preventive and therapeutic properties in laboratory studies and clinical trials. Antioxid. Redox Signal. 2008, 10, 511–546. [Google Scholar] [CrossRef]
- Nabavi, S.F.; Thiagarajan, R.; Rastrelli, L.; Daglia, M.; Sobarzo-Sánchez, E.; Alinezhad, H.; Nabavi, S.M. Curcumin: A natural product for diabetes and its complications. Curr. Top Med. Chem. 2015, 15, 2445–2455. [Google Scholar] [CrossRef]
- Mathew, D.; Hsu, W.L. Antiviral potential of curcumin. J. Funct. Foods 2018, 40, 692–699. [Google Scholar] [CrossRef]
- Sahebkar, A.; Henrotin, Y. Analgesic efficacy and safety of curcuminoids in clinical practice: A systematic review and meta-analysis of randomized controlled trials. Pain Med. 2016, 17, 1192–1202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osawa, T. Nephroprotective and hepatoprotective effects of curcuminoids. Adv. Exp. Med. Biol. 2007, 595, 407–423. [Google Scholar] [PubMed]
- Miriyala, S.; Panchatcharam, M.; Rengarajulu, P. Cardioprotective effects of curcumin. Adv. Exp. Med. Biol. 2007, 595, 359–377. [Google Scholar] [PubMed]
- Shishodia, S. Molecular mechanisms of curcumin action: Gene expression. Biofactors 2013, 39, 37–55. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.C.; Prasad, S.; Kim, J.H.; Patchva, S.; Webb, L.J.; Priyadarsini, I.K.; Aggarwal, B.B. Multitargeting by curcumin as revealed by molecular interaction studies. Nat. Prod. Rep. 2011, 28, 1937–1955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bordoloi, D.; Roy, N.K.; Monisha, J.; Padmavathi, G.; Kunnumakkara, A.B. Multi-targeted agents in cancer cell chemosensitization: What we learn from curcumin thus far. Recent Pat. Anti-Canc. 2016, 11, 67–97. [Google Scholar] [CrossRef] [PubMed]
- Kunnumakkara, A.B.; Anand, P.; Aggarwal, B.B. Curcumin inhibits proliferation, invasion, angiogenesis and metastasis of different cancers through interaction with multiple cell signaling proteins. Cancer Lett. 2008, 269, 199–225. [Google Scholar] [CrossRef]
- Nelson, K.M.; Dahlin, J.L.; Bisson, J.; Graham, J.; Pauli, G.F.; Walters, M.A. The essential medicinal chemistry of curcumin. J. Med. Chem. 2017, 60, 1620–1637. [Google Scholar] [CrossRef] [PubMed]
- Schieffer, G.W. Pressurized liquid extraction of curcuminoids and curcuminoid degradation products from turmeric (curcuma longa) with subsequent HPLC assays. J. Liq. Chromatogr. Relat. Technol. 2002, 25, 3033–3044. [Google Scholar] [CrossRef]
- Schneider, C.; Gordon, O.N.; Edwards, R.L.; Luis, P.B. Degradation of curcumin: From mechanism to biological implications. J. Agric. Food Chem. 2015, 63, 7606–7614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Priyadarsini, K.I.; Maity, D.K.; Naik, G.H.; Kumar, M.S.; Unnikrishnan, M.K.; Satav, J.G.; Mohan, H. Role of phenolic O-H and methylene hydrogen on the free radical reactions and antioxidant activity of curcumin. Free Radic. Biol. Med. 2003, 35, 475–484. [Google Scholar] [CrossRef]
- Sharma, R.A.; Steward, W.P.; Gescher, A.J. Pharmacokinetics and pharmacodynamics of curcumin. Adv. Exp. Med. Biol. 2007, 595, 453–470. [Google Scholar] [PubMed] [Green Version]
- Ireson, C.; Orr, S.; Jones, D.J.; Verschoyle, R.; Lim, C.K.; Luo, J.L.; Howells, L.; Plummer, S.; Jukes, R.; Williams, M.; et al. Characterization of metabolites of the chemopreventive agent curcumin in human and rat hepatocytes and in the rat in vivo, and evaluation of their ability to inhibit phorbol ester-induced prostaglandin E2 production. Cancer Res. 2001, 61, 1058–1064. [Google Scholar] [PubMed]
- Rodrigues, F.C.; Anil Kumar, N.A.; Thakur, G. Developments in the anticancer activity of structurally modified curcumin: An up-to-date review. Eur. J Med. Chem. 2019, 177, 76–104. [Google Scholar] [CrossRef]
- Tomeh, M.A.; Hadianamrei, R.; Zhao, X. A Review of Curcumin and Its Derivatives as Anticancer Agents. Int. J. Mol. Sci. 2019, 20, 1033. [Google Scholar] [CrossRef] [Green Version]
- Di Martino, R.M.C.; De Simone, A.; Andrisano, V.; Bisignano, P.; Bisi, A.; Gobbi, S.; Rampa, A.; Fato, R.; Bergamini, C.; Perez, D.I.; et al. Versatility of the curcumin scaffold: Discovery of potent and balanced dual BACE-1 and GSK-3b inhibitors. J. Med. Chem. 2016, 59, 531–544. [Google Scholar] [CrossRef]
- Noureddin, S.A.; El-Shishtawy, R.M.; Al-Footy, K.O. Curcumin analogues and their hybrid molecules as multifunctional drugs. Eur. J. Med. Chem. 2019, 82, 11631–11671. [Google Scholar]
- Barzegar, A.; Moosavi-Movahedi, A.A. Intracellular ROS protection efficiency and free radical-scavenging activity of curcumin. PLoS ONE 2011, 6, e26012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Epstein, J.; Sanderson, I.R.; Macdonald, T.T. Curcumin as a therapeutic agent: The evidence of in vitro, animal and human studies. Br. J. Nutr. 2010, 103, 1545–1557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- For the clinical progress of curcumin, refer to https://clinicaltrials.gov/.
- Hsieh, M.T.; Chang, L.C.; Hung, H.Y.; Lin, H.Y.; Shih, M.H.; Tsai, C.H.; Kuo, S.C.; Lee, K.H. New bis(hydroxymethyl) alkanoate curcuminoid derivatives exhibit activity against triple-negative breast cancer in vitro and in vivo. Eur. J Med. Chem. 2017, 131, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Berry, L.M.; Wollenberg, L.; Zhao, Z. Esterase activities in the blood, liver and intestine of several preclinical species and humans. Drug Metab. Lett. 2009, 3, 70–77. [Google Scholar] [CrossRef]
- Shi, Q.; Shih, C.C.; Lee, K.H. Novel anti-prostate cancer curcumin analogues that enhance androgen receptor degradation activity. Anticancer Agents Med. Chem. 2009, 9, 904–912. [Google Scholar] [CrossRef]
- 2,2,5-trimethyl-1,3-dioxane-5-carbonyl chloride 13 was formed by treating the acid counterpart with thionyl chloride. For detailed procedure, see: R. Giri, X. Chen, J.Q. Yu, Palladium-catalyzed asymmetric iodination of unactivated C-H bonds under mild conditions. Angew Chem. Int. Ed. Engl. 2005, 29, 2112–2115.
- Basile, V.; Ferrari, E.; Lazzari, S.; Belluti, S.; Pignedoli, F.; Imbriano, C. Curcumin derivatives: Molecular basis of their anti-cancer activity. Biochem. Pharmacol. 2009, 78, 1305–1315. [Google Scholar] [CrossRef] [Green Version]
- Prasad, S.; Tyagi, A.K.; Aggarwal, B.B. Recent developments in delivery, bioavailability, absorption and metabolism of curcumin: The golden pigment from golden spice. Cancer Res. Treat. 2014, 46, 2–18. [Google Scholar] [CrossRef] [Green Version]
- Liederer, B.M.; Borchardt, R. Enzymes involved in the bioconversion of ester-based prodrugs. J. Pharm. Sci. 2006, 95, 1177–1195. [Google Scholar] [CrossRef]
- Kunihiro, A.G.; Luis, P.B.; Brickey, J.A.; Julia, A.; Jen, B.F.; Chow, H.H.S.; Schmeider, C.; Funk, J.L. Beta-glucuronidase catalyzes deconjugation and activation of curcumin-glucuronide in bone. J. Nat. Prod. 2019, 82, 500–509. [Google Scholar] [CrossRef]
- Ozawa, H.; Imaizumi, A.; Sumi, Y.; Hashimoto, T.; Kanai, M.; Makino, Y.; Tsuda, T.; Takahashi, N.; Kakeya, H. Curcumin β-D-glucuronide plays an important role to keep high levels of free-form curcumin in the blood. Biol. Pharm. Bull. 2017, 40, 1515–1524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ge, S.; Tu, Y.; Hu, M. Challenges and opportunities with predicting in vivo phase II metabolism via glucuronidation from in vitro data. Curr. Pharmacol. Rep. 2016, 2, 326–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, K.Y.; Lin, L.C.; Tseng, T.Y.; Wang, S.C.; Tsai, T.H. Oral bioavailability of curcumin in rat and the herbal analysis from Curcuma longa by LC−MS/MS. J. Chromatogr. B Anal. Technol.Biomed. Life Sci. 2007, 853, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, Y.W.; Huang, C.Y.; Yang, S.Y.; Peng, Y.H.; Yu, C.P.; Lee Chao, P.D.; Chi, H.Y. Oral intake of curcumin markedly activated CYP 3A4: In vivo and ex-vivo studies. Sci. Rep. 2014, 4, 6587–6594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Sample Availability: Samples of the compounds are not available from the authors. |










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Compound | R1 | R2 | IC50 a (M)/ 48 h | |
|---|---|---|---|---|
| MDA-MB-231 | HCT-116 | |||
| Curcumin | OH | H | 22.05 ± 2.97 | 26.33 ± 1.38 |
| 1 |  | H | 3.06 ± 0.18 | 6.25 ± 0.60 |
| 2 | | methyl | 2.25 ± 0.10 | 3.10 ± 0.29 |
| 3 | | ethyl | 3.02 ± 0.15 | 1.93 ± 0.01 |
| 4 | | benzyl | 2.45 ± 0.24 | 7.66 ± 0.61 |
| 5 | | propargyl | 5.87 ± 0.10 | 5.81 ± 0.23 |
| 6 | | allyl | 3.13 ± 0.54 | 0.92 ± 0.06 |
| 9 | | CH2OH | >100 | >100 |
| 10 | | CH2OAc | 6.55 ± 0.04 | 6.53 ± 0.01 |
| 11 | | CH2OMe | 84.92 ± 1.67 | 69.08 ± 3.11 |
| 12 | OCH3 | CH2OH | 53.50 ± 3.45 | 58.00 ± 0.60 |
| 14 | OCH3 |  | 57.43 ± 2.06 | 40.10 ± 1.61 |
| 2m | OH | methyl | 4.17 ± 0.15 | 2.17 ± 0.16 |
| 3m | OH | ethyl | 9.12 ± 0.40 | 8.18 ± 0.57 |
| 4m | OH | benzyl | 36.52 ± 1.66 | 9.40 ± 0.02 |
| 5m | OH | propargyl | 3.10 ± 0.15 | 1.38 ± 0.06 |
| 6m | OH | allyl | 6.88 ± 0.36 | 4.40 ± 0.53 |
| 15 | OH | propyl | 18.17 ± 0.53 | 10.73 ± 1.63 |
| 16 | OH | butyl | >100 | 67.00 ± 3.18 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, D.-Y.; Hou, Y.-C.; Yang, J.-S.; Lin, H.-Y.; Chang, T.-Y.; Lee, K.-H.; Kuo, S.-C.; Hsieh, M.-T. Synthesis, Anticancer Activity, and Preliminary Pharmacokinetic Evaluation of 4,4-Disubstituted Curcuminoid 2,2-bis(Hydroxymethyl)Propionate Derivatives. Molecules 2020, 25, 479. https://doi.org/10.3390/molecules25030479
Lee D-Y, Hou Y-C, Yang J-S, Lin H-Y, Chang T-Y, Lee K-H, Kuo S-C, Hsieh M-T. Synthesis, Anticancer Activity, and Preliminary Pharmacokinetic Evaluation of 4,4-Disubstituted Curcuminoid 2,2-bis(Hydroxymethyl)Propionate Derivatives. Molecules. 2020; 25(3):479. https://doi.org/10.3390/molecules25030479
Chicago/Turabian StyleLee, Der-Yen, Yu-Chi Hou, Jai-Sing Yang, Hui-Yi Lin, Tsu-Yuan Chang, Kuo-Hsiung Lee, Sheng-Chu Kuo, and Min-Tsang Hsieh. 2020. "Synthesis, Anticancer Activity, and Preliminary Pharmacokinetic Evaluation of 4,4-Disubstituted Curcuminoid 2,2-bis(Hydroxymethyl)Propionate Derivatives" Molecules 25, no. 3: 479. https://doi.org/10.3390/molecules25030479