
Evaluation of Cyclic Peptide Inhibitors of the Grb7 Breast Cancer Target: Small Change in Cargo Results in Large Change in Cellular Activity

 , , and
, , and 
Abstract
:1. Introduction
2. Results
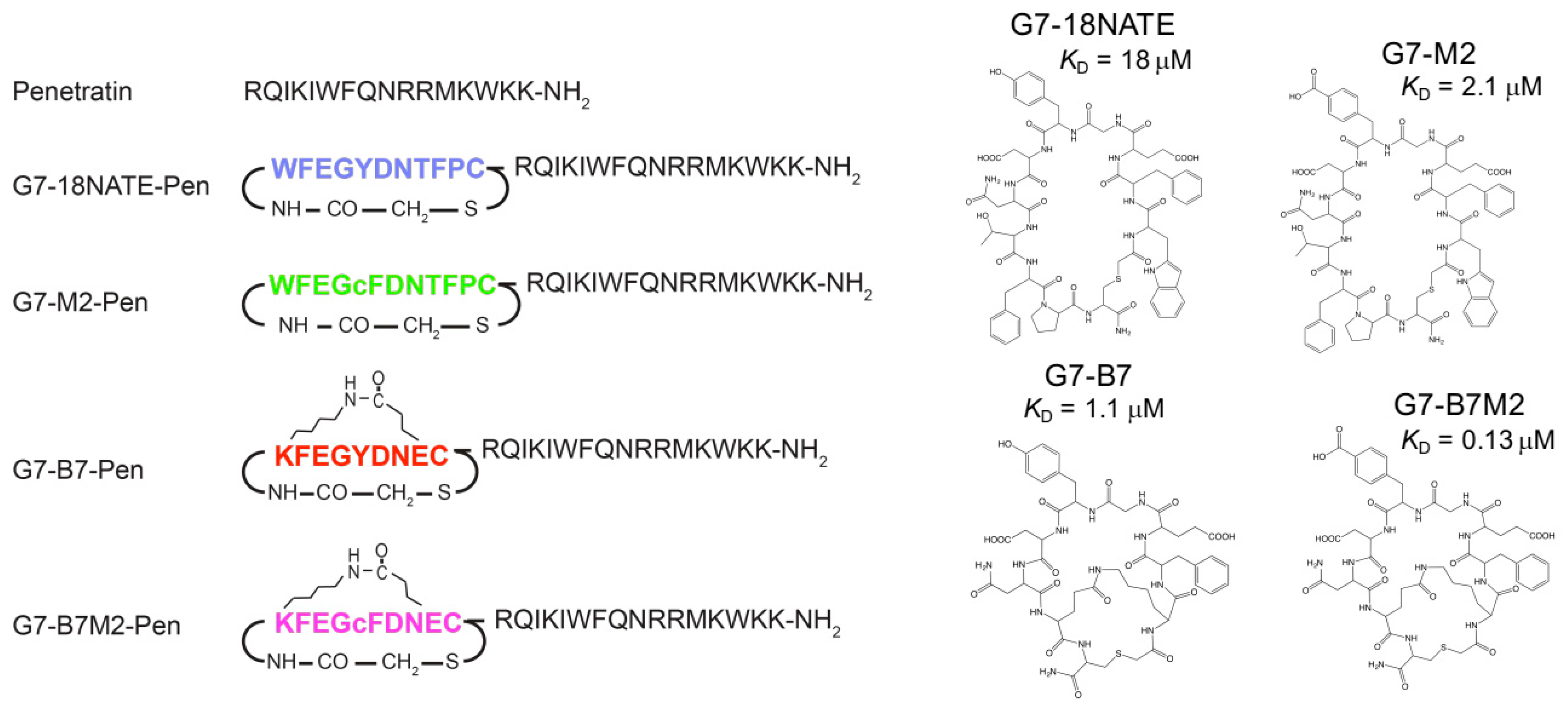
2.1. Preparation of G7-Peptides
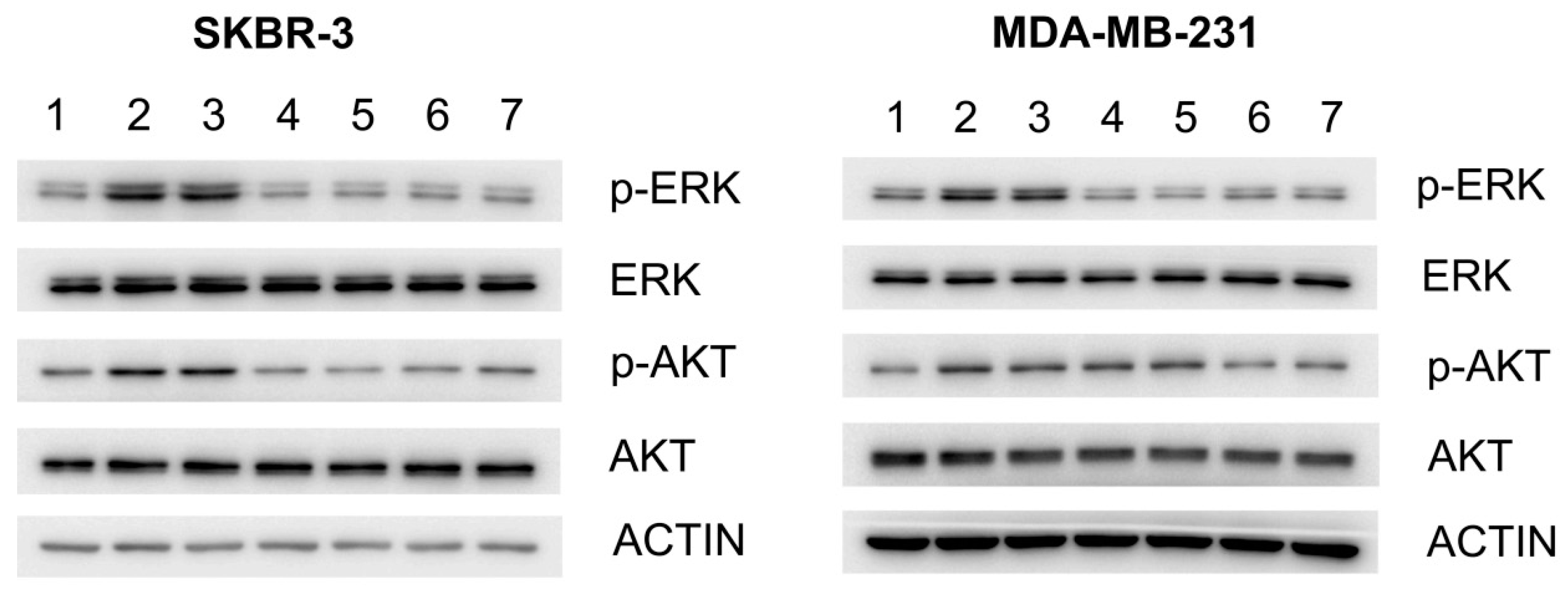
2.2. Effect of G7-Peptides on Signaling
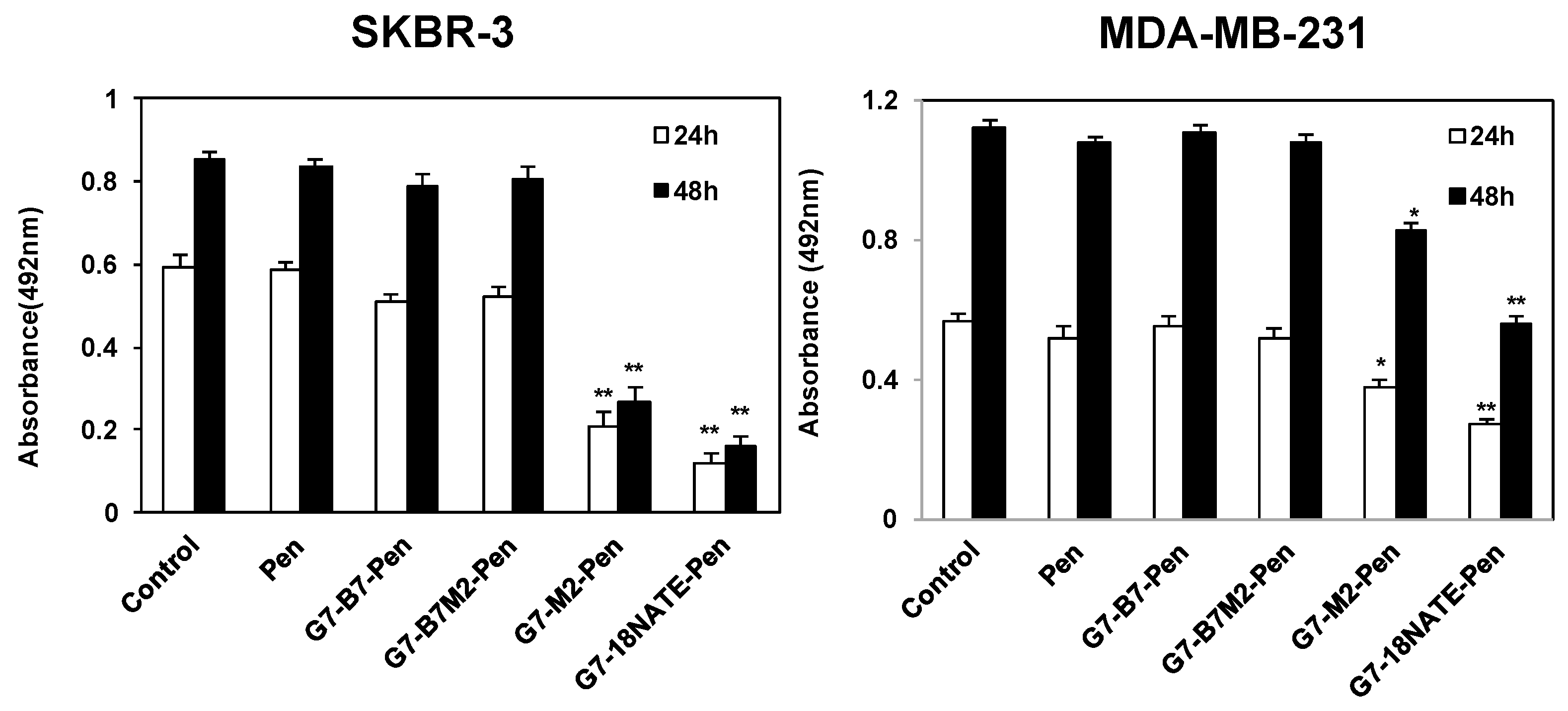
2.3. Effect of G7-Peptides on Cell Proliferation
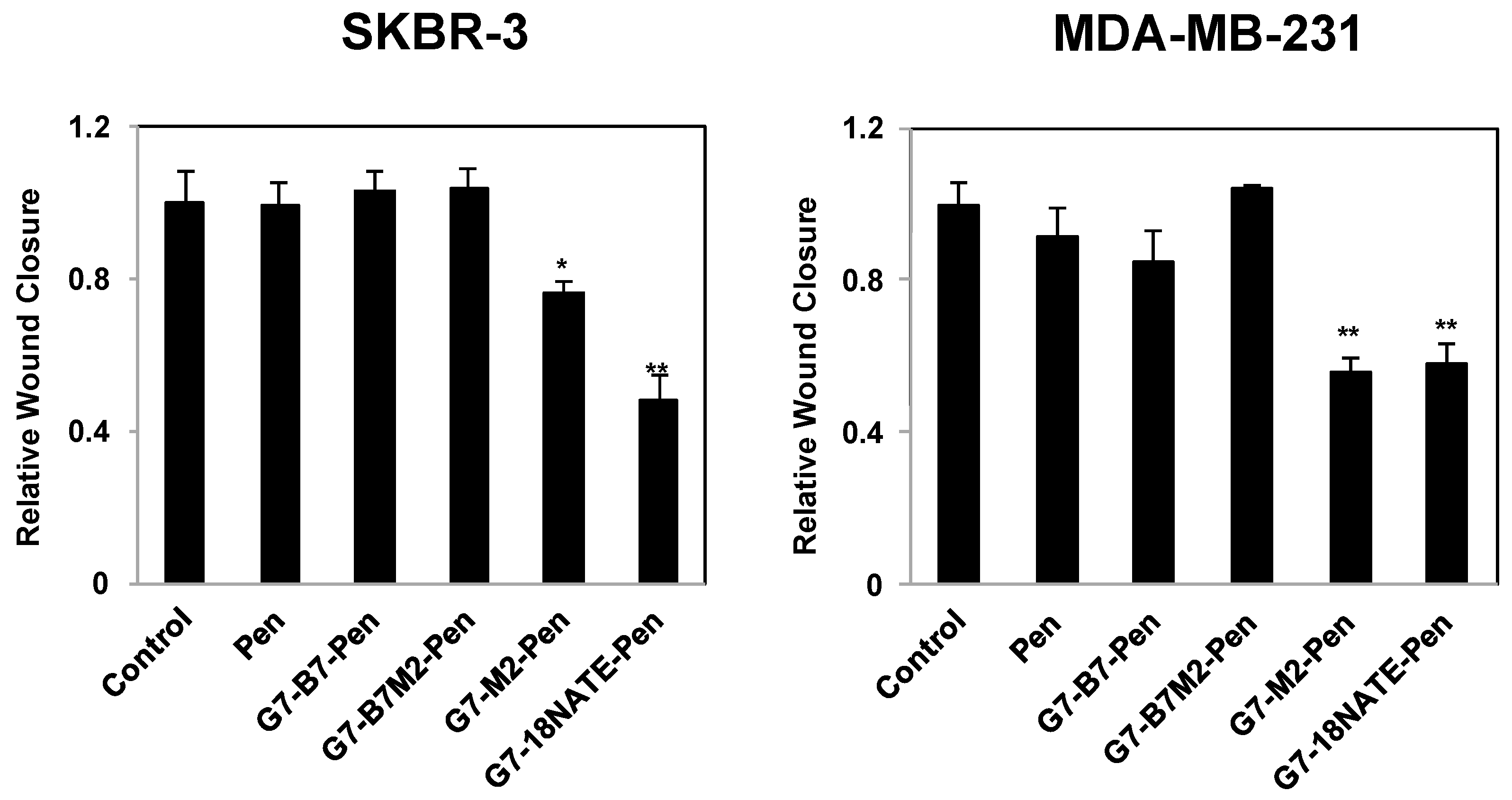
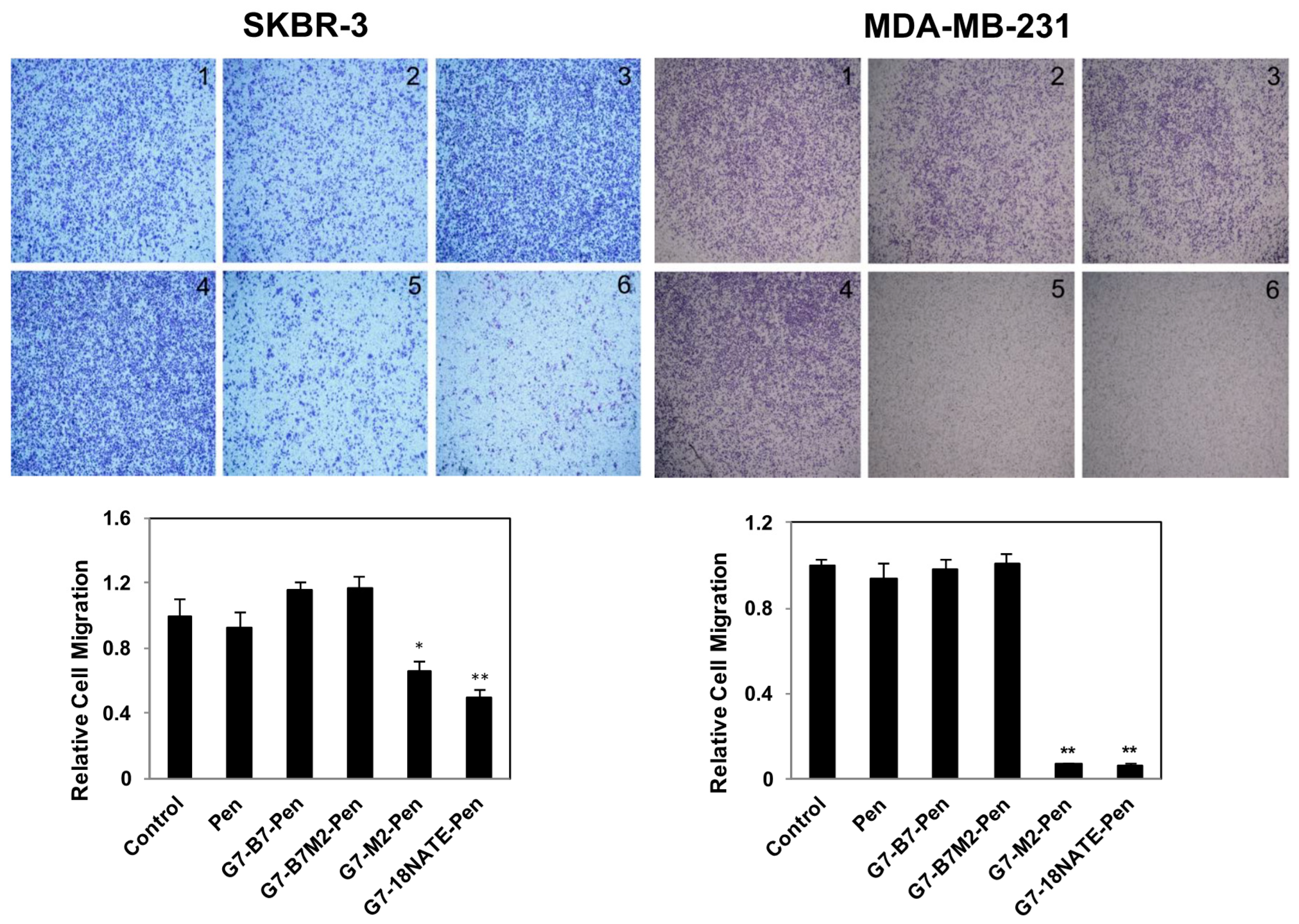
2.4. Effect of G7-Peptides on Cell Migration
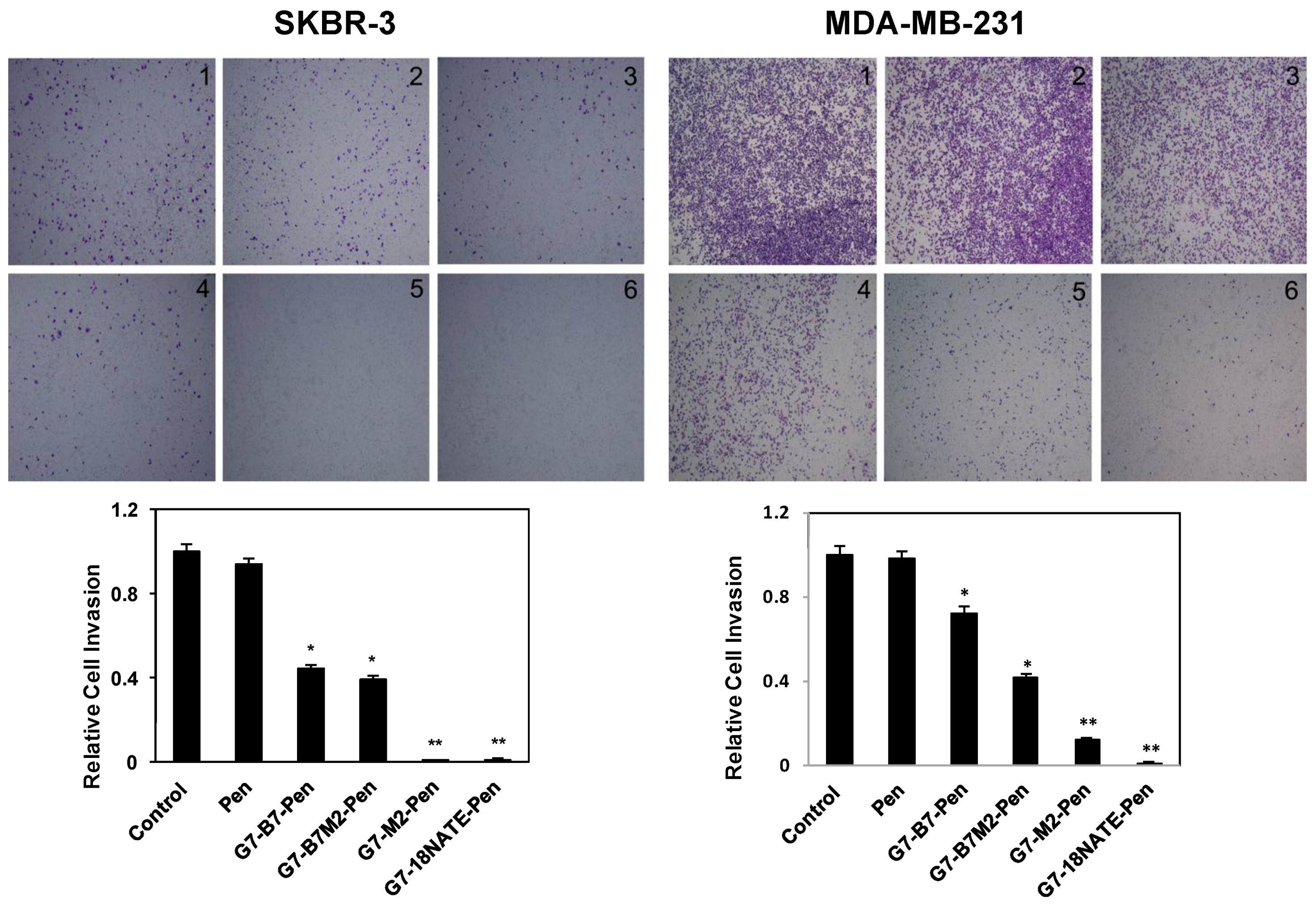
2.5. Effect of G7-Peptides on Invasion
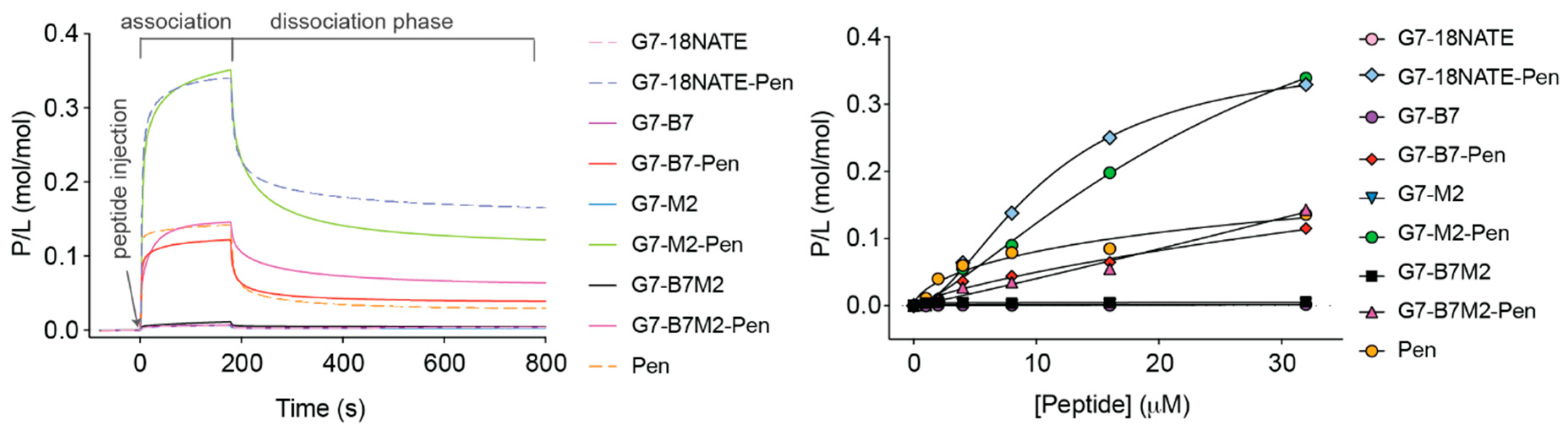
2.6. Potential Differential Effect of the Penetratin Cell Permeability Tag
2.7. Investigation of G7-Peptide Cellular Uptake
3. Discussion
4. Materials and Methods
4.1. Cell Line and Reagents
4.2. Western Blotting
4.3. Cell Proliferation Assay
4.4. Wound Healing Assay
4.5. Transwell Motility Assay
4.6. Transwell Invasion Assay
4.7. ITC Experiments
4.8. SPR Measurement of Lipid Bilayer Interactions
4.9. Internalization of Peptide Inside Cells Quantified Using Mass Spectrometry
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Grb7 | growth receptor bound protein |
| SH2 domain | Src homology domain 2 |
| TNBC | triple negative breast cancer |
| cmF | carboxymethylphenylalanine |
| cF | carboxyphenylalanine |
| pY | phosphotyrosine |
| CPP | cell penetrating peptide |
| SPR | surface plasmon resonance |
| ITC | Isothermal titration calorimetry |
References
- Siveen, K.S.; Prabhu, K.S.; Achkar, I.W.; Kuttikrishnan, S.; Shyam, S.; Khan, A.Q.; Merhi, M.; Dermime, S.; Uddin, S. Role of Non Receptor Tyrosine Kinases in Hematological Malignances and its Targeting by Natural Products. Mol. Cancer 2018, 17, 31. [Google Scholar] [CrossRef] [PubMed]
- Montor, W.R.; Salas, A.; Melo, F.H.M. Receptor tyrosine kinases and downstream pathways as druggable targets for cancer treatment: The current arsenal of inhibitors. Mol. Cancer 2018, 17, 55. [Google Scholar] [CrossRef] [PubMed]
- Rimawi, M.F.; Schiff, R.; Osborne, C.K. Targeting HER2 for the treatment of breast cancer. Annu. Rev. Med. 2015, 66, 111–128. [Google Scholar] [CrossRef] [PubMed]
- Lucas-Fernandez, E.; Garcia-Palmero, I.; Villalobo, A. Genomic organization and control of the Grb7 gene family. Curr. Genomics 2008, 9, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Han, D.C.; Guan, J.L. Association of focal adhesion kinase with Grb7 and its role in cell migration. J. Biol Chem. 1999, 274, 24425–24430. [Google Scholar] [CrossRef] [PubMed]
- Han, D.C.; Shen, T.L.; Guan, J.L. Role of Grb7 targeting to focal contacts and its phosphorylation by focal adhesion kinase in regulation of cell migration. J. Biol. Chem. 2000, 275, 28911–28917. [Google Scholar] [CrossRef]
- Stein, D.; Wu, J.; Fuqua, S.A.; Roonprapunt, C.; Yajnik, V.; D′Eustachio, P.; Moskow, J.J.; Buchberg, A.M.; Osborne, C.K.; Margolis, B. The SH2 domain protein GRB-7 is co-amplified, overexpressed and in a tight complex with HER2 in breast cancer. EMBO J. 1994, 13, 1331–1340. [Google Scholar] [CrossRef]
- Nadler, Y.; Gonzalez, A.M.; Camp, R.L.; Rimm, D.L.; Kluger, H.M.; Kluger, Y. Growth factor receptor-bound protein-7 (Grb7) as a prognostic marker and therapeutic target in breast cancer. Ann. Oncol. 2010, 21, 466–473. [Google Scholar] [CrossRef]
- Ramsey, B.; Bai, T.; Hanlon Newell, A.; Troxell, M.; Park, B.; Olson, S.; Keenan, E.; Luoh, S.W. GRB7 protein over-expression and clinical outcome in breast cancer. Breast Cancer Res. Treat. 2011, 127, 659–669. [Google Scholar] [CrossRef]
- Bai, T.; Luoh, S.W. GRB-7 facilitates HER-2/Neu-mediated signal transduction and tumor formation. Carcinogenesis 2007, 29, 473–479. [Google Scholar] [CrossRef] [Green Version]
- Chan, D.W.; Hui, W.W.; Cai, P.C.; Liu, M.X.; Yung, M.M.; Mak, C.S.; Leung, T.H.; Chan, K.K.; Ngan, H.Y. Targeting GRB7/ERK/FOXM1 signaling pathway impairs aggressiveness of ovarian cancer cells. PLoS ONE 2012, 7, e52578. [Google Scholar] [CrossRef] [PubMed]
- Pero, S.C.; Daly, R.J.; Krag, D.N. Grb7-based molecular therapeutics in cancer. Expert Rev. Mol. Med. 2003, 5, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Chu, P.Y.; Tai, Y.L.; Shen, T.L. Grb7, a Critical Mediator of EGFR/ErbB Signaling, in Cancer Development and as a Potential Therapeutic Target. Cells 2019, 8, 435. [Google Scholar] [CrossRef] [PubMed]
- Shen, T.L.; Guan, J.L. Grb7 in intracellular signaling and its role in cell regulation. Front. Biosci. 2004, 9, 192–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, D.C.; Shen, T.L.; Guan, J.L. The Grb7 family proteins: Structure, interactions with other signaling molecules and potential cellular functions. Oncogene 2001, 20, 6315–6321. [Google Scholar] [CrossRef] [PubMed]
- Fiddes, R.J.; Campbell, D.H.; Janes, P.W.; Sivertsen, S.P.; Sasaki, H.; Wallasch, C.; Daly, R.J. Analysis of Grb7 recruitment by heregulin-activated erbB receptors reveals a novel target selectivity for erbB3. J. Biol. Chem. 1998, 273, 7717–7724. [Google Scholar] [CrossRef] [PubMed]
- Machida, K.; Mayer, B.J. The SH2 domain: Versatile signaling module and pharmaceutical target. Biochim. Biophys. Acta 2005, 1747, 1–25. [Google Scholar] [CrossRef]
- Morlacchi, P.; Robertson, F.M.; Klostergaard, J.; McMurray, J.S. Targeting SH2 domains in breast cancer. Future Med. Chem. 2014, 6, 1909–1926. [Google Scholar] [CrossRef] [Green Version]
- Pero, S.C.; Oligno, L.; Daly, R.J.; Soden, A.L.; Liu, C.; Roller, P.P.; Li, P.; Krag, D.N. Identification of novel non-phosphorylated ligands, which bind selectively to the SH2 domain of Grb7. J. Biol. Chem. 2002, 277, 11918–11926. [Google Scholar] [CrossRef]
- Gunzburg, M.J.; Ambaye, N.D.; Hertzog, J.T.; Borgo, M.P.; Pero, S.C.; Krag, D.N.; Wilce, M.C.J.; Aguilar, M.I.; Perlmutter, P.; Wilce, J.A. Use of SPR to Study the Interaction of G7-18NATE Peptide with the Grb7-SH2 Domain. Int. J. Pept. Res. Ther. 2010, 16, 177–184. [Google Scholar] [CrossRef]
- Gunzburg, M.J.; Ambaye, N.D.; Del Borgo, M.P.; Pero, S.C.; Krag, D.N.; Wilce, M.C.; Wilce, J.A. Interaction of the non-phosphorylated peptide G7-18NATE with Grb7-SH2 domain requires phosphate for enhanced affinity and specificity. J. Mol. Recognit. 2012, 25, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Watson, G.M.; Lucas, W.A.H.; Gunzburg, M.J.; Wilce, J.A. Insight into the Selectivity of the G7-18NATE Inhibitor Peptide for the Grb7-SH2 Domain Target. Front Mol. Biosci. 2017, 4, 64. [Google Scholar] [CrossRef] [PubMed]
- Deshayes, S.; Morris, M.C.; Divita, G.; Heitz, F. Cell-penetrating peptides: Tools for intracellular delivery of therapeutics. Cell Mol. Life Sci. 2005, 62, 1839–1849. [Google Scholar] [CrossRef] [PubMed]
- Pradip, D.; Bouzyk, M.; Dey, N.; Leyland-Jones, B. Dissecting GRB7-mediated signals for proliferation and migration in HER2 overexpressing breast tumor cells: GTP-ase rules. Am. J. Cancer Res. 2013, 3, 173–195. [Google Scholar]
- Giricz, O.; Calvo, V.; Pero, S.C.; Krag, D.N.; Sparano, J.A.; Kenny, P.A. GRB7 is required for triple-negative breast cancer cell invasion and survival. Breast Cancer Res. Treat. 2012, 133, 607–615. [Google Scholar] [CrossRef]
- Pero, S.C.; Shukla, G.S.; Cookson, M.M.; Flemer, S., Jr.; Krag, D.N. Combination treatment with Grb7 peptide and Doxorubicin or Trastuzumab (Herceptin) results in cooperative cell growth inhibition in breast cancer cells. Br. J. Cancer 2007, 96, 1520–1525. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, S.; Pero, S.C.; Taguchi, K.; Shimada, M.; Mori, M.; Krag, D.N.; Arii, S. Specific peptide ligand for Grb7 signal transduction protein and pancreatic cancer metastasis. J. Natl. Cancer Inst. 2006, 98, 491–498. [Google Scholar] [CrossRef]
- Ambaye, N.D.; Pero, S.C.; Gunzburg, M.J.; Yap, M.; Clayton, D.J.; Del Borgo, M.P.; Perlmutter, P.; Aguilar, M.I.; Shukla, G.S.; Peletskaya, E.; et al. Structural basis of binding by cyclic nonphosphorylated peptide antagonists of Grb7 implicated in breast cancer progression. J. Mol. Biol. 2011, 412, 397–411. [Google Scholar] [CrossRef]
- Watson, G.M.; Gunzburg, M.J.; Ambaye, N.D.; Lucas, W.A.; Traore, D.A.; Kulkarni, K.; Cergol, K.M.; Payne, R.J.; Panjikar, S.; Pero, S.C.; et al. Cyclic peptides incorporating phosphotyrosine mimetics as potent and specific inhibitors of the Grb7 breast cancer target. J. Med. Chem. 2015, 58, 7707–7718. [Google Scholar] [CrossRef]
- Gunzburg, M.J.; Ambaye, N.D.; Del Borgo, M.P.; Perlmutter, P.; Wilce, J.A. Design and testing of bicyclic inhibitors of Grb7—Are two cycles better than one? J. Pept. Sci. 2013, 100, 543–549. [Google Scholar] [CrossRef]
- Gunzburg, M.J.; Kulkarni, K.; Watson, G.M.; Ambaye, N.D.; Del Borgo, M.P.; Brandt, R.; Pero, S.C.; Perlmutter, P.; Wilce, M.C.; Wilce, J.A. Unexpected involvement of staple leads to redesign of selective bicyclic peptide inhibitor of Grb7. Sci. Rep. 2016, 6, 27060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watson, G.M.; Kulkarni, K.; Sang, J.; Ma, X.; Gunzburg, M.J.; Perlmutter, P.; Wilce, M.C.J.; Wilce, J.A. Discovery, Development, and Cellular Delivery of Potent and Selective Bicyclic Peptide Inhibitors of Grb7 Cancer Target. J. Med. Chem. 2017, 60, 9349–9359. [Google Scholar] [CrossRef] [PubMed]
- Lim, R.C.; Price, J.T.; Wilce, J.A. Context-dependent role of Grb7 in HER2+ve and triple-negative breast cancer cell lines. Breast Cancer Res. Treat. 2014, 143, 593–603. [Google Scholar] [CrossRef] [PubMed]
- Kalafatovic, D.; Giralt, E. Cell-Penetrating Peptides: Design Strategies beyond Primary Structure and Amphipathicity. Molecules 2017, 22, 1929. [Google Scholar] [CrossRef] [PubMed]
- Maiolo, J.R.; Ferrer, M.; Ottinger, E.A. Effects of cargo molecules on the cellular uptake of arginine-rich cell-penetrating peptides. Biochim. Biophys. Acta 2005, 1712, 161–172. [Google Scholar] [CrossRef] [Green Version]
- Oba, M.; Kato, T.; Furukawa, K.; Tanaka, M. A Cell-Penetrating Peptide with a Guanidinylethyl Amine Structure Directed to Gene Delivery. Sci. Rep. 2016, 6, 19913. [Google Scholar] [CrossRef] [Green Version]
- Watson, G.M.; Kulkarni, K.; Brandt, R.; Del Borgo, M.P.; Aguilar, M.I.; Wilce, J.A. Shortened Penetratin Cell-Penetrating Peptide Is Insufficient for Cytosolic Delivery of a Grb7 Targeting Peptide. ACS Omega 2017, 2, 670–677. [Google Scholar] [CrossRef] [Green Version]
- Hedegaard, S.F.; Derbas, M.S.; Lind, T.K.; Kasimova, M.R.; Christensen, M.V.; Michaelsen, M.H.; Campbell, R.A.; Jorgensen, L.; Franzyk, H.; Cardenas, M.; et al. Fluorophore labeling of a cell-penetrating peptide significantly alters the mode and degree of biomembrane interaction. Sci. Rep. 2018, 8, 6327. [Google Scholar] [CrossRef]
- Birch, D.; Christensen, M.V.; Staerk, D.; Franzyk, H.; Nielsen, H.M. Fluorophore labeling of a cell-penetrating peptide induces differential effects on its cellular distribution and affects cell viability. Biochim. Biophys. Acta 2017, 1859, 2483–2494. [Google Scholar] [CrossRef]
- Kauffman, W.B.; Fuselier, T.; He, J.; Wimley, W.C. Mechanism Matters: A Taxonomy of Cell Penetrating Peptides. Trends Biochem. Sci. 2015, 40, 749–764. [Google Scholar] [CrossRef] [Green Version]
- Henriques, S.T.; Huang, Y.H.; Chaousis, S.; Sani, M.A.; Poth, A.G.; Separovic, F.; Craik, D.J. The Prototypic Cyclotide Kalata B1 Has a Unique Mechanism of Entering Cells. Chem. Biol. 2015, 22, 1087–1097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, K.; Lee, T.H.; Aguilar, M.I. The role of electrostatic interactions in the membrane binding of melittin. J. Mol. Recognit. 2011, 24, 108–118. [Google Scholar] [CrossRef] [PubMed]
- Cascales, L.; Henriques, S.T.; Kerr, M.C.; Huang, Y.H.; Sweet, M.J.; Daly, N.L.; Craik, D.J. Identification and characterization of a new family of cell-penetrating peptides: Cyclic cell-penetrating peptides. J. Biol. Chem. 2011, 286, 36932–36943. [Google Scholar] [CrossRef] [PubMed]
- Henriques, S.T.; Melo, M.N.; Castanho, M.A. Cell-penetrating peptides and antimicrobial peptides: How different are they? Biochem. J. 2006, 399, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Watson, H. Biological membranes. Essays Biochem. 2015, 59, 43–69. [Google Scholar] [CrossRef] [PubMed]
- Ingolfsson, H.I.; Melo, M.N.; van Eerden, F.J.; Arnarez, C.; Lopez, C.A.; Wassenaar, T.A.; Periole, X.; de Vries, A.H.; Tieleman, D.P.; Marrink, S.J. Lipid organization of the plasma membrane. J. Am. Chem. Soc. 2014, 136, 14554–14559. [Google Scholar] [CrossRef]
- van Meer, G.; Voelker, D.R.; Feigenson, G.W. Membrane lipids: Where they are and how they behave. Nat. Rev. Mol. Cell Biol. 2008, 9, 112–124. [Google Scholar] [CrossRef]
- Utsugi, T.; Schroit, A.J.; Connor, J.; Bucana, C.D.; Fidler, I.J. Elevated expression of phosphatidylserine in the outer membrane leaflet of human tumor cells and recognition by activated human blood monocytes. Cancer Res. 1991, 51, 3062–3066. [Google Scholar]
- Zwaal, R.F.; Comfurius, P.; Bevers, E.M. Surface exposure of phosphatidylserine in pathological cells. Cell Mol. Life Sci. 2005, 62, 971–988. [Google Scholar] [CrossRef]
- El-Andaloussi, S.; Jarver, P.; Johansson, H.J.; Langel, U. Cargo-dependent cytotoxicity and delivery efficacy of cell-penetrating peptides: A comparative study. Biochem. J. 2007, 407, 285–292. [Google Scholar] [CrossRef]
- Chu, P.Y.; Li, T.K.; Ding, S.T.; Lai, I.R.; Shen, T.L. EGF-induced Grb7 recruits and promotes Ras activity essential for the tumorigenicity of Sk-Br3 breast cancer cells. J. Biol. Chem. 2010, 285, 29279–29285. [Google Scholar] [CrossRef] [PubMed]
- Al Soraj, M.; He, L.; Peynshaert, K.; Cousaert, J.; Vercauteren, D.; Braeckmans, K.; De Smedt, S.C.; Jones, A.T. siRNA and pharmacological inhibition of endocytic pathways to characterize the differential role of macropinocytosis and the actin cytoskeleton on cellular uptake of dextran and cationic cell penetrating peptides octaarginine (R8) and HIV-Tat. J. Controlled Release 2012, 161, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, C.A.; Pei, D. Bicyclic Peptides as Next-Generation Therapeutics. Chemistry 2017, 23, 12690–12703. [Google Scholar] [CrossRef] [PubMed]
- Lian, W.; Jiang, B.; Qian, Z.; Pei, D. Cell-permeable bicyclic peptide inhibitors against intracellular proteins. J. Am. Chem. Soc. 2014, 136, 9830–9833. [Google Scholar] [CrossRef] [PubMed]
- Yau, W.M.; Wimley, W.C.; Gawrisch, K.; White, S.H. The preference of tryptophan for membrane interfaces. Biochemistry 1998, 37, 14713–14718. [Google Scholar] [CrossRef]
- Habault, J.; Poyet, J.L. Recent Advances in Cell Penetrating Peptide-Based Anticancer Therapies. Molecules 2019, 24, 927. [Google Scholar] [CrossRef]
- Ramaker, K.; Henkel, M.; Krause, T.; Rockendorf, N.; Frey, A. Cell penetrating peptides: A comparative transport analysis for 474 sequence motifs. Drug Deliv. 2018, 25, 928–937. [Google Scholar] [CrossRef]
- Geback, T.; Schulz, M.M.; Koumoutsakos, P.; Detmar, M. TScratch: A novel and simple software tool for automated analysis of monolayer wound healing assays. Biotechniques 2009, 46, 265–274. [Google Scholar] [CrossRef]
- Brautigam, C.A.; Zhao, H.; Vargas, C.; Keller, S.; Schuck, P. Integration and global analysis of isothermal titration calorimetry data for studying macromolecular interactions. Nat. Protoc. 2016, 11, 882–894. [Google Scholar] [CrossRef]
- Henriques, S.T.; Pattenden, L.K.; Aguilar, M.I.; Castanho, M.A. PrP(106–126) does not interact with membranes under physiological conditions. Biophys. J. 2008, 95, 1877–1889. [Google Scholar] [CrossRef]
- Henriques, S.T.; Huang, Y.H.; Castanho, M.A.; Bagatolli, L.A.; Sonza, S.; Tachedjian, G.; Daly, N.L.; Craik, D.J. Phosphatidylethanolamine binding is a conserved feature of cyclotide-membrane interactions. J. Biol. Chem. 2012, 287, 33629–33643. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Limited amounts of samples of the compounds are available from the authors. |









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
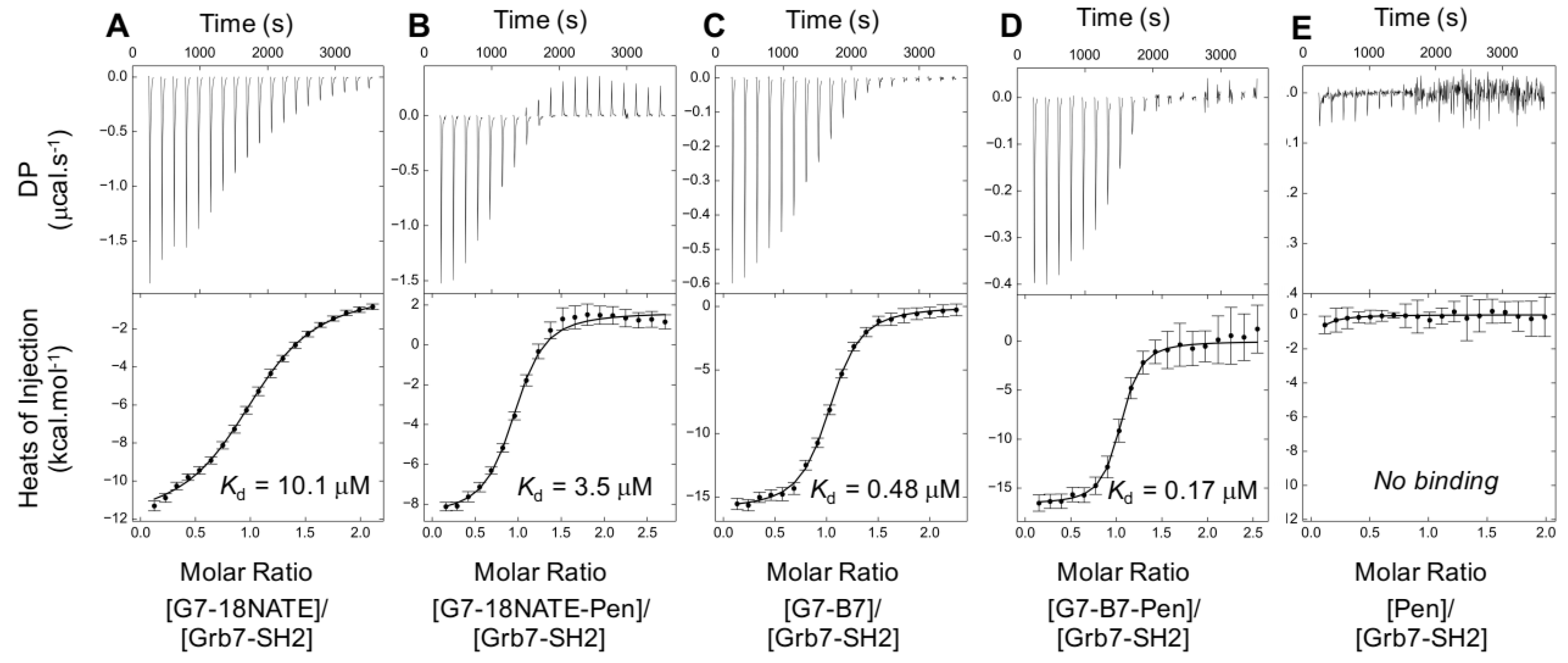
| Binding Parameter | G7-B7 | G7-B7-Pen | G7-18NATE | G7-18NATE-Pen |
|---|---|---|---|---|
| Kb (106 M−1) | 2.06 | 5.95 | 1.00 | 0.290 |
| ΔH (kcal·mol−1) | −16.0 (−15.8–−16.2) | −16.6 (−16.4–−17.0) | −12.7 (−12.5–−12.9) | −10.2 (−9.5–−10.9) |
| ΔS (kcal·mol−1·K−1) | −24.6 | −24.8 | −19.8 | −9.19 |
| −TΔS (kcal·mol−1) | 7.35 | 7.41 | 5.91 | 2.74 |
| ΔG (kcal·mol−1) | −8.62 | −9.24 | −6.82 | −7.45 |
| KD (μM) | 0.48 (0.43–0.55) | 0.17 (0.13–0.22) | 10.1 (9.24–11.0) | 3.45 (2.29–5.15) |
| Peptide | Total Concentration of Peptide Associated with Cells (nM) | Concentration of Peptide in Supernatant (nM) | Concentration of Peptide Associated with Pellet (nM) | |||
|---|---|---|---|---|---|---|
| Average | SEM | Average | SEM | Average | SEM | |
| G7-18NATE | n.d.* | n.d. | n.d. | n.d. | n.d. | n.d. |
| G7-18NATE-Pen | 696.4 | 112.2 | 221.1 | 49.2 | 312.8 | 60.8 |
| G7-B7M2-Pen | 208.0 | 12.7 | n.d. | n.d. | 86.7 | 36.1 |
| Peptide | % Unbound Peptide Associated with Total Cell | % Unbound Peptide Associated with Supernatant | % Unbound Peptide Associated with Pellet | |||
|---|---|---|---|---|---|---|
| Average | SEM | Average | SEM | Average | SEM | |
| G7-18NATE | n.d.* | n.d. | n.d. | n.d. | n.d. | n.d. |
| G7-18NATE-Pen | 6.2% | 1.0% | 2.0% | 0.4% | 2.8% | 0.5% |
| G7-B7M2-Pen | 1.5% | 0.1% | n.d. | n.d. | 0.6% | 0.3% |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sang, J.; Kulkarni, K.; Watson, G.M.; Ma, X.; Craik, D.J.; Henriques, S.T.; Poth, A.G.; Benfield, A.H.; Wilce, J.A. Evaluation of Cyclic Peptide Inhibitors of the Grb7 Breast Cancer Target: Small Change in Cargo Results in Large Change in Cellular Activity. Molecules 2019, 24, 3739. https://doi.org/10.3390/molecules24203739
Sang J, Kulkarni K, Watson GM, Ma X, Craik DJ, Henriques ST, Poth AG, Benfield AH, Wilce JA. Evaluation of Cyclic Peptide Inhibitors of the Grb7 Breast Cancer Target: Small Change in Cargo Results in Large Change in Cellular Activity. Molecules. 2019; 24(20):3739. https://doi.org/10.3390/molecules24203739
Chicago/Turabian StyleSang, Jianrong, Ketav Kulkarni, Gabrielle M. Watson, Xiuquan Ma, David J. Craik, Sónia T. Henriques, Aaron G. Poth, Aurélie H. Benfield, and Jacqueline A. Wilce. 2019. "Evaluation of Cyclic Peptide Inhibitors of the Grb7 Breast Cancer Target: Small Change in Cargo Results in Large Change in Cellular Activity" Molecules 24, no. 20: 3739. https://doi.org/10.3390/molecules24203739