
Catalyst-Free Synthesis of Polysubstituted 5-Acylamino-1,3-Thiazoles via Hantzsch Cyclization of α-Chloroglycinates

 ,
, 
Abstract
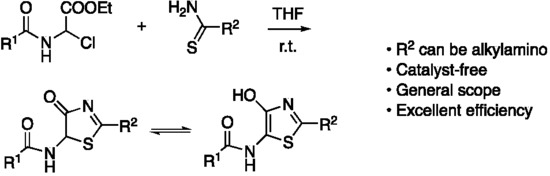
:
1. Introduction
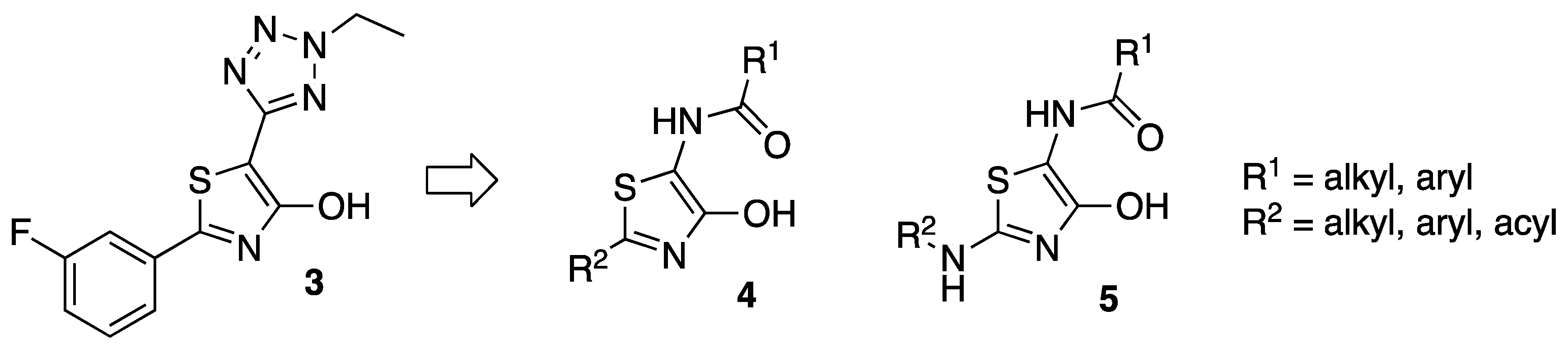
2. Results
3. Materials and Methods
3.1. General
3.2. General Procedure for the Synthesis of α-Hydroxyglycinates (7)
3.3. General Procedure for the Synthesis of α-Chloroglycinates (8)
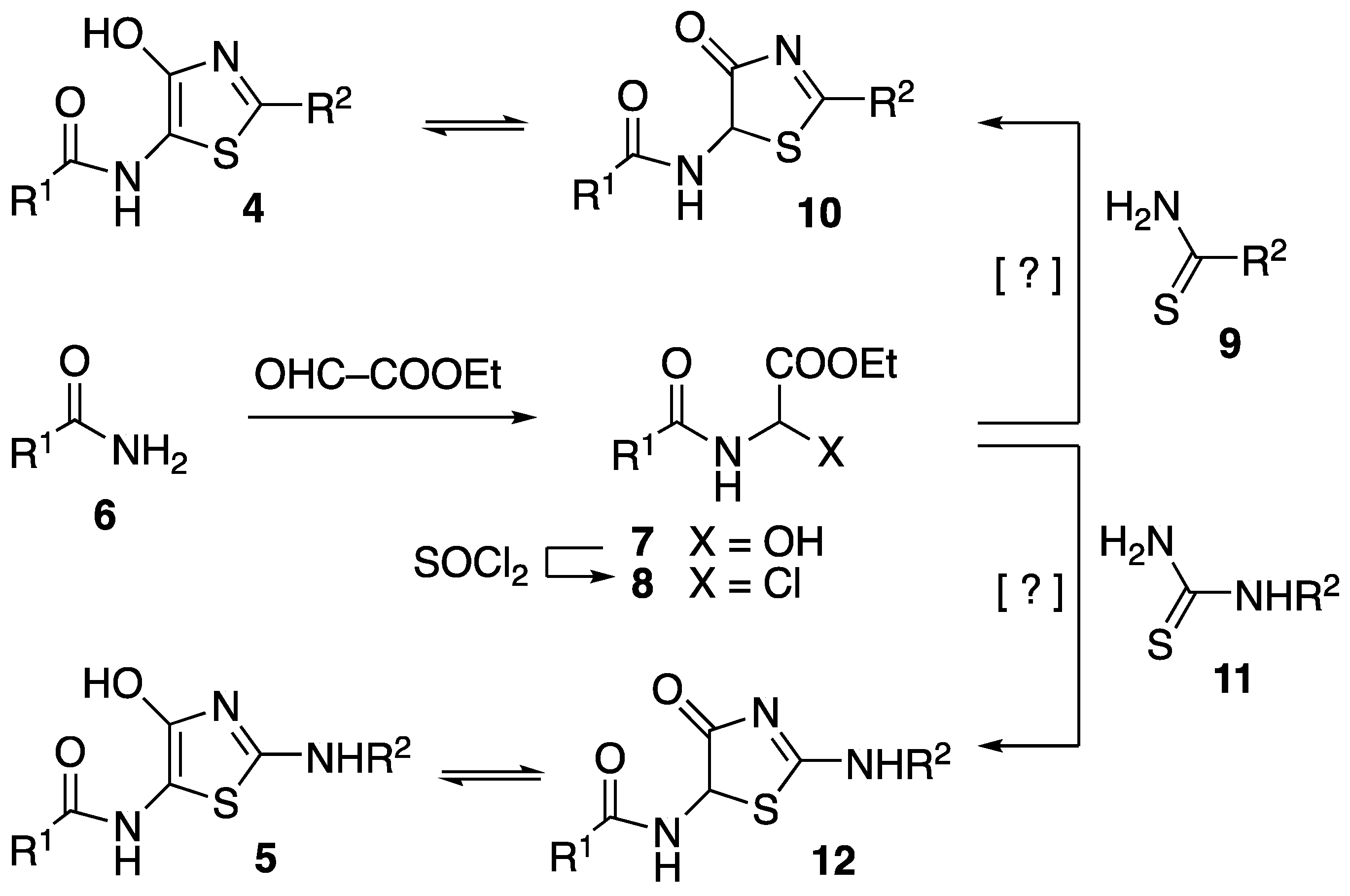
3.4. General Procedure for the Synthesis of 5-Amido-4-Hydroxy Thiazoles 4 and Their Keto Tautomers 10
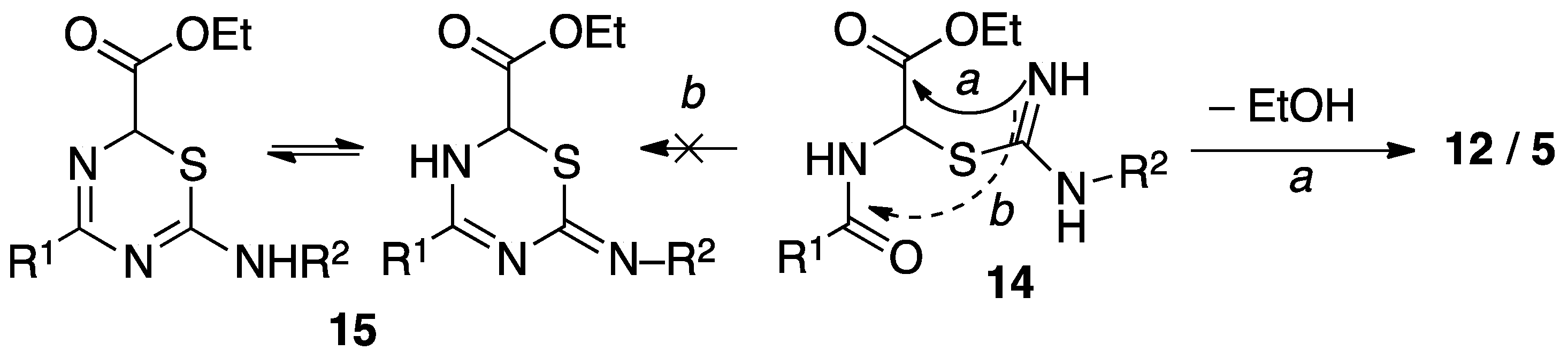
3.5. General Procedure for the Synthesis of 5-Amido-2-Amino Thiazoles 5 and Their Keto Tautomers (12)
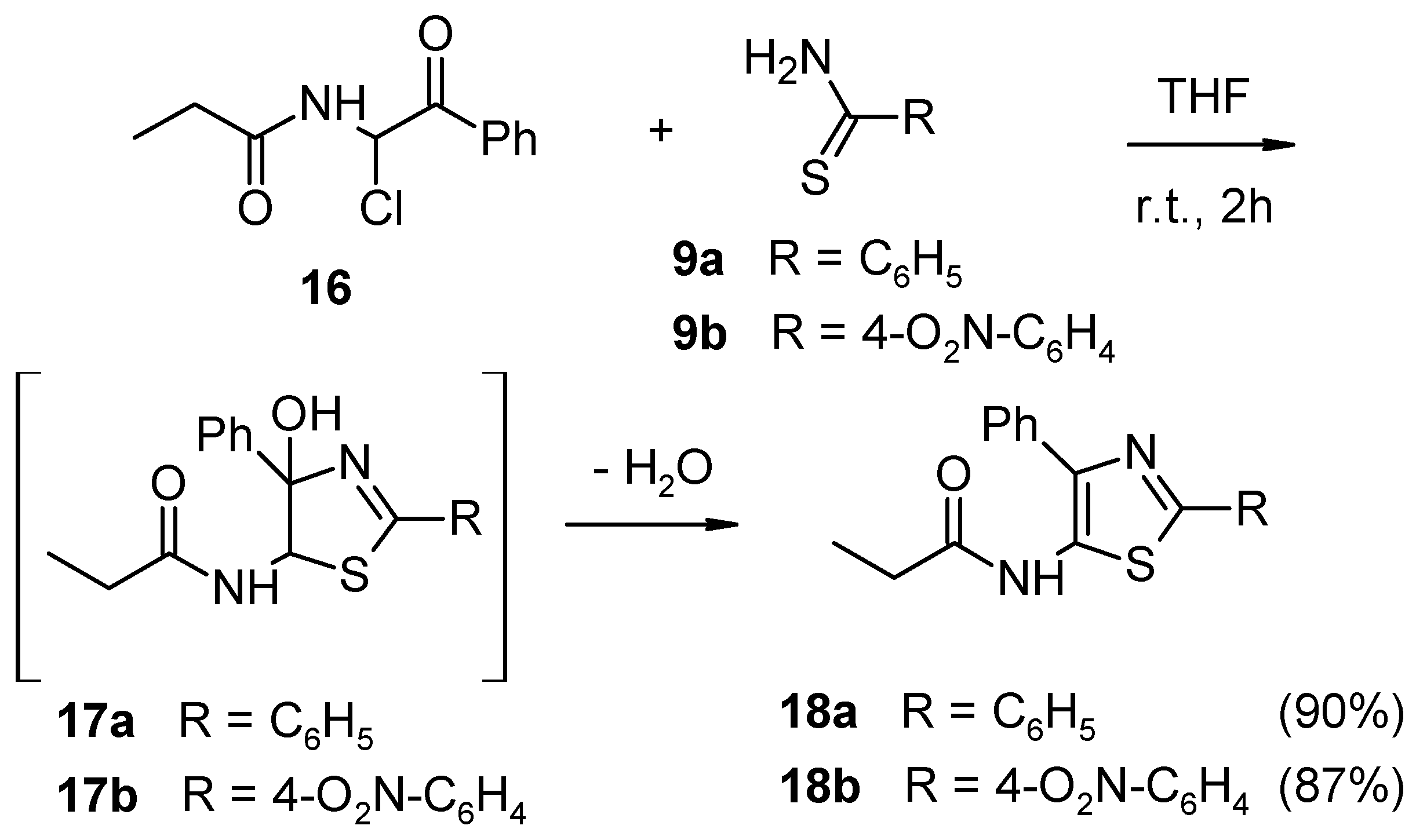
3.6. General Procedure for the Synthesis of 5-Amido-4-Phenyl Thiazoles (18)
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Taylor, A.P.; Robinson, R.P.; Fobian, Y.M.; Blakemore, D.C.; Jones, L.H.; Fadeyi, O. Modern advances in heterocyclic chemistry in drug discovery. Org. Biomol. Chem. 2016, 14, 6611–6637. [Google Scholar] [CrossRef] [PubMed]
- Li, J.J. Heterocyclic Chemistry in Drug Discovery; John Wiley and Sons: Hoboken, NJ, USA, 2013. [Google Scholar]
- Pola, S. Significance of Thiazole-Based Hetrocycles for Bioactive Systems, Scope of Selective Hetrocycles from Organic and Pharmaceutical Perspective; Varala, R., Ed.; InTech: Rijeka, Croatia, 2016. [Google Scholar]
- De Caro, C.; Russo, R.; Avagliano, C.; Cristiano, C.; Calignano, A.; Aramini, A.; Bianchini, G.; Allegretti, M.; Brandolini, L. Antinociceptive effect of two novel transient receptor potential melastatin 8 antagonists in acute and chronic pain models in rat. Brit. J. Pharmac. 2018, 175, 1691–1706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moriconi, A.; Bianchini, G.; Cologioia, S.; Brandolini, L.; Aramini, A.; Liberati, C.; Bovolenta, S. TRPM8 Antagonists. Patent WO092711, June 2013. [Google Scholar]
- Feng, M.; Tang, B.; Liang, S.H.; Jiang, X. Sulfur containing scaffolds in drugs: Synthesis and application in medicinal chemistry. Curr. Top. Med. Chem. 2016, 16, 1200–1216. [Google Scholar] [CrossRef] [PubMed]
- Colella, M.; Musci, P.; Carlucci, C.; Lillini, S.; Tomassetti, M.; Aramini, A.; Degennaro, L.; Luisi, R. 1, 3-Dibromo-1,1-difluoro-2-propanone as a Useful Synthon for a Chemoselective Preparation of 4-Bromodifluoromethyl Thiazoles. ACS Omega 2018, 3, 14841–14848. [Google Scholar] [CrossRef]
- Radhakrishnan, R.; Sreejalekshni, K.G.J. Computational Design, Synthesis, and Structure Property Evaluation of 1, 3-Thiazole-Based Color-Tunable Multi-heterocyclic Small Organic Fluorophores as Multifunctional Molecular Materials. Org. Chem. 2018, 83, 3453–3466. [Google Scholar] [CrossRef]
- Patil, R.V.; Chavan, J.U.; Beldar, A.G. Synthesis of aminothiazoles: Polymer-supported approaches. RSC Adv. 2017, 7, 23765–23778. [Google Scholar] [CrossRef]
- Das, D.; Sidkar, P.; Bairagi, M. Recent developments of 2-aminothiazoles in medicinal chemistry. Eur. J. Med. Chem. 2016, 109, 89–98. [Google Scholar] [CrossRef]
- Yurttas, L.; Ciftci, G.A.; Temel, H.E.; Saglik, B.N.; Demir, B.; Levent, S. Biological Activity Evaluation of Novel 1, 2, 4-Triazine Derivatives Containing Thiazole/Benzothiazole Rings. Anticancer Agents Med. Chem. 2017, 17, 1846–1853. [Google Scholar] [CrossRef]
- Budak, Y.; Kocyigit, U.M.; Gürdere, M.B.; Özcan, K.; Taslimi, P.; Gülçin, I.; Ceylan, M. Synthesis and investigation of antibacterial activities and carbonic anhydrase and acetyl cholinesterase inhibition profiles of novel 4, 5-dihydropyrazol and pyrazolyl-thiazole derivatives containing methanoisoindol-1,3-dion unit. Synth. Commun. 2017, 47, 2313–2323. [Google Scholar] [CrossRef]
- Li, L.; Zhang, C.L.; Song, H.R.; Tan, C.Y.; Ding, H.W.; Jiang, Y.Y. Discovery of novel dual inhibitors of VEGFR and PI3K kinases containing 2-ureidothiazole scaffold. Chin. Chem. Lett. 2016, 27, 1–6. [Google Scholar] [CrossRef]
- Gaikwad, N.D.; Patil, S.V.; Bobade, V.D. Synthesis and biological evaluation of some novel thiazole substituted benzotriazole derivatives. Bioorg. Med. Chem. Lett. 2012, 22, 3449–3459. [Google Scholar] [CrossRef] [PubMed]
- Gallardo-Godoy, A.; Gever, J.; Fife, K.L.; Silber, B.M.; Prusiner, S.B.; Renslo, A.R.J. 2-Aminothiazoles as therapeutic leads for prion diseases. Med. Chem. 2011, 54, 1010–1021. [Google Scholar] [CrossRef] [PubMed]
- Pirotte, B.; Delarge, J.; Coyette, J.; Frere, J.M. Antibacterial activity of 5-acylaminothiazole derivatives, synthetic drugs related to beta-lactam antibiotics. J. Antib. 1991, 44, 844–853. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Ji, J.; Deng, R.; Tang, J.; Yang, F.; Feng, G.K.; Chen, W.D.; Wu, X.Q.; Qian, X.J.; Ding, K.; et al. DC120, a novel AKT inhibitor, preferentially suppresses nasopharyngeal carcinoma cancer stem-like cells by downregulating Sox2. Oncotarget 2015, 6, 6944–6958. [Google Scholar] [CrossRef]
- Chang, S.; Zhang, Z.; Zhuang, X.; Luo, J.; Cao, X.; Li, H.; Tu, Z.; Lu, X.; Ren, X.; Ding, K. New thiazole carboxamides as potent inhibitors of Akt kinases. Bioorg. Med. Chem. Lett. 2012, 22, 1208–1212. [Google Scholar] [CrossRef]
- Abdelazeem, A.H.; Habash, M.; Maghrabi, I.A.; Taha, M.O. Synthesis and evaluation of novel diphenylthiazole derivatives as potential anti-inflammatory agents. Med. Chem. Res. 2015, 24, 3681–3695. [Google Scholar] [CrossRef]
- Shivaprasad, C.M.; Jagadish, S.; Swaroop, T.R.; Ashwini, N.; Harsha, K.B.; Rangappa, K.S. Synthesis, antibacterial, antioxidant and anti-inflammatory activities of new benzimidazole derivatives. Asian J. Biochem. Pharmac. Res. 2014, 4, 316–327. [Google Scholar]
- Giles, K.; Berry, D.B.; Condello, C.; Hawley, R.C.; Gallardo-Godoy, A.; Bryant, C.; Oehler, A.; Elepano, M.; Bhardway, S.; Patel, S.; et al. Different 2-aminothiazole therapeutics produce distinct patterns of scrapie prion neuropathology in mouse brains. Pharmacol. Exp. Ther. 2015, 355, 2–12. [Google Scholar] [CrossRef]
- Larsen, S.D.; Stachew, C.F.; Clare, P.M.; Cubbage, J.W.; Leach, K.L. A catch-and-release strategy for the combinatorial synthesis of 4-acylamino-1,3-thiazoles as potential CDK5 inhibitors. Bioorg. Med. Chem. Lett. 2003, 13, 3491–3495. [Google Scholar] [CrossRef]
- Shih, A.; Nazi, I.; Kelton, J.G.; Arnold, D.M. Novel treatments for immune thrombocytopenia. Press Med. 2014, 43, e87–e95. [Google Scholar] [CrossRef] [Green Version]
- Mistretta, F.A.; Russo, A.; Castiglione, F.; Battiga, A.; Calciago, G.; Montorsi, F.; Brandolini, L.; Bianchini, G.; Aramini, A.; Allegretti, M.; et al. DFL23448, A Novel Transient Receptor Potential Melastin 8–Selective Ion Channel Antagonist, Modifies Bladder Function and Reduces Bladder Overactivity in Awake Rats. Pharmacol. Exp. Ther. 2016, 356, 200–211. [Google Scholar] [CrossRef] [PubMed]
- Baranak-Stojanovic, M.; Klaumunzer, U.; Markovic, R.; Kleinpeter, E. Structure, configuration, conformation and quantification of the push–pull effect of 2-alkylidene-4-thiazolidinones and 2-alkylidene-4, 5-fused bicyclic thiazolidine derivatives. Tetrahedron 2010, 66, 8958–8967. [Google Scholar] [CrossRef]
- Baranac-Stojanovic, M.; Tatar, J.; Kleinpeter, E.; Markovic, R. High-yield synthesis of substituted and unsubstituted pyridinium salts containing a 4-oxothiazolidine moiety. Synthesis 2008, 13, 2117–2121. [Google Scholar] [CrossRef]
- Stadelmann, B.; Scholl, S.; Muller, J.; Hemphill, A.J. Application of an in vitro drug screening assay based on the release of phosphoglucose isomerase to determine the structure–activity relationship of thiazolides against Echinococcus multilocularis metacestodes. Antimicrob. Chemother. 2010, 65, 512–519. [Google Scholar] [CrossRef]
- Esposito, M.; Muller, N.; Hemphill, A. Structure–activity relationships from in vitro efficacies of the thiazolide series against the intracellular apicomplexan protozoan Neospora caninum. Int. J. Parasitol. 2007, 37, 183–190. [Google Scholar] [CrossRef]
- Cimarelli, C.; Bordi, S.; Piermattei, P.; Pellei, M.; Del Bello, F.; Marcantoni, E. An efficient Lewis acid catalyzed Povarov reaction for the one-pot stereocontrolled synthesis of polyfunctionalized tetrahydroquinolines. Synthesis 2017, 49, 5387–5395. [Google Scholar] [CrossRef]
- Cimarelli, C.; Di Nicola, M.; Diomedi, S.; Giovannini, R.; Hamprecht, D.; Properzi, R.; Sorana, F.; Marcantoni, E. An efficient one-pot two catalyst system in the construction of 2-substituted benzimidazoles: Synthesis of benzimidazo [1,2-c] quinazolines. Org. Biomol. Chem. 2015, 13, 11687–11695. [Google Scholar] [CrossRef]
- Properzi, R.; Marcantoni, E. Construction of heterocyclic structures by trivalent cerium salts promoted bond forming reactions. Chem. Soc. Rev. 2014, 43, 779–791. [Google Scholar] [CrossRef]
- Rehm, F.B.H.; Jackson, M.A.; De Geyter, E.; Yap, K.; Gilding, E.K.; Durek, T.; Craik, D.J. Papain-like cysteine proteases prepare plant cyclic peptide precursors for cyclization. PNAS 2019, 116, 7831–7836. [Google Scholar] [CrossRef] [Green Version]
- Ambhaikar, N.B. Thiazoles and Benzothiazoles. In Heterocyclic Chemistry in Drug Discovery; Li, J.K., Ed.; Wiley: Hoboken, NJ, USA, 2013; pp. 283–322. [Google Scholar]
- Metzger, J.V. Thiazoles and their Benzo Derivatives. In Comprehensive Heterocyclic Chemistry; Katritzky, A.R., Rees, C.W., Eds.; Pergamon Press: Oxford, UK, 1984; Volume 6, pp. 235–331. [Google Scholar]
- Rajer, V.N.; Swaroop, T.R.; Anil, S.M.; Bommegowda, Y.K.; Rangappa, K.S.; Sadashiva, M.P. Base-Induced Cyclization of Active Methylene Isocyanides with Xanthate Esters: An Efficient Method for the Synthesis of 5-Alkoxy-4-(tosyl/ethoxycarbonyl)-1,3-thiazoles. Synlett 2017, 28, 2281–2284. [Google Scholar]
- Hantzsch, A.; Weber, J.H. Ueber verbindungen des thiazols (pyridins der thiophenreihe). Ber. Dtsch. Chem. Ges. 1887, 20, 3118. [Google Scholar] [CrossRef]
- Merritt, E.A.; Bagley, M.C. Holzapfel-Meyers-Nicolaou Modification of the Hantzsch Thiazole Synthesis. Synthesis 2007, 22, 3535–3541. [Google Scholar]
- Facchinetti, V.; Avellar, M.N.; Nery, A.C.S.; Gomes, C.R.B.; Vasconcelos, T.R.A.; de Souza, M.V.N. An Eco-friendly, Hantzsch-Based, Solvent-Free Approach to 2-Aminothiazoles and 2-Aminoselenazoles. Synthesis 2016, 48, 437–440. [Google Scholar]
- Ben-Ishai, D.; Sataty, I.; Bernstein, Z. A new synthesis of n-acyl aromatic α-amino acids—Amidoalkylation of aromatic and heterocyclic compounds with glyoxylic acid derivatives. Tetrahedron 1976, 32, 1571–1573. [Google Scholar] [CrossRef]
- Zoller, U.; Ben-Ishai, D. Amidoalkylation of mercaptans with glyoxylic acid derivatives. Tetrahedron 1975, 31, 863–866. [Google Scholar] [CrossRef]
- Bernstein, Z.; Ben-Ishai, D. Synthesis of N-substituted aziridine-2-carboxylates. Tetrahedron 1977, 33, 881–883. [Google Scholar] [CrossRef]
- Samantha, S.S.; Roche, S.P.J. In Situ-Generated Glycinyl Chloroaminals for a One-Pot Synthesis of Non-proteinogenic α-Amino Esters. Org. Chem. 2017, 82, 8514–8526. [Google Scholar] [CrossRef]
- Zhang, J.; Coqueron, P.Y.; Ciufolini, M.A. Development and applications of an oxazole-forming reaction. Heterocycles 2011, 82, 949–980. [Google Scholar]
- Zhang, J.; Ciufolini, M.A. An approach to the bis-oxazole macrocycle of diazonamides. Org. Lett. 2011, 13, 390–393. [Google Scholar] [CrossRef]
- Zhang, J.; Coqueron, P.Y.; Vors, J.P.; Ciufolini, M.A. Synthesis of 5-Amino-Oxazole-4-Carboxylates from α-Chloroglycinates. Org. Lett. 2010, 12, 3942–3945. [Google Scholar] [CrossRef]
- Zhang, J.; Polishchuk, E.A.; Chen, J.; Ciufolini, M.A.J. Development of an oxazole conjunctive reagent and application to the total synthesis of siphonazoles. Org. Chem. 2009, 74, 9140–9151. [Google Scholar] [CrossRef] [PubMed]
- Coqueron, P.Y.; Didier, C.; Ciufolini, M.A. Iterative Oxazole Assembly via α-Chloroglycinates: Total Synthesis of (−)-Muscoride A. Angew. Chem. Int. Ed. 2003, 42, 1411–1414. [Google Scholar] [CrossRef] [PubMed]
- Raczynska, E.D.; Kosinska, W.; Osmialowski, B.; Gawinecki, R. Tautomeric equilibria in relation to pi-electron delocalization. Chem. Rev. 2005, 105, 3561–3621. [Google Scholar]
- Di Deo, M.; Bartoli, G.; Bellucci, M.C.; Bosco, M.; Marcantoni, E.; Sambri, L.; Torregiani, E.J. A Simple, Efficient, and General Method for the Conversion of Alcohols into Alkyl Iodides by a CeCl3 × 7H2O/NaI System in Acetonitrile. Org. Chem. 2000, 65, 2830–2833. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Andersen, K.K. On the Tautomerism of 2,4-Disubstituted Thiazolones. Eur. J. Org. Chem. 2002, 3, 557–563. [Google Scholar] [CrossRef]
- Billi, K.; Cosimelli, B.; Leoni, A.; Spinelli, D.J. Ring-ring interconversions. Part 3†. On the effect of the substituents on the thiazole moiety in the ring-opening/ring-closing reactions of nitrosoimidazo[2,1-b][1,3]thiazoles with hydrochloric acid. J. Heterocycl. Chem. 2000, 37, 875–878. [Google Scholar] [CrossRef]
- Pil’o, S.G.; Brovarets, V.S.; Vinogradova, T.K.; Golovchenko, A.V.; Drach, B.S. Synthesis of new 5-mercapto-1,3-oxazole derivatives on the basis of 2-acylamino-3,3-dichloroacrylonitriles and their analogs. Russ. J. Gen. Chem. 2002, 72, 1714–1723. [Google Scholar] [CrossRef]
- Ishida, T.; Hirata, F.; Sato, H.; Kato, S.J. Molecular Theory of Solvent Effect on Keto−Enol Tautomers of Formamide in Aprotic Solvents: RISM-SCF Approach. Phys. Chem. B 1998, 102, 2045–2050. [Google Scholar] [CrossRef]
- Dudding, T.; Hafez, A.M.; Taggi, A.E.; Wagerle, T.R.; Lectka, T. A catalyst that plays multiple roles: Asymmetric synthesis of β-substituted aspartic acid derivatives through a four-stage, one-pot procedure. Org. Lett. 2002, 4, 387–390. [Google Scholar] [CrossRef]
- Eberlin, M.N. Electrospray ionization mass spectrometry: A major tool to investigate reaction mechanisms in both solution and the gas phase. Eur. J. Mass Spectrom. 2007, 13, 19–28. [Google Scholar] [CrossRef]
- Silva Santos, L.; Knaack, L.; Metzger, J.O. Investigation of chemical reactions in solution using API-MS. Int. J. Mass Spectrom. 2005, 246, 84–104. [Google Scholar] [CrossRef]
- Chau, J.; Zhang, J.; Ciufolini, M.A. A Peterson avenue to 5-alkenyloxazoles. Tetrahedron Lett. 2009, 50, 6163–6165. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are available from the authors, compounds (7a–d) and 7b, 7c, 7d, 4aa, 4ac, 4ad, 4bb, 4bc, 4cb, 12 ab, 12cd. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||
|---|---|---|---|---|
| Entry | R1 | R2 | Product (10 + 4) b | Yield (%) c |
| 1 | Ph (8a) | Ph (9a) | 10aa + 4aa d | 88 |
| 2 | Piperonyl (8b) | Ph (9a) | 10ab + 4ab d | 76 |
| 3 | Et (8c) | Ph (9a) | 10ac + 4ac d | 94 |
| 4 | PhCH=CH (8d) | Ph (9a) | 10ad + 4ad d | 81 |
| 5 | Ph (8a) | 4-NO2-C6H4 (9b) | 10ba + 4ba d | 74 |
| 6 | Piperonyl (8b) | 4-NO2-C6H4 (9b) | 10bb + 4bb d | 87 |
| 7 | Et (8c) | 4-NO2-C6H4 (9b) | 10bc + 4bc d | 94 |
| 8 | Ph (8a) | 4-MeO-C6H4 (9c) | 10ca + 4ca | 94 |
| 9 | Piperonyl (8b) | 4-MeO-C6H4 (9c) | 10cb + 4cb d | 68 |
| 10 | Ph (8a) | 4-Cl-C6H4 (9d) | 10da + 4da d | 78 |
| 11 | Piperonyl (8b) | 4-Cl-C6H4 (9d) | 10db + 4db d | 95 |
| 12 | Et (8c) | 4-Cl-C6H4 (9d) | 10dc + 4dc d | 90 |
 | ||||
|---|---|---|---|---|
| Entry | R1 | R2 | Product (12 + 5) b | Yield (%) c |
| 1 | Ph (8a) | H (11a) | 12aa d + 5aa | 65 |
| 2 | Ph (8a) | 4-NO2-C6H4 (11b) | 12ab d + 5ab | 96 |
| 3 | Ph (8a) | 4-CH3O-C6H4 (11c) | 12ac d + 5ac | 97 |
| 4 | Ph (8a) | 4-CH3CO-C6H4 (11d) | 12ad d + 5ad | 77 |
| 5 | Ph (8a) | CH3CO (11e) | 12ae d + 5ae | 62 |
| 6 | Piperonyl (8b) | 4-NO2-C6H4 (11b) | 12bb + 5bb d | 75 |
| 7 | Piperonyl (8b) | 4-CH3CO-C6H4 (11d) | 12bd + 5bd d | 76 |
| 8 | Et (8c) | 4-NO2-C6H4 (11b) | 12cb d + 5cb | 81 |
| 9 | Et (8c) | 4-CH3CO-C6H4 (11d) | 12cd d + 5cd | 80 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tomassetti, M.; Lupidi, G.; Piermattei, P.; Rossi, F.V.; Lillini, S.; Bianchini, G.; Aramini, A.; Ciufolini, M.A.; Marcantoni, E. Catalyst-Free Synthesis of Polysubstituted 5-Acylamino-1,3-Thiazoles via Hantzsch Cyclization of α-Chloroglycinates. Molecules 2019, 24, 3846. https://doi.org/10.3390/molecules24213846
Tomassetti M, Lupidi G, Piermattei P, Rossi FV, Lillini S, Bianchini G, Aramini A, Ciufolini MA, Marcantoni E. Catalyst-Free Synthesis of Polysubstituted 5-Acylamino-1,3-Thiazoles via Hantzsch Cyclization of α-Chloroglycinates. Molecules. 2019; 24(21):3846. https://doi.org/10.3390/molecules24213846
Chicago/Turabian StyleTomassetti, Mara, Gabriele Lupidi, Pamela Piermattei, Federico V. Rossi, Samuele Lillini, Gianluca Bianchini, Andrea Aramini, Marco A. Ciufolini, and Enrico Marcantoni. 2019. "Catalyst-Free Synthesis of Polysubstituted 5-Acylamino-1,3-Thiazoles via Hantzsch Cyclization of α-Chloroglycinates" Molecules 24, no. 21: 3846. https://doi.org/10.3390/molecules24213846