3.2. Synthetic Procedures
3.2.1. Synthesis of TPP Carboxylic Acids
Triphenylphosphine (11.5 mmol) was added in a solution of bromoalkyl carboxylic acid (12 mmol) in dry acetonitrile (20 mL) and the mixture was stirred at reflux under argon for 48 h. After cooling, the solvent was evaporated and the product precipitated out upon addition of ethyl acetate (or diethyl ether). Filtration and washing with the same solvent furnished pure products [
38].
(5-Carboxypentyl)triphenylphosphonium bromide (2a). White powder (4.83 g, 92%). 1H NMR (200 MHz, CDCl3) δ: 8.46 (bs, 1H, COOH), 7.91–7.57 (m, 15H, ArH), 3.73–3.56 (m, 2H, CH2P), 2.41–2.33 (m, 2H, CH2COOH), 1.67–1.55 (m, 6H, CH2). 13C NMR (50 MHz, CDCl3) δ: 176.09, 135.21 (d, J = 2.8 Hz, PPh3 para), 133.66 (d, J = 10.0 Hz, PPh3 ortho), 130.65 (d, J = 12.5 Hz, PPh3 meta), 118.11 (d, J = 86.0 Hz, PPh3 ipso), 34.25, 29.58 (d, J = 16.2 Hz, CH2CH2CH2P), 24.06, 22.56 (d, J = 51.0 Hz, CH2P), 21.99 (d, J = 4.1 Hz, CH2CH2P). 31P NMR (81 MHz, CDCl3) δ: 25.23. ES-MS m/z for C24H26O2P [M]+: calcd. 377.2, found 377.2.
(10-Carboxydecyl)triphenylphosphonium bromide (2b). White powder (5.88 g, 97%). 1H NMR (200 MHz, CDCl3) δ: 8.82 (bs, 1H, COOH), 7.80–7.60 (m, 15H, ArH), 3.66–3.44 (m, 2H, CH2P), 2.29 (t, J = 7.0 Hz, 2H, CH2COOH), 1.69–1.38 (m, 6H, CH2), 1.31–1.00 (m, 10H, CH2). 13C NMR (50 MHz, CDCl3) δ: 177.66 (C=O), 135.10 (d, J = 2.8 Hz, PPh3 para), 133.52 (d, J = 9.9 Hz, PPh3 ortho), 130.53 (d, J = 12.5 Hz, PPh3 meta), 118.09 (d, J = 85.9 Hz, PPh3 ipso), 34.40, 30.28 (d, J = 15.9 Hz, CH2CH2CH2P), 28.99, 28.86, 28.84, 28.82, 28.73, 24.65, 22.56 (d, J = 50.7 Hz, CH2P), 22.44 (d, J = 4.5 Hz, CH2CH2P). 31P NMR (81 MHz, CDCl3) δ: 25.08. ES-MS m/z for C29H36O2P [M]+: calcd. 447.2, found 447.2.
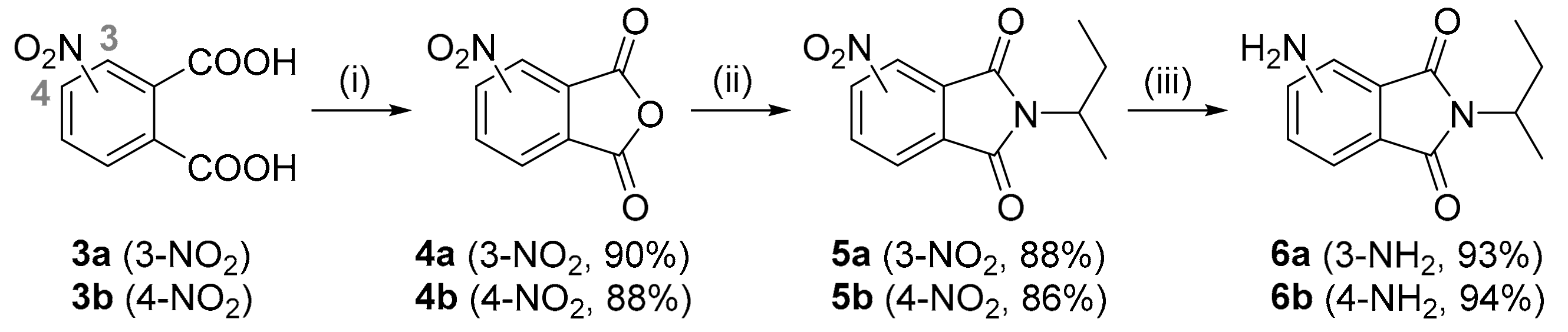
3.2.2. Synthesis of the Nitrophthalic Anhydrides 4a,b
A mixture of 3- or 4-nitrophthalic acid (10 g, 0.047 mol) and acetic anhydride (24 mL) was stirred at reflux for 1h. After cooling, volatiles were evaporated (repeated addition of toluene and evaporation facilitated the procedure). The anhydride precipitated out of the residue upon addition of diethyl ether as sub-white powder (3-nitrophthalic anhydride 4a: 7.90 g (90%), 4-nitrophthalic anhydride 4b: 8.00 g (88%)). The anhydrides were used in the next step without characterization.
3.2.3. Synthesis of the Nitrophthalimides 5a,b
A mixture of 3-nitrophthalic anhydride (7 g, 36.25 mmol), sec-butylamine (5.3 g, 72.50 mmol) and acetic acid (60 mL) was refluxed for 18 h. After cooling, volatiles were evaporated and dichloromethane (200 mL) was added. The solution was washed with aq. NaHCO3 (2 × 60 mL) and water (2 × 60 mL), dried (Na2SO4) and the solvent evaporated, affording the desired phthalimides.
2-(sec-Butyl)-4-nitroisoindoline-1,3-dione (5a). From 3-nitrophthalic anhydride 4a. Beige solid, 7.92 g (88%). 1H NMR (200 MHz, CDCl3) δ: 8.10–8.03 (m, 2H, H-5, H-7), 7.94–7.85 (m, 1H, H-6), 4.35–4.17 (m, 1H, NCH), 2.13–1.67 (m, 2H, CH2), 1.45 (d, J = 7.0 Hz, 3H, CHCH3), 0.86 (t, J = 7.4 Hz, 3H, CH3). 13C NMR (50 MHz, CDCl3) δ: 165.98, 163.08, 144.91, 135.34, 133.85, 128.86, 126.82, 123.33, 49.91, 26.58, 18.10, 11.20. ES-MS m/z for C12H12N2O4 [M]+: calcd. 248.0, found 248.0. ES-HRMS m/z for C12H12N2NaO4 [M + Na]+: calcd. 248.0797, found 248.0801.
2-(sec-Butyl)-5-nitroisoindoline-1,3-dione (5b). From 3-nitrophthalic anhydride 4b. Beige solid, 7.74 g (86%). 1H NMR (200 MHz, CDCl3) δ: 8.59 (s, 1H, H-4), 8.57 (d, J = 7.5 Hz, 1H, H-6), 8.00 (d, J = 7.5 Hz, 1H, H-7), 4.38–4.14 (m, 1H, NCH), 2.15–1.66 (m, 2H, CH2), 1.46 (d, J = 7.0 Hz, 3H, CHCH3), 0.85 (t, J = 7.4 Hz, 3H, CH3). 13C NMR (50 MHz, CDCl3) δ: 166.38, 166.07, 151.62, 136.35, 133.28, 129.14, 124.28, 118.43, 49.92, 26.69, 18.19, 11.20. ES-HRMS m/z for C12H12N2NaO4 [M + Na]+: calcd. 248.0797, found 248.0803.
3.2.4. Synthesis of Aminophthalimides 6a,b
A stirred solution of the nitrophthalimide (1.83 g, 7.37 mmol) in methanol (30 mL) was degassed (Ar) for 30 min. 10% Pd/C (200 mg) was added, then bubbled with H2 for a while and the mixture was stirred under an H2 atmosphere (20 bar) for 18 h. The mixture was filtered through celite, washed with methanol, and the filtrate was concentrated, leaving the corresponding aminophthalimide.
4-Amino-2-(sec-butyl)isoindoline-1,3-dione (6a). From 4-nitrophthalimide 5a. Yellow solid, 1.50 g (93%). 1H NMR (200 MHz, CDCl3) δ: 7.36 (dd, J = 8.3, 7.1 Hz, 1H, H-6), 7.08 (d, J = 7.1 Hz, 1H, H-5), 6.87 (d, J = 8.3 Hz, 1H, H-7), 5.41 (bs, 2H, NH2), 4.28–4.01 (m, 1H, NCH), 2.17–1.64 (m, 2H, CH2), 1.45 (d, J = 7.0 Hz, 3H, CHCH3), 0.87 (t, J = 7.4 Hz, 3H, CH3). 13C NMR (50 MHz, CDCl3) δ: 170.51, 168.84, 145.27, 134.82, 132.52, 120.88, 112.10, 110.92, 48.48, 26.81, 18.40, 11.23. ES-HRMS m/z for C12H14N2NaO2 [M + Na]+: calcd. 241.0947, found 241.0948.
5-Amino-2-(sec-butyl)isoindoline-1,3-dione (6b). From 5-nitrophthalimide 5b. Yellow solid, 1.51 g (94%). 1H NMR (200 MHz, DMSO-d6) δ: 7.45 (d, J = 8.1 Hz, 1H, H-7), 6.89 (s, 1H, H-4), 6.78 (d, J = 7.9 Hz, 1H, H-6), 6.45 (s, 2H, NH2), 4.09–3.95 (m, 1H, CHCH2), 1.98–1.58 (m, 2H, CH2), 1.34 (d, J = 6.8 Hz, 3H, CHCH3), 0.76 (t, J = 7.2 Hz, 3H, CH3). 13C NMR (50 MHz, DMSO-d6): 168.55, 168.27, 155.02, 134.29, 124.81, 116.65, 116.49, 106.83, 47.77, 26.45, 18.45, 11.22. ES-HRMS m/z for C12H14N2NaO2 [M + Na]+: calcd. 241.0947, found 241.0948.
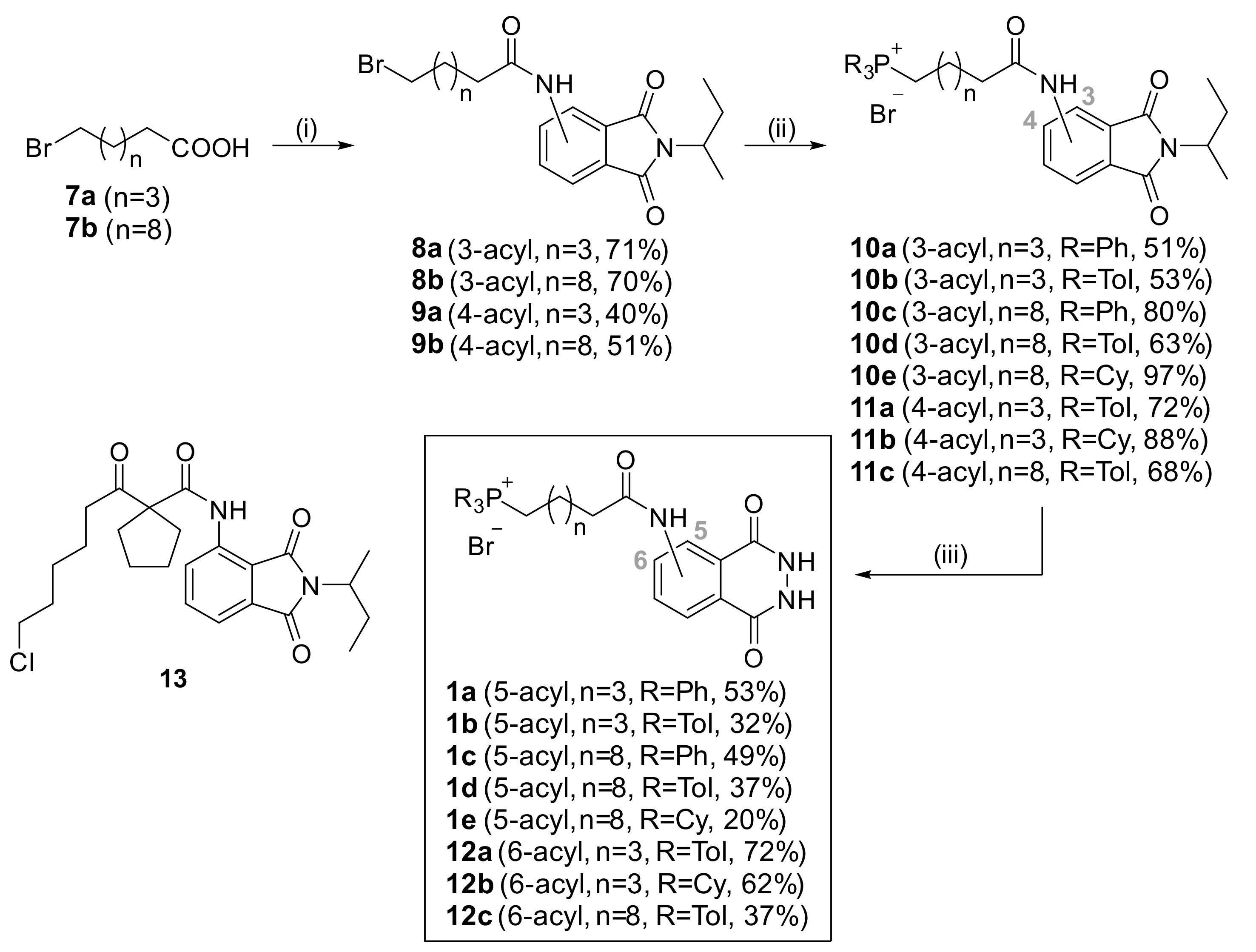
3.2.5. General Procedure for the Acylation of Phthalimides
A solution of the carboxylic acid (11 mmol) in oxalyl chloride (10 mL) was stirred for 5 h under Ar. Then, the volatiles were evaporated to dryness under reduced pressure at room temperature. The residue was dissolved in dry dichloromethane (8 mL) and added dropwise to a cooled (0 °C) solution of the aminophthalimide (2.18 g, 10 mmol) and pyridine (1.61 mL, 20 mmol) in dichloromethane (24 mL) under Ar. The resulting mixture was stirred at r.t. for 18 h. Water (100 mL) was added, the layers were separated and the aqueous was washed with dichloromethane (2 × 40 mL). The combined organic layers were dried (Na2SO4), solvent was evaporated and the residue was subjected to column chromatography, affording the corresponding acylated phthalimide.
6-Bromo-N-(2-(sec-butyl)-1,3-dioxoisoindolin-4-yl)hexanamide (8a). From 6-bromohexanoic acid and 4-aminophthalimide 6a. Chromatography with EtOAc/petroleum ether 8:1 to 4:1. Brownish oil (2.8 g, 71%). 1H NMR (200 MHz, CDCl3) δ: 9.60 (bs, 1H, NH), 8.73 (d, J = 8.4 Hz, 1H, H-5), 7.63 (t, J = 7.9 Hz, 1H, H-6), 7.45 (d, J = 7.2 Hz, 1H, H-7), 4.28–4.09 (m, 1H, CH), 3.41 (t, J = 6.7 Hz, 2H, BrCH2), 2.47 (t, J = 7.4 Hz, 2H, CH2CO), 2.12–1.65 (m, 6H), 1.60–1.42 (m, 5H), 0.86 (t, J = 7.4 Hz, 3H, CH2CH3). 13C NMR (50 MHz, CDCl3) δ: 172.01, 170.71, 168.05, 137.24, 135.78, 131.37, 124.61, 117.83, 115.55, 49.19, 37.71, 33.63, 32.45, 27.72, 26.90, 24.40, 18.54, 11.39. ES-HRMS m/z for C18H22BrN2O3 [M − H]−: calcd. 393.0819, found 393.0821.
11-Bromo-N-(2-(sec-butyl)-1,3-dioxoisoindolin-4-yl)undecanamide (8b). From 11-bromoundecanoic acid and 4-aminophthalimide 6a. Chromatography with EtOAc/petroleum ether 8:1 to 4:1. Brownish oil (3.3 g, 70%). 1H NMR (200 MHz, CDCl3) δ: 9.58 (bs, 1H, NH), 8.74 (d, J = 8.4 Hz, 1H, H-5), 7.62 (t, J = 7.9 Hz, 1H, H-6), 7.44 (d, J = 7.3 Hz, 1H, H-7), 4.27–4.10 (m, 1H, CH), 3.37 (t, J = 6.8 Hz, 2H, BrCH2), 2.44 (t, J = 7.5 Hz, 2H, CH2CO), 2.08–1.65 (m, 6H), 1.45–1.19 (m, 15H), 0.86 (t, J = 7.4 Hz, 3H, CH2CH3). 13C NMR (50 MHz, CDCl3) δ: 172.49, 170.69, 167.93, 137.40, 135.71, 131.43, 124.64, 117.70, 115.57, 77.16, 49.20, 38.06, 34.08, 32.87, 29.41, 29.38, 29.29, 29.19, 28.78, 28.20, 26.93, 25.33, 18.50, 11.36. ES-HRMS m/z for C23H32BrN2O3 [M − H]−: calcd. 463.1602, found 463.1613.
6-Bromo-N-(2-(sec-butyl)-1,3-dioxoisoindolin-4-yl)hexanamide (9a). From 6-bromohexanoic acid and 5-aminophthalimide 6b. Chromatography with methanol/DCM 0% to 5%. Brownish oil (2.7 g, 40%, mixture with 10 mol% of the corresponding chloride). 1H NMR (200 MHz, CDCl3) δ: 8.01 (dd, J = 8.1, 1.8 Hz, 1H, H-6), 7.96 (d, J = 1.5 Hz, 1H, H-4), 7.94 (bs, 1H, NH), 7.75 (d, J = 8.1 Hz, 1H, H-7), 4.32–4.14 (m, 1H, CH), 3.42 (t, J = 6.6 Hz, 2H, BrCH2), 2.47 (t, J = 7.3 Hz, 2H, CH2CO), 2.15–1.71 (m, 6H), 1.61–1.44 (m, 5H), 0.86 (t, J = 7.4 Hz, 3H, CH2CH3). 13C NMR (50 MHz, CDCl3) δ: 172.38, 168.10, 168.02, 143.71, 132.97, 126.00, 123.82, 123.75, 113.82, 48.85, 37.00, 33.33, 32.06, 27.39, 26.57, 24.29, 18.11, 11.04. ES-HRMS m/z for C18H22BrN2O3 [M − H]−: calcd. 393.0819, found 393.0811.
11-Bromo-N-(2-(sec-butyl)-1,3-dioxoisoindolin-5-yl)undecanamide (9b). From 11-bromoundecanoic acid and 5-aminophthalimide 6b. Chromatography with methanol/DCM 0% to 5%. Brownish oil (2.7 g, 58%). 1H NMR (200 MHz, CDCl3) δ: 8.25 (bs, 1H, NH), 8.09 (d, J = 8.2 Hz, 1H, H-6), 7.97 (s, 1H, H-4), 7.74 (d, J = 8.1 Hz, 1H, H-7), 4.32–4.13 (m, 1H, CH), 3.38 (t, J = 6.7 Hz, 2H, BrCH2), 2.45 (t, J = 6.8 Hz, 2H, CH2CO), 2.08–1.65 (m, 6H), 1.47–1.19 (m, 15H), 0.86 (t, J = 7.3 Hz, 3H, CH2CH3). 13C NMR (50 MHz, CDCl3) δ: 172.16, 168.60, 168.29, 143.79, 133.45, 126.43, 124.46, 123.84, 113.86, 49.25, 37.91, 34.25, 32.86, 29.45, 29.42 (2C), 29.32, 28.80, 28.21, 26.96, 25.49, 18.54, 11.43. ES-HRMS m/z for C23H32BrN2O3 [M − H]−: calcd. 463.1602, found 463.1610.
3.2.6. General Procedure for the Synthesis of Phosphonium Phthalimides
A solution of the bromide (1 mmol) and the phosphine (2 mmol) in dry acetonitrile (5 mL) was refluxed under Ar for 3 days. After cooling, the solvent was evaporated and the residue was subjected to column chromatography, yielding the corresponding phosphonium cation.
(6-((2-(sec-Butyl)-1,3-dioxoisoindolin-4-yl)amino)-6-oxohexyl)triphenylphosphonium bromide (10a). From bromide 8a and triphenylphosphine. Chromatography with methanol/DCM 3 to 10%. White solid (335 mg, 51%). 1H NMR (200 MHz, CDCl3) δ: 9.53 (bs, 1H, NH), 8.62 (d, J = 8.4 Hz, H-5), 7.85–7.60 (m, 15H, ArH), 7.55 (t, J = 8.0 Hz, 1H, H-6), 7.39 (d, J = 7.2 Hz, 1H, H-7), 4.23–4.05 (m, 1H, CH), 3.80–3.65 (m, 2H, PCH2), 2.40 (t, J = 6.1 Hz, 2H, CH2CO), 2.07–1.60 (m, 8H), 1.39 (d, J = 6.9 Hz, 3H, CH3), 0.81 (t, J = 7.3 Hz, 3H, CH2CH3). 13C NMR (50 MHz, CDCl3) δ: 171.16, 169.35, 167.10, 136.11, 134.80, 134.34 (d, J = 2.7 Hz, PPh3 para), 132.74 (d, J = 10.0 Hz, PPh3 ortho),130.45, 129.76 (d, J = 12.5 Hz, PPh3 meta), 123.89, 117.21 (d, J = 86.0 Hz, PPh3 ipso), 116.90, 114.88, 48.90, 36.26, 28.82 (d, J = 16.0 Hz, CH2CH2CH2P), 25.94, 23.52, 21.44 (d, J = 50.0 Hz, CH2P), 21.40 (d, J = 4.0 Hz, CH2CH2P), 17.60, 10.49. 31P NMR (81 MHz, CDCl3) δ: 24.89. ES-HRMS m/z for C36H38N2O3P [M]+: calcd. 577.2615, found 577.2608.
(6-((2-(sec-Butyl)-1,3-dioxoisoindolin-4-yl)amino)-6-oxohexyl)tri-p-tolylphosphonium bromide (10b). From bromide 8a and tri(p-tolyl)phosphine. Chromatography with methanol/DCM 5% to 20%. White solid (371 mg, 53%). 1H NMR (200 MHz, CDCl3) δ: 9.56 (bs, 1H, NH), 8.67 (d, J = 8.4 Hz, H-5), 7.70–7.57 (m, 7H, ArH), 7.50–7.43 (m, 7H, ArH, H-6, H-7), 4.26–4.08 (m, 1H, CH), 3.64–3.50 (m, 2H, PCH2), 2.46–2.34 (m, 11H, ArCH3, CH2CO), 2.07–1.60 (m, 8H), 1.43 (d, J = 6.9 Hz, 3H, CH3), 0.85 (t, J = 7.4 Hz, 3H, CH2CH3). 13C NMR (50 MHz, CDCl3) δ: 171.88, 170.24, 167.92, 146.15 (d, J = 3.0 Hz, PAr3 para), 136.94, 135.47, 133.34 (d, J = 10.3 Hz, PAr3 ortho), 131.26, 131.09 (d, J = 12.9 Hz, PAr3 meta), 124.57, 117.64, 115.59, 114.83 (d, J = 88.7 Hz, PAr3 ipso), 48.98, 37.03, 29.63 (d, J = 16.6 Hz, CH2CH2CH2P), 26.70, 24.35, 22.66 (d, J = 53.0 Hz, CH2P), 22.18 (d, J = 4.5 Hz, CH2CH2P), 21.79 (d, J = 1.2 Hz, ArCH3), 18.36, 11.23. 31P NMR (81 MHz, CDCl3) δ: 23.97. ES-HRMS m/z for C39H44N2O3P [M]+: calcd. 619.3084, found 619.3080.
(11-((2-(sec-Butyl)-1,3-dioxoisoindolin-4-yl)amino)-11-oxoundecyl)triphenylphosphonium bromide (10c). From bromide 8b and triphenylphosphine. Chromatography with methanol/DCM 5% to 10%. White solid (582 mg, 80%). 1H NMR (200 MHz, CDCl3) δ: 9.55 (bs, 1H, NH), 8.71 (d, J = 8.4 Hz, H-5), 7.84–7.55 (m, 16H, ArH, H-6), 7.41 (d, J = 7.2 Hz, 1H, H-7), 4.24–4.07 (m, 1H, CH), 3.77–3.61 (m, 2H, PCH2), 2.39 (t, J = 7.5 Hz, 2H, CH2CO), 2.09–1.58 (m, 6H), 1.41 (d, J = 6.9 Hz, 3H, CH3), 1.34–1.10 (m, 12H), 0.83 (t, J = 7.4 Hz, 3H, CH2CH3). 13C NMR (50 MHz, CDCl3) δ: 172.55, 170.70, 168.12, 137.42, 135.71, 135.08 (d, J = 3.0 Hz, PPh3 para), 133.79 (d, J = 9.9 Hz, PPh3 ortho), 131.48, 130.58 (d, J = 12.6 Hz, PPh3 meta), 124.69, 118.25 (d, J = 85.9 Hz, PPh3 ipso), 117.72, 115.63, 49.24, 38.06, 30.49 (d, J = 15.5 Hz, CH2CH2CH2P), 29.37, 29.27 (2C), 29.18 (2C), 26.95, 25.32, 22.87 (d, J = 49.6 Hz, CH2P), 22.75 (d, J = 4.4 Hz, CH2CH2P), 18.52, 11.37. 31P NMR (81 MHz, CDCl3) δ: 25.31. ES-HRMS m/z for C41H48N2O3P [M]+: calcd. 647.3397, found 647.3397.
(11-((2-(sec-Butyl)-1,3-dioxoisoindolin-4-yl)amino)-11-oxoundecyl)tri-p-tolylphosphonium bromide (10d). From bromide 8b and tri(p-tolyl)phosphine. Chromatography with methanol/DCM 5% to 10%. White solid (485 mg, 63%). 1H NMR (200 MHz, CDCl3) δ: 9.57 (bs, 1H, NH), 8.73 (d, J = 8.4 Hz, H-5), 7.68–7.42 (m, 14H, ArH, H-6, H-7), 4.26–4.09 (m, 1H, CH), 3.57–3.42 (m, 2H, PCH2), 2.44 (s, 9H, ArCH3), 2.39 (t, J = 7.9 Hz, 2H, CH2CO), 2.07–1.57 (m, 6H), 1.43 (d, J = 7.0 Hz, 3H, CH3), 1.36–1.13 (m, 12H), 0.88 (t, J = 7.4 Hz, 3H, CH2CH3). 13C NMR (50 MHz, CDCl3) δ: 172.55, 170.67, 168.10, 146.25 (d, J = 3.0 Hz, PAr3 para), 137.36, 135.70, 133.53 (d, J = 10.3 Hz, PAr3 ortho), 131.41, 131.21 (d, J = 12.9 Hz, PAr3 meta), 124.64, 117.71, 115.56, 115.19 (d, J = 88.6 Hz, PAr3 ipso), 49.20, 38.04, 30.55 (d, J = 15.6 Hz, CH2CH2CH2P), 29.76, 29.38, 29.27, 29.22, 29.17, 26.91, 25.30, 23.06 (d, J = 51.1 Hz, CH2P), 22.67 (d, J = 4.3 Hz, CH2CH2P), 21.94 (d, J = 1.2 Hz, ArCH3) 18.52, 11.37. 31P NMR (81 MHz, CDCl3) δ: 24.13. ES-MS m/z for C44H54N2O3P [M]+: calcd. 689.3867, found 689.3869.
(11-((2-(sec-Butyl)-1,3-dioxoisoindolin-4-yl)amino)-11-oxoundecyl)tricyclohexylphosphonium bromide (10e). From bromide 8b and tricyclohexylphosphine. Chromatography with methanol/DCM 5% to 10%. White solid (723 mg, 97%). 1H NMR (200 MHz, CDCl3) δ: 9.51 (bs, 1H, NH), 8.65 (d, J = 8.3 Hz, H-5), 7.55 (t, J = 8.1 Hz, 1H, H-6), 7.36 (d, J = 7.2 Hz, 1H, H-7), 4.20–4.02 (m, 1H, CH), 2.67–2.22 (m, 7H, PCH, COCH2), 1.96–1.15 (m, 51H, CH), 0.78 (t, J = 7.3 Hz, 3H, CH2CH3). 13C NMR (50 MHz, CDCl3) δ: 172.07, 170.18, 167.63, 136.92, 135.27, 130.98, 124.19, 117.24, 115.14, 48.73, 37.59, 30.91 (d, J = 13.6 Hz, CH2CH2CH2P), 29.46 (d, J = 40.3 Hz, PCy3-C1), 28.91, 28.88, 28.86, 28.73, 28.61, 26.87 (d, J = 3.5 Hz, PCy3-C2), 26.46, 26.08 (d, J = 11.8 Hz, PCy3-C3), 25.08, 24.86, 22.44 (d, J = 5.2 Hz, CH2CH2P), 18.08, 15.45 (d, J = 43.0 Hz, CH2P), 10.94. 31P NMR (81 MHz, CDCl3) δ: 32.62. ES-HRMS m/z for C41H66N2O3P [M]+: calcd. 665.4806, found 665.4804.
(6-((2-(sec-Butyl)-1,3-dioxoisoindolin-5-yl)amino)-6-oxohexyl)tri-p-tolylphosphonium bromide (11a). From bromide 9a and tri(p-tolyl)phosphine. Chromatography with methanol/DCM 3 to 10%. White solid (504 mg, 72%). 1H NMR (200 MHz, CDCl3) δ: 10.98 (bs, 1H, NH), 8.59 (s, 1H, H-4), 8.13 (d, J = 7.9 Hz, H-6), 7.67–7.43 (m, 13H, ArH, H-7), 4.29–4.10 (m, 1H, CH), 3.45–3.26 (m, 2H, PCH2), 2.65 (t, J = 7.2 Hz, CH2CO), 2.46 (s, 9H, ArCH3), 2.06–1.70 (m, 8H), 1.43 (d, J = 6.9 Hz, 3H, CH3), 0.84 (t, J = 7.4 Hz, 3H, CH2CH3). 13C NMR (50 MHz, CDCl3) δ: 173.10, 168.45, 168.28, 146.26 (d, J = 3.0 Hz, PAr3 para), 145.00, 133.09 (d, J = 10.3 Hz, PAr3 ortho), 132.66, 131.03 (d, J = 12.9 Hz, PAr3 meta), 125.20, 124.14, 123.14, 114.49 (d, J = 88.9 Hz, PAr3 ipso), 113.83, 48.47, 36.72, 29.70 (d, J = 15.9 Hz, CH2CH2CH2P), 26.61, 24.32, 22.87 (d, J = 52.5 Hz, CH2P), 21.64 (d, J = 1.2 Hz, ArCH3), 21.39 (d, J = 4.0 Hz, CH2CH2P) 18.22, 11.07. 31P NMR (81 MHz, CDCl3) δ: 22.24. ES-HRMS m/z for C39H44N2O3P [M]+: calcd. 619.3084, found 619.3094.
(6-((2-(sec-Butyl)-1,3-dioxoisoindolin-5-yl)amino)-6-oxohexyl)tricyclohexylphosphonium bromide (11b). From bromide 9a and tricyclohexylphosphine. Chromatography with methanol/DCM 3% to 10%. White solid (595 mg, 88%, contaminated with 25 mol% tricyclohexylphosphinoxide). 1H NMR (400 MHz, CDCl3) δ: 10.87 (bs, 1H, NH), 8.42 (d, J = 1.8 Hz, 1H, H-4), 8.04 (dd, J = 8.2, 1.9 Hz, H-6), 7.45 (d, J = 8.1 Hz, 1H, H-7), 4.06–3.97 (m, 1H, NCH), 2.57 (t, J = 7.3 Hz, CH2CO), 2.48–2.39 (m, 3H, PCH), 2.18–2.11 (m, 2H, PCH2), 1.97–1.05 (m, 41H, CH, O=CCy3*), 0.67 (t, J = 7.4 Hz, 3H, CH2CH3). 13C NMR (100 MHz, CDCl3) δ: 172.85, 168.33, 168.20, 144.95, 132.66, 125.25, 123.98, 123.09, 113.64, 48.45, 36.11, 35.02* (d, J = 60.8 Hz, O=PCy3-C1), 30.00 (d, J = 14.1 Hz, CH2CH2CH2P), 29.64 (d, J = 40.4 Hz, PCy3-C1), 26.90 (d, J = 3.8 Hz, PCy3-C2), 26.59* (d, J = 11.6 Hz, O=PCy3-C3), 26.55, 26.12 (d, J = 11.9 Hz, PCy3-C3), 26.02* (d, J = 3.0 Hz, O=PCy3-C2), 25.82* (d, J = 1.3 Hz, O=PCy3-C4), 25.07 (d, J = 1.2 Hz, PCy3-C4), 24.20 (d, J = 1.0 Hz, CH2CH2CH2CH2P), 21.43 (d, J = 4.7 Hz, CH2CH2P), 18.13, 15.48 (d, J = 42.8 Hz, CH2P), 10.96. 31P NMR (81 MHz, CDCl3) δ: 33.64. ES-HRMS m/z for C36H56N2O3P [M]+: calcd. 595.4023, found 595.4032. *signals attributed to tricyclohexylphosphinoxide.
(11-((2-(sec-Butyl)-1,3-dioxoisoindolin-5-yl)amino)-11-oxoundecyl)tri-p-tolylphosphonium bromide (11c). From bromide 9b and tri(p-tolyl)phosphine. Chromatography with methanol/DCM 3 to 10%. White solid (523 mg, 68%). 1H NMR (400 MHz, CDCl3) δ: 10.74 (bs, 1H, NH), 8.52 (d, J = 1.8 Hz, 1H, H-4), 8.14 (dd, J = 8.2, 1.8 Hz, H-6), 7.51 (dd, J = 12.4, 8.1 Hz, PTol-Hortho), 7.46 (d, J = 8.1 Hz, 1H, H-7), 7.41 (dd, J = 8.3, 3.2 Hz, PTol-Hmeta), 4.13–4.04 (m, 1H, NCH), 3.29–3.22 (m, 1H, PCH), 2.58 (t, J = 7.5 Hz, CH2CO), 2.39 (s, PTol-CH3), 1.97–1.85 (m, 1H, CH2CH3), 1.70–1.43 (m, 7H), 1.32 (t, J = 7.0, CHCH3), 1.27–1.10 (m, 10H), 0.74 (t, J = 7.4 Hz, 3H, CH2CH3). 13C NMR (100 MHz, CDCl3) δ: 173.82, 168.69, 168.51, 146.48 (d, J = 2.9 Hz, PAr3 para), 145.42, 133.26 (d, J = 10.4 Hz, PAr3 ortho), 132.85, 131.20 (d, J = 12.8 Hz, PAr3 meta), 125.24, 124.28, 123.29, 114.84 (d, J = 88.7 Hz, PAr3 ipso), 114.14, 48.64, 37.10, 30.28 (d, J = 15.5 Hz, CH2CH2CH2P), 28.75, 28.71, 28.70, 28.65, 28.39, 26.82, 25.29, 23.07 (d, J = 51.9 Hz, CH2P), 22.39 (d, J = 4.4 Hz, CH2CH2P), 21.79 (d, J = 1.5 Hz, ArCH3), 18.37, 11.21. 31P NMR (162 MHz, CDCl3) δ: 22.81. ES-HRMS m/z for C44H54N2O3P [M]+: calcd. 689.3867, found 689.3886.
3.2.7. General Procedure for the Synthesis of Phosphonium Phthalhydrazides
A solution of the phosphonium phthalimide (0.4 mmol) and hydrazine hydrate (6 mmol) in ethanol (15 mL) was refluxed for 3 h. After cooling, the volatiles were evaporated and the residue was subjected to column chromatography, yielding the corresponding phthalhydrazide.
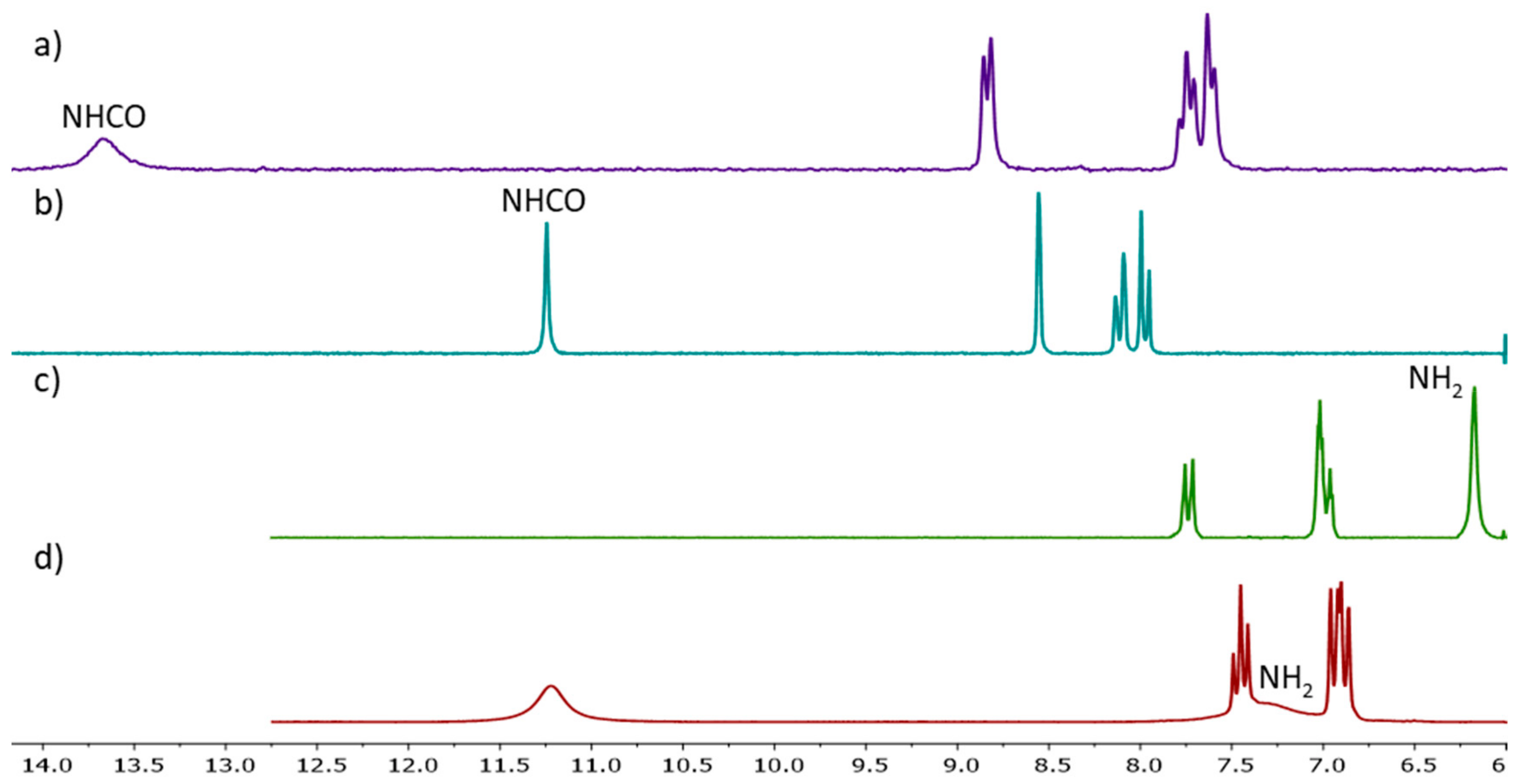
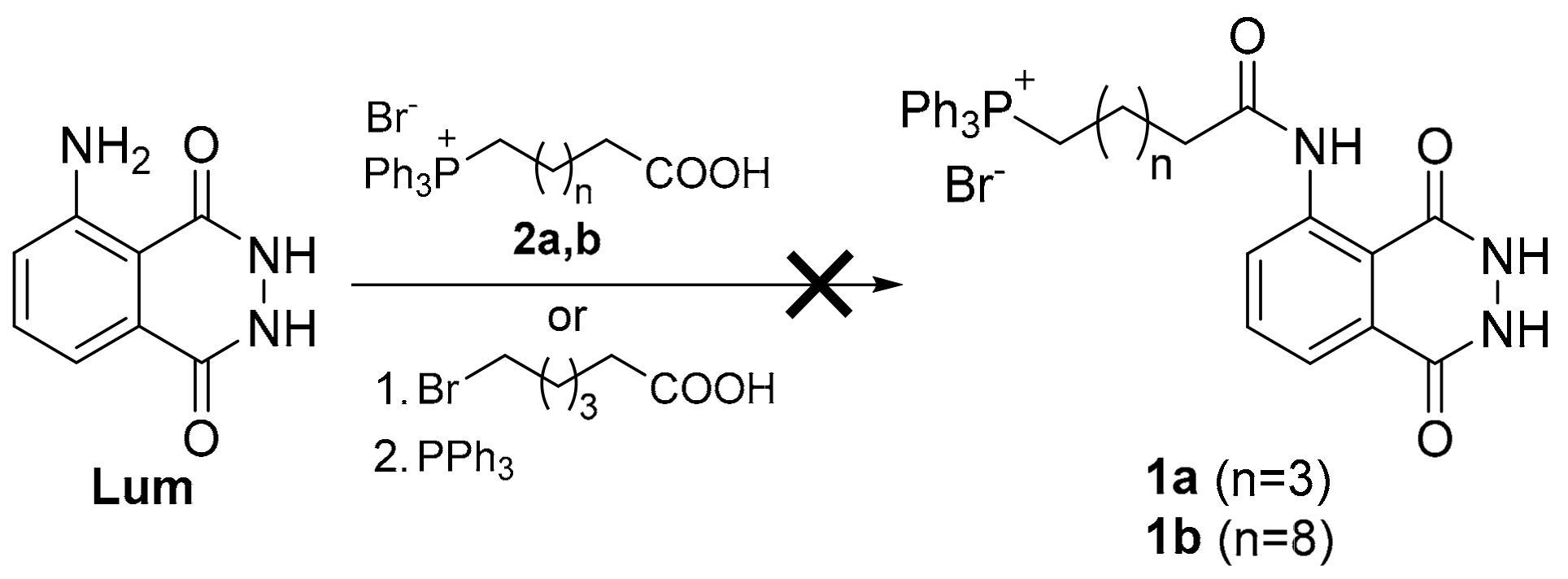
(6-((1,4-Dioxo-1,2,3,4-tetrahydrophthalazin-5-yl)amino)-6-oxohexyl)triphenylphosphonium bromide (1a). From phthalimide 10a. Chromatography with methanol/DCM 5% to 20%. White solid (139 mg, 53%). 1H NMR (200 MHz, CDCl3) δ: 12.53 (bs, 1H, NH), 12.06 (bs, 2H, NH),8.78 (d, J = 8.1 Hz, 1H, H-6), 7.80–7.45 (m, 17H, Ar-H, H-7, H-8), 3.71–3.50 (m, 2H, CH2P), 2.39–2.22 (m, 2H, CH2CO), 1.78–1.54 (m, 6H, CH2). 13C NMR (50 MHz, CDCl3) δ: 172.02, 159.90, 154.47, 140.90, 135.13 (d, J = 1.8 Hz, PAr3 para), 134.21, 133.52 (d, J = 9.9 Hz, PAr3 ortho), 130.54 (d, J = 12.5 Hz, PAr3 meta), 127.91, 122.22, 119.59, 118.03 (d, J = 85.9 Hz, ipso), 114.80, 37.95, 29.71 (d, J = 17.4 Hz, CH2CH2CH2P), 24.40, 22.25 (d, J = 49.6 Hz, CH2P), 22.23 (d, J = 3.2 Hz, CH2CH2P). 31P NMR (81 MHz, CDCl3) δ: 25.33. ES-HRMS m/z for C32H31N3O3P [M]+: calcd. 536.2098, found 536.2097.
(6-((1,4-Dioxo-1,2,3,4-tetrahydrophthalazin-5-yl)amino)-6-oxohexyl)tri-p-tolylphosphonium bromide (1b). From phthalimide 10b. Chromatography with methanol/DCM 5% to 20%. White solid (84 mg, 32%). 1H NMR (200 MHz, CDCl3) δ: 12.53 (bs, 1H, NH), 12.66 (bs, 2H, NH), 8.80 (d, J = 8.0 Hz, 1H, H-6), 7.70 (d, J = 7.0 Hz, 1H, H-8), 7.57–7.30 (m, 13H, Ar-H, H-7), 3.46–3.25 (m, 2H, CH2P), 2.41–2.18 (m, 11H, ArCH3, CH2CO), 1.78–1.52 (m, 6H, CH2). 13C NMR (50 MHz, CDCl3) δ: 172.34, 160.21, 155.19, 146.48 (d, J = 1.6 Hz, PAr3 para), 140.93, 134.08, 133.44 (d, J = 10.4 Hz, PAr3 ortho), 131.30 (d, J = 12.9 Hz, PAr3 meta),128.51, 122.31, 119.94, 114.98 (d, J = 88.8 Hz, PAr3 ipso), 115.25, 38.29, 29.93 (d, J = 16.0 Hz, CH2CH2CH2P), 24.58, 22.74 (d, J = 49.2 Hz, CH2P), 22.30 (d, J = 4.8 Hz, CH2CH2P), 21.97. 31P NMR (81 MHz, CDCl3) δ: 25.23. ES-HRMS m/z for C35H37N3O3P [M]+: calcd. 578.2657, found 578.2619.
(11-((1,4-Dioxo-1,2,3,4-tetrahydrophthalazin-5-yl)amino)-11-oxoundecyl)triphenylphosphonium bromide (1c). From phthalimide 10c. Chromatography with methanol/DCM 5% to 20%. White solid (135 mg, 49%). 1H NMR (200 MHz, DMSO-d6) δ: 15.58 (bs, 1H, NH), 8.74 (d, J = 7.7 Hz, 1H, H-6), 7.90–7.70 (m, 15H, ArH), 7.62 (d, J = 7.7 Hz, 1H, H-8), 7.49 (t, J = 7.7 Hz, 1H, H-7), 3.60–3.45 (m, 2H, CH2P), 2.28 (t, J = 7.1 Hz, 2H, CH2CO), 1.65–1.10 (m, 16H, CH2). 13C NMR (50 MHz, DMSO-d6) δ: 171.09, 160.37, 156.81, 140.29, 134.84 (d, J = 2.6 Hz, PAr3 para), 133.58 (d, J = 10.1 Hz, PAr3 ortho), 130.34, 130.22 (d, J = 12.4 Hz, PAr3 meta), 129.99, 119.50, 118.62 (d, J = 85.6 Hz, PAr3 ipso), 118.59, 117.36, 38.10, 30.72, 29.67 (d, J = 16.5 Hz, CH2CH2CH2P), 28.59 (2C), 28.51, 27.95, 25.03, 21.67 (d, J = 4.2 Hz, CH2CH2P), 20.09 (d, J = 49.4 Hz, CH2P). 31P NMR (162 MHz, CDCl3) δ: 23.39. ES-HRMS m/z for C37H41N3O3P [M]+: calcd. 606.2880, found 606.2888.
(11-((1,4-Dioxo-1,2,3,4-tetrahydrophthalazin-5-yl)amino)-11-oxoundecyl)tri-p-tolylphosphonium bromide (1d). From phthalimide 10d. Chromatography with methanol/DCM 5% to 20%. White solid (108 mg, 37%). 1H NMR (200 MHz, CDCl3) δ: 12.74 (bs, 1H, NH), 8.95 (d, J = 8.2 Hz, 1H, H-6), 7.77 (d, J = 7.7 Hz, 1H, H-8), 7.62 (t, J = 8.1 Hz, 1H, H-7), 7.55–7.36 (m, 15H, ArH), 3.36–3.22 (m, 2H, CH2P), 2.42–2.27 (m, 11H, ArCH3, CH2CO), 1.72–1.04 (m, 16H, CH2). 13C NMR (50 MHz, CDCl3) δ: 172.80, 159.66, 154.97, 146.34 (d, J = 3.0 Hz, PAr3 para), 141.21, 134.06, 133.24 (d, J = 10.3 Hz, PAr3 ortho), 131.15 (d, J = 12.9 Hz, PAr3 meta), 128.28, 122.41, 119.66, 115.12, 114.86 (d, J = 88.7 Hz, PAr3 ipso), 38.62, 30.38 (d, J = 15.7 Hz, CH2CH2CH2P), 28.91 (2C), 28.87, 28.83, 28.77, 25.31, 22.82 (d, J = 51.6 Hz, CH2P), 22.49 (d, J = 4.2 Hz, CH2CH2P), 21.81 (d, J = 1.2 Hz, ArCH3). 31P NMR (81 MHz, CDCl3) δ: 23.82. ES-HRMS m/z for C40H47N3O3P [M]+: calcd. 648.3350, found 648.3437.
Tricyclohexyl(11-((1,4-dioxo-1,2,3,4-tetrahydrophthalazin-5-yl)amino)-11-oxoundecyl)phosphonium bromide (1e). From phthalimide 10e. Chromatography with methanol/DCM 5% to 20%. White solid (56 mg, 20%). 1H NMR (400 MHz, CDCl3) δ: 12.67 (bs, 1H, NH), 8.98 (d, J = 8.3 Hz, 1H, H-6), 7.81 (d, J = 7.8 Hz, 1H, H-8), 7.69 (t, J = 8.1 Hz, 1H, H-7), 2.53 (q, J = 11.5 Hz, 3H, PCyCH), 2.40 (t, J = 7.5 Hz, 2H, COCH2), 2.38–2.30 (m, 2H, PCH2), 2.01–1.19 (m, 46H, CH2). 13C NMR (100 MHz, CDCl3) δ: 172.81, 159.82, 154.77, 141.40, 134.35, 128.09, 122.63, 119.67, 115.09, 38.73, 31.18 (d, J = 13.6 Hz, CH2CH2CH2P), 30.02 (d, J = 40.3 Hz, PCy3-C1), 29.15, 28.98, 28.94, 28.86, 28.82, 27.25 (d, J = 3.9 Hz, PCy3-C2), 26.53 (d, J = 11.7 Hz, PCy3-C3), 25.47, 25.35, 22.79 (d, J = 5.2 Hz, CH2CH2P), 15.82 (d, J = 42.5 Hz, CH2P). 31P NMR (81 MHz, CDCl3) δ: 32.71. ES-HRMS m/z for C37H59N3O3P [M]+: calcd. 624.4289, found 624.4295.
(6-((1,4-dioxo-1,2,3,4-tetrahydrophthalazin-6-yl)amino)-6-oxohexyl)tri-p-tolylphosphonium bromide (12a). From phthalimide 11a. Chromatography with methanol/DCM 5% to 20%. White solid (190 mg, 72%). 1H NMR (200 MHz, DMSO-d6) δ: 11.46 (bs, 2H, NHNH), 10.63 (bs, 1H, NHCO), 8.42 (s, 1H, H-5), 8.03–7.94 (m, 2H, H-7, H-8), 7.68–7.55 (m, 12H, ArH), 3.56–3.45 (m, 2H, CH2P), 2.46–2.30 (m, 11H, ArCH3, CH2CO), 1.60–1.45 (m, 6H, CH2). 13C NMR (50 MHz, DMSO-d6) δ: 172.13, 155.00, 154.50, 145.53 (d, J = 2.8 Hz, PAr3 para), 143.10, 133.46 (d, J = 10.4 Hz, PAr3 ortho), 130.80 (d, J = 12.8 Hz, PAr3 meta),128.18, 126.24, 123.29, 122.33, 115.51 (d, J = 88.2 Hz, PAr3 ipso), 113.28, 36.08, 29.51 (d, J = 17.5 Hz, CH2CH2CH2P), 24.30, 21.67 (d, J = 5.0 Hz, CH2CH2P), 21.28 (d, J = 1.4 Hz, ArCH3), 20.52 (d, J = 52.0 Hz, CH2P). 31P NMR (81 MHz, DMSO-d6) δ: 24.04. ES-HRMS m/z for C35H37N3O3P [M]+: calcd. 578.2567, found 578.2627.
Tricyclohexyl(6-((1,4-dioxo-1,2,3,4-tetrahydrophthalazin-6-yl)amino)-6-oxohexyl)phosphonium bromide (12b). From phthalimide 11b. Chromatography with methanol/DCM 5% to 20%. White solid (157 mg, 62%). 1H NMR (200 MHz, DMSO-d6) δ: 11.25 (bs, 1H, NHCO), 8.56 (s, 1H, H-5), 8.11 (d, J = 8.7 Hz, 1H, H-7), 7.97 (d, J = 8.6 Hz, 1H, H-8), 2.59–2.43 (m, 5H, PCH), 2.32–2.15 (m, 2H, COCH2), 2.03–1.26 (m, 36H, CH2). 13C NMR (50 MHz, DMSO-d6) δ: 172.31, 155.00, 154.39, 143.20, 128.18, 126.15, 123.34, 122.35, 113.32, 36.03, 30.11 (d, J = 13.3 Hz, CH2CH2CH2P), 28.54 (d, J = 41.2 Hz, CH2P-cyclo), 26.11 (d, J = 4.2 Hz, CH2CH2P-cyclo), 25.94 (d, J = 12.8 Hz, CH2CH2CH2P-cyclo), 25.02, 24.26, 21.44 (d, J = 2.8 Hz, CH2CH2P), 14.28 (d, J = 44.0 Hz, CH2P). 31P NMR (81 MHz, DMSO-d6) δ: 32.47. ES-HRMS m/z for C32H49N3O3P [M]+: calcd. 554.3506, found 554.3556.
(11-((1,4-Dioxo-1,2,3,4-tetrahydrophthalazin-6-yl)amino)-11-oxoundecyl)tri-p-tolylphosphonium bromide (12c). From phthalimide 11c. Chromatography with methanol/DCM 5% to 20%. White solid (108 mg, 37%). 1H NMR (200 MHz, DMSO-d6) δ: 11.44 (bs, 2H, NHNH), 10.47 (bs, 1H, NHCO), 8.40 (s, 1H, H-5), 8.03–7.94 (m, 2H, H-7, H-8), 7.68–7.53 (m, 12H, ArH), 3.52–3.43 (m, 2H, CH2P), 2.44–2.33 (m, 11H, ArCH3, CH2CO), 1.65–1.16 (m, 16H, CH2). 13C NMR (50 MHz, DMSO-d6) δ: 172.32, 154.93, 154.38, 145.55 (d, J = 3.0 Hz, PAr3 para), 143.17, 133.44 (d, J = 10.4 Hz, PAr3 ortho), 130.80 (d, J = 12.8 Hz, PAr3 meta), 128.15, 126.24, 123.31, 122.23, 115.54 (d, J = 88.2 Hz, PAr3 ipso), 113.26, 36.45, 29.79 (d, J = 16.6 Hz, CH2CH2CH2P), 28.75, 28.68, 28.63, 28.13, 25.02, 21.76 (d, J = 3.5 Hz, CH2CH2P), 21.28 (d, J = 1.5 Hz, ArCH3), 20.46 (d, J = 50.4 Hz, CH2P). 31P NMR (81 MHz, DMSO-d6) δ: 24.13. ES-HRMS m/z for C40H47N3O3P [M]+: calcd. 648.3350, found 648.3401.
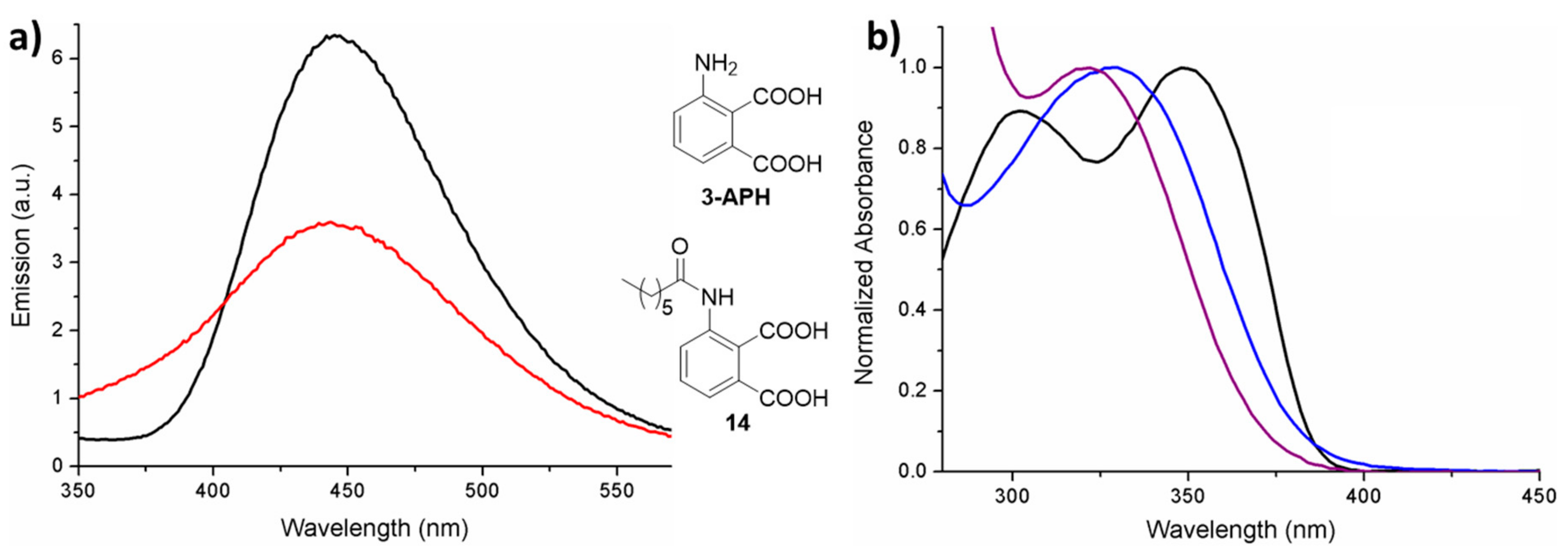
3.2.8. Synthesis of 3-Heptanamidophthalic Acid (14)
Synthesis of 4-aminoisobenzofuran-1,3-dione15 [
50]. A stirred solution of 3-nitrophthalic anhydride
4a (4.5 g, 23 mmol) in THF (40 mL) was degassed (Ar) for 30 min. 10% Pd/C (200 mg) was added, then bubbled with H
2 for a while and the mixture was stirred under H
2 atmosphere (20 bar) for 18 h. The mixture was filtered through celite, washed with ethyl acetate and the filtrate was concentrated. The solid residue was washed with dichloromethane (extracting ≈ 1 g of a mixture containing the product), silica gel was added in the combined washings, the solvent was evaporated and the residue was dry-loaded onto column chromatography (dichloromethane), affording 3-aminophthalic anhydride
15 as yellow solid (488 mg, 13%).
1H NMR (200 MHz, DMSO-
d6)
δ: 7.57 (t,
J = 7.8 Hz, 1H, H-6), 7.11 (d,
J = 7.0 Hz, 1H, H-5), 7.09 (d,
J = 8.5 Hz, 1H, H-4), 6.83 (s, 2H, NH
2).
13C NMR (50 MHz, DMSO-
d6)
δ: 164.00, 163.92, 148.33, 137.21, 131.47, 122.25, 112.68, 108.01. ES-MS
m/
z for C
8H
5NO
3 [M]
+: calcd. 163.0, found 163.0. A solution of heptanoic acid (263 mg, 2.024 mmol) in oxalyl chloride (1 mL) was stirred for 5 h under Ar. Then, the volatiles were evaporated to dryness under reduced pressure at room temperature. The residue was dissolved in dry dichloromethane (1 mL) and added dropwise to a cooled (0 °C) solution of the anhydride
15 (300 mg, 1.84 mmol) and pyridine (474 μL, 5.52 mmol) in dichloromethane (5 mL) under Ar. The resulting mixture was stirred at r.t. for 48 h. Water (40 mL) was added and the aqueous phase was washed with dichloromethane (2 × 20 mL) and ethyl acetate (3 × 20 mL). The combined ethyl acetate washings were dried (Na
2SO
4), solvent was evaporated and the residue was subjected to column chromatography (0% to 20% methanol/DCM) yielding phthalic acid
14 as off-white solid (17 mg, 3%).
1H NMR (200 MHz, DMSO-
d6)
δ: 11.92 (bs, 1H, NH), 8.40 (d,
J = 8.2, Hz, 1H, H-4), 7.38–7.27 (m, 2H, H-5, H-6), 2.28 (t,
J = 7.3 Hz, 2H, COCH
2), 1.64–1.52 (m, 2H, COCH
2C
H2), 1.33–1.24 (m, 6H), 0.86 (t,
J = 6.3 Hz, 3H, hexyl-CH
3).
13C NMR (50 MHz, DMSO-
d6)
δ: 173.29, 173.20, 172.82, 155.51, 135.85, 128.47, 127.95, 123.88, 123.34, 37.27, 31.31, 28.58, 25.34, 22.18, 12.98. ES-HRMS
m/
z for C
15H
18NO
5 [M − H]
−: calcd. 292.1185, found 292.1188.












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}