
Incorporation of Putative Helix-Breaking Amino Acids in the Design of Novel Stapled Peptides: Exploring Biophysical and Cellular Permeability Properties

 , , , ,
, , , ,
Abstract
:
1. Introduction
2. Methods
2.1. Peptide Synthesis
2.2. Protein Production
2.3. Crystallization and Data Collection
2.4. X-ray Structure and Refinement
2.5. Mdm2 Binding Assay
2.6. p53 Beta-Lactamase Reporter Gene Cellular Functional Assay
2.7. Lactate Dehydrogenase (LDH) Release Assay
2.8. Tetracycline Beta-Lactamase Reporter Gene Cellular Assay (Counterscreen)
2.9. Isothermal Titration Calorimetry (ITC)
2.10. Circular Dichroism (CD)
2.11. Surface Plasmon Resonance (SPR)
2.12. Computational Chemistry and Molecular Dynamics (MD) Studies
2.13. Whole Cell Homogenate Stability
2.14. Stapled Peptide Lipophilicity Analysis and Correlation with Cellular/Target Ratios
2.15. Statistics
3. Results and Discussion
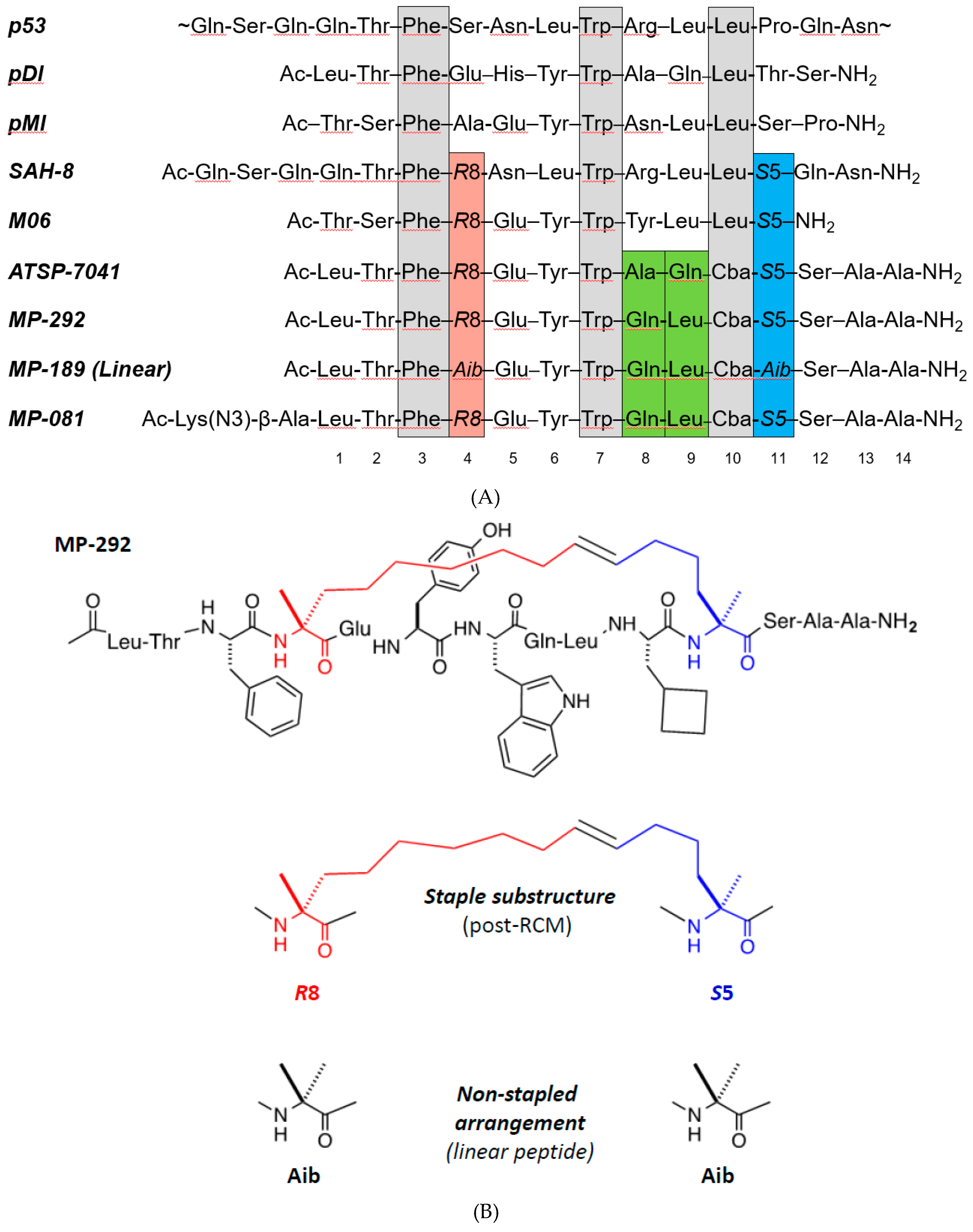
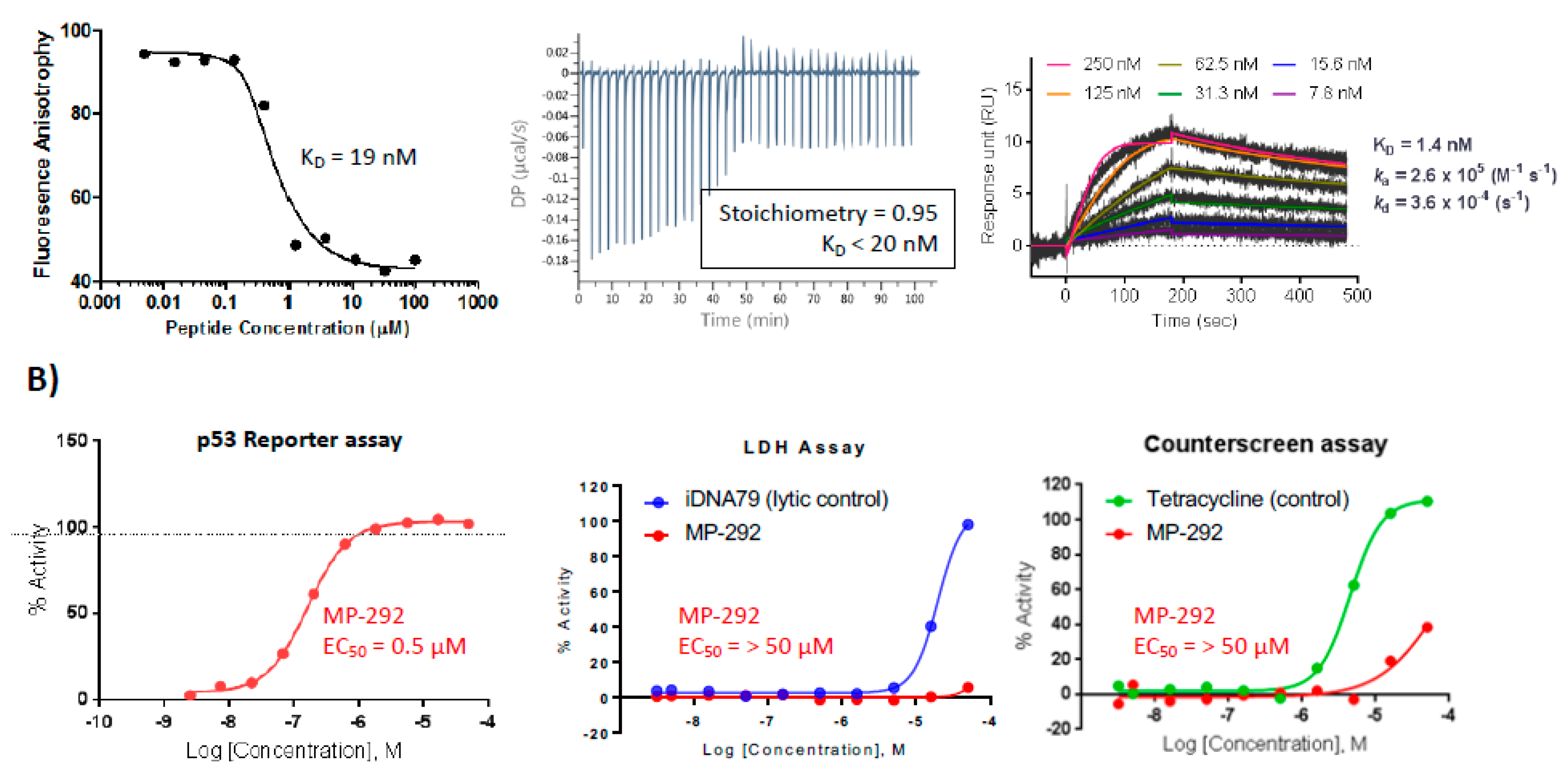
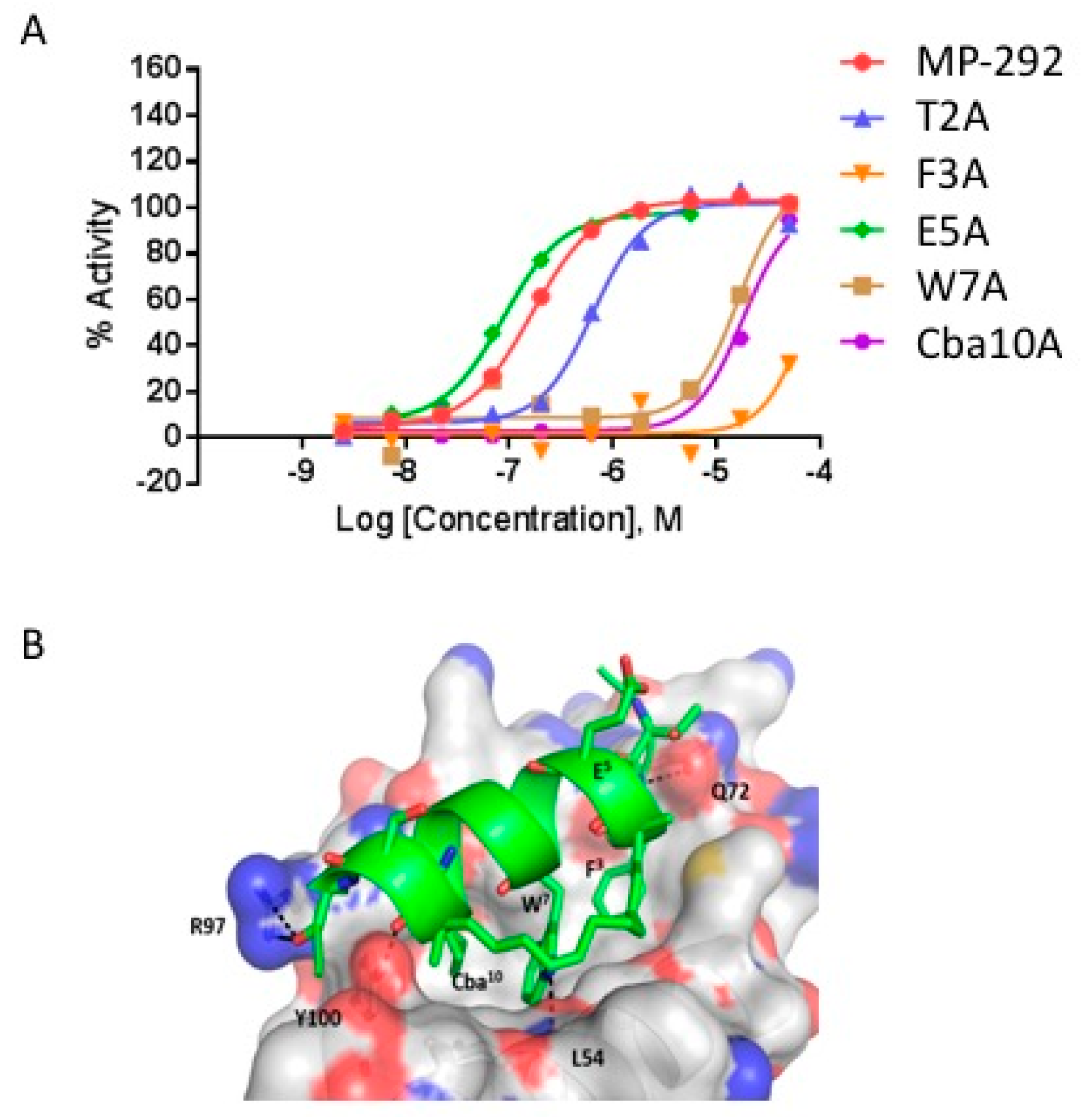
3.1. MP-292 is a High Affinity Mdm2 Binder with On-Target Cellular Activity
3.2. Ala Scanning of Stapled Peptide Analogs of MP-292 Confirms Key Residues for Target Binding
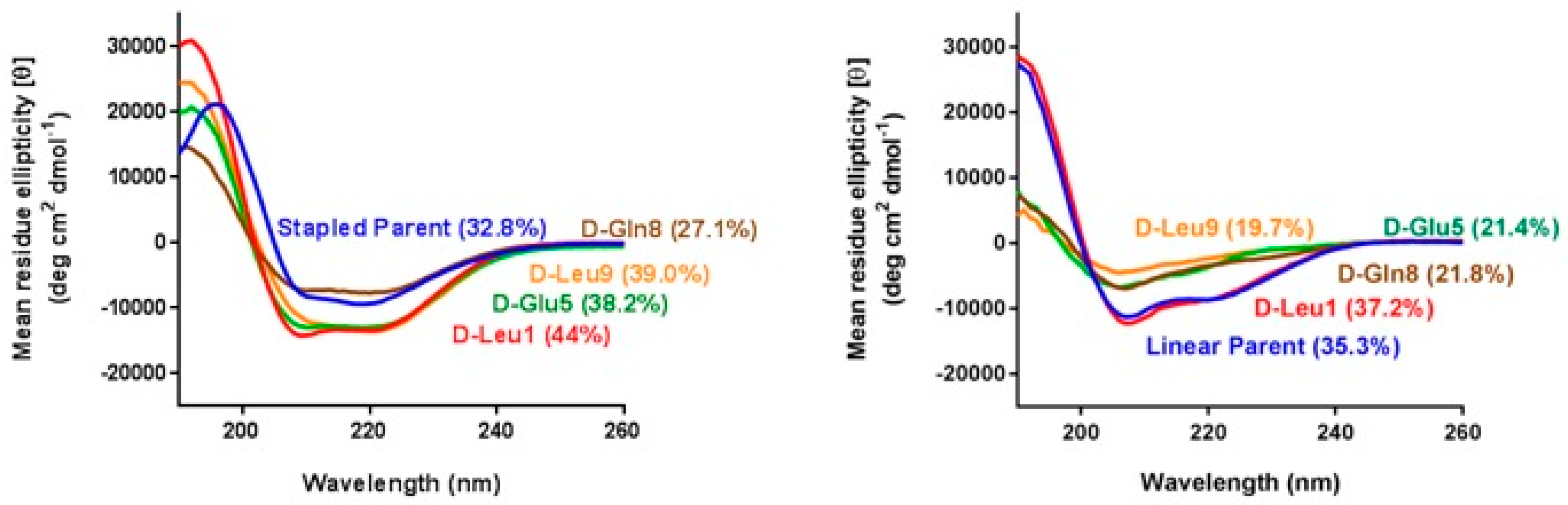
3.3. α-Helicity, Target Binding, and Cellular Activity Were Generally Unaffected by d-Amino Acid Substitutions into MP-292
3.4. d-Amino Acid Scanning of Linear Peptides Results in Decreased α-Helicity, Target Binding, and Cellular Activity
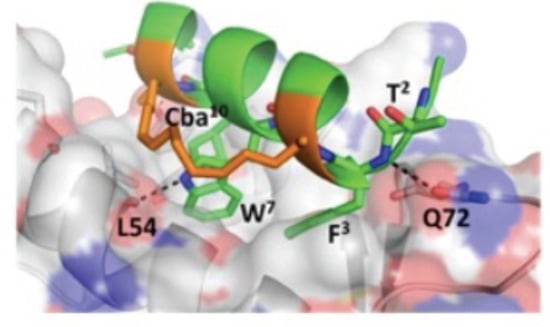
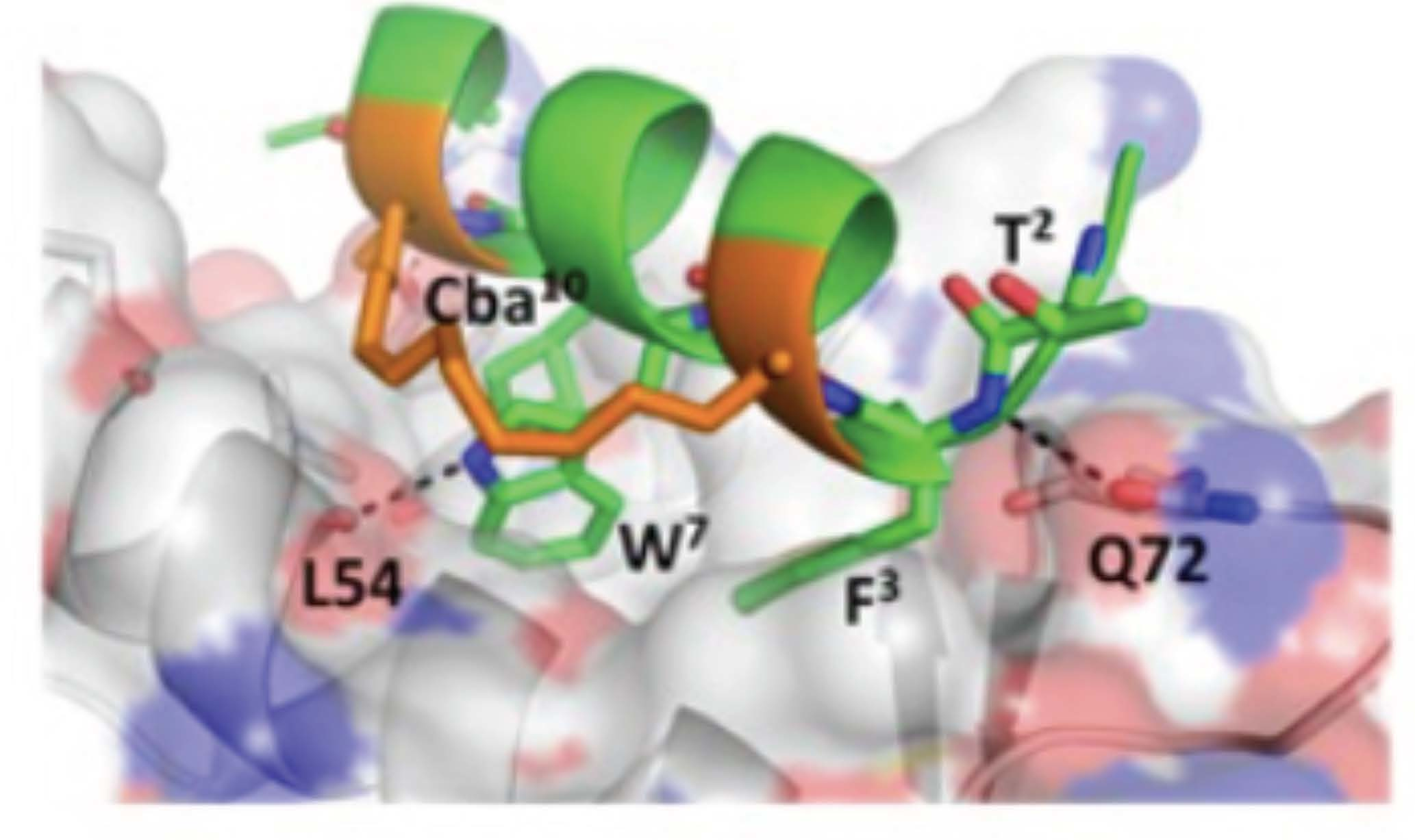
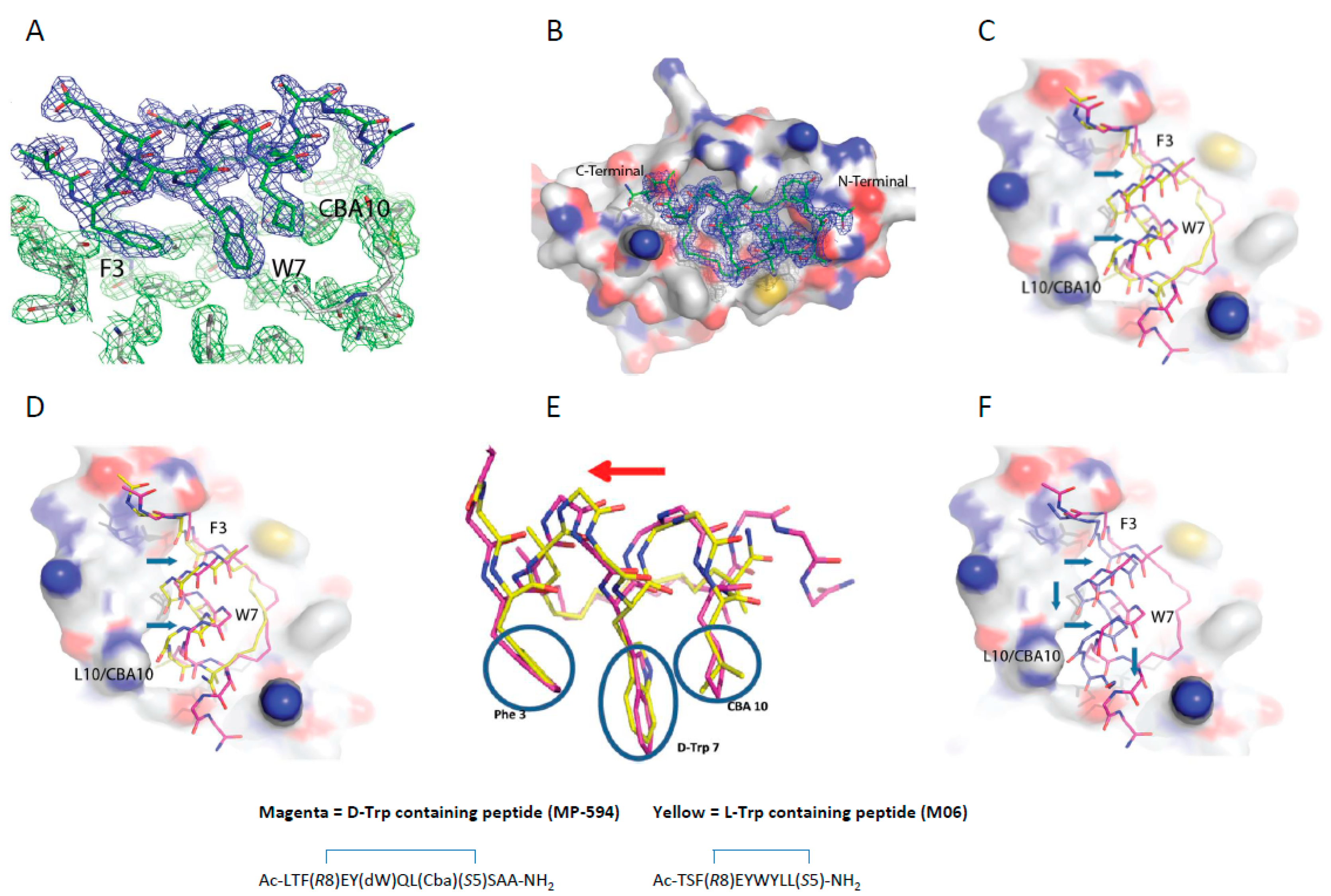
3.5. d-Trp7 Modified Stapled Peptide: Computational Modeling and X-Ray Crystallographic Studies with Mdm2
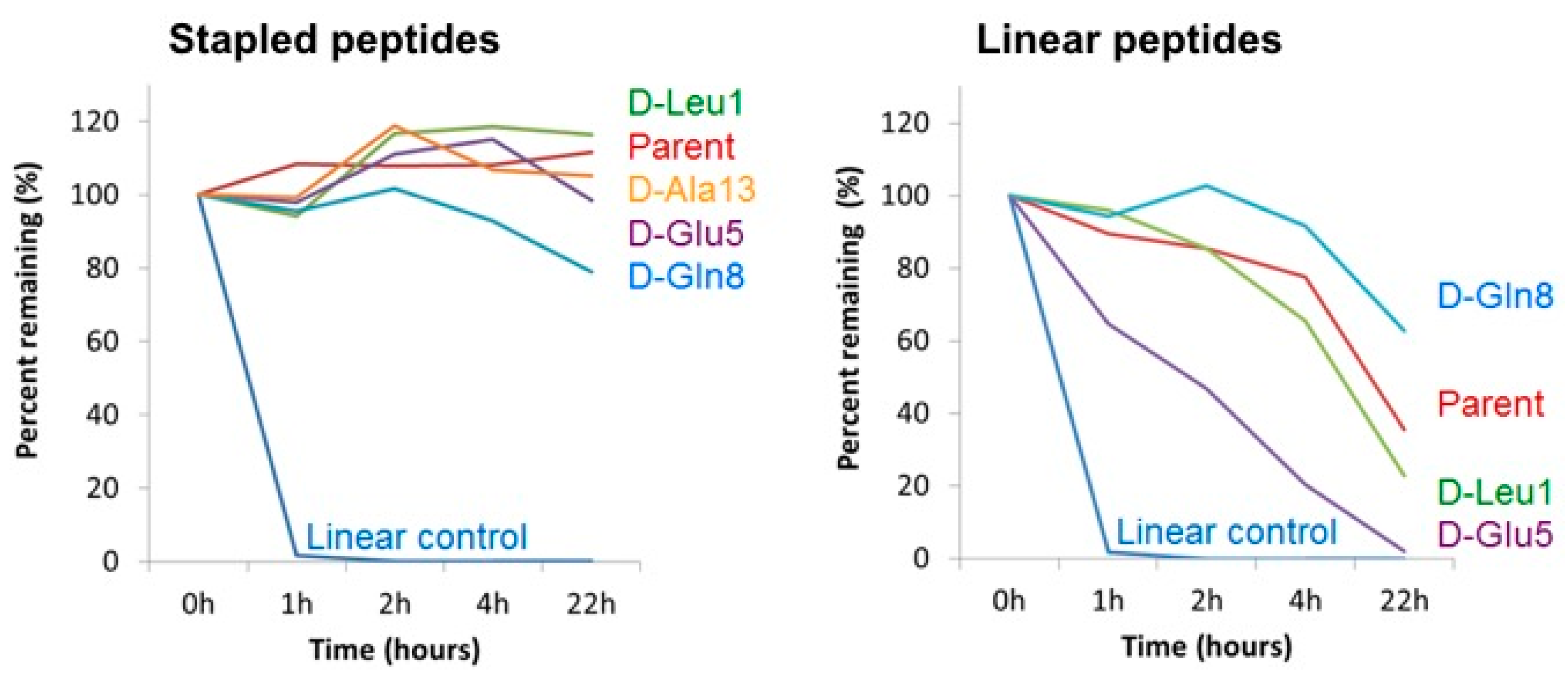
3.6. Cell Homogenate Proteolytic Stability of d-Amino Acid Modified Stapled Versus Linear Peptides
3.7. Aib, N-Me-Amino Acid, Pro, and Gly Substitutions Had Minimal Effects on a-Helicity, Target Binding, and Cellular Activity
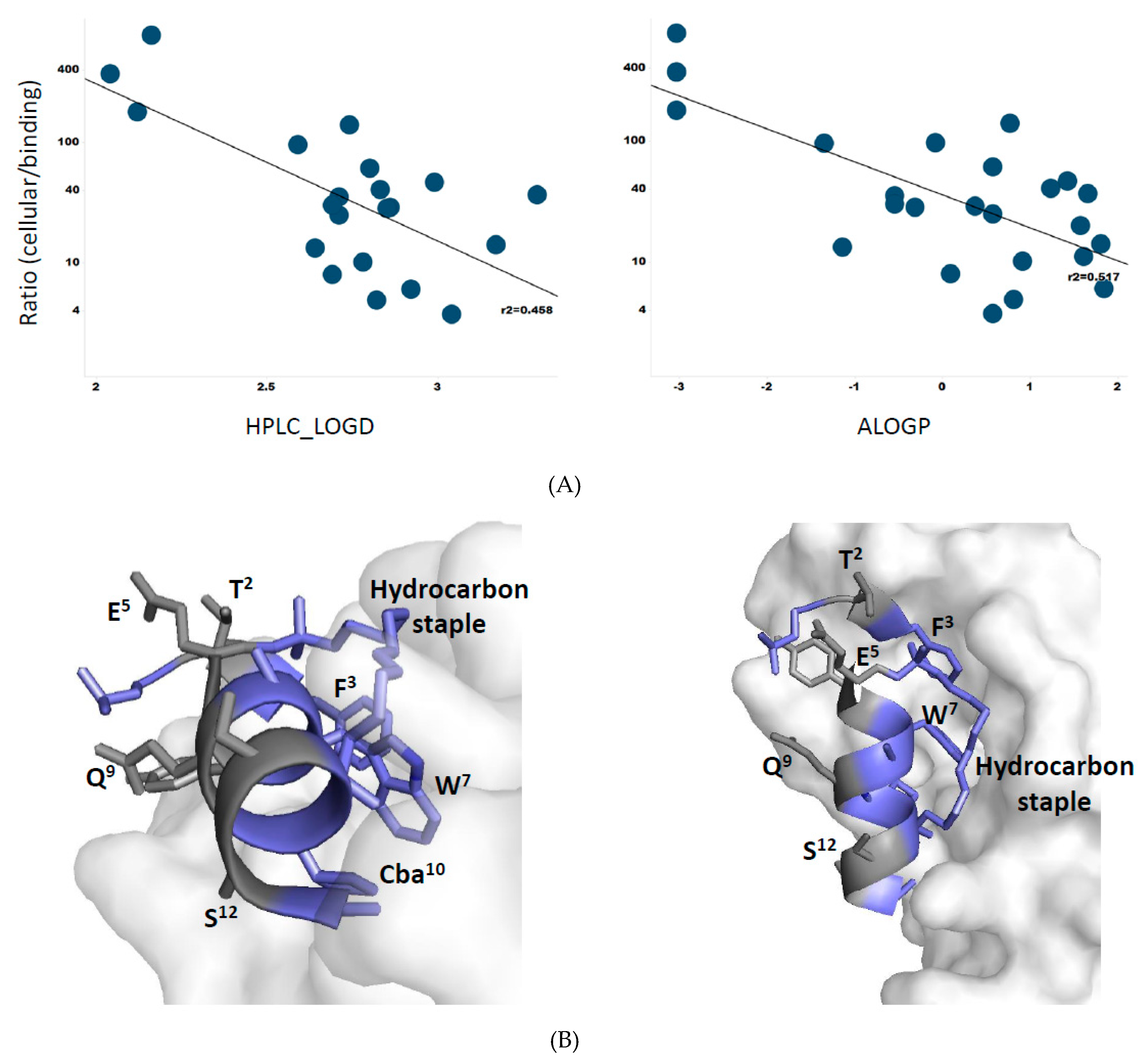
3.8. Membrane Permeability Correlates with Peptide Lipophilicity
3.9. Conclusions: A Macrocyclic α-Helical Peptide Model System to Explore Biophysical and Cellular Permeability Properties
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Arkin, M.R.; Tang, Y.; Wells, J.A. Small-molecule inhibitors of protein-protein interactions: Progressing toward the reality. Chem. Biol. 2014, 21, 1102–1114. [Google Scholar] [CrossRef] [PubMed]
- Sousa, F.; Castro, P.; Fonte, P.; Kennedy, P.J.; Neves-Petersen, M.T.; Sarmento, B. Nanoparticles for the delivery of therapeutic antibodies: Dogma or promising strategy? Expert Opin. Drug Deliv. 2017, 14, 1163–1176. [Google Scholar] [CrossRef] [PubMed]
- Vlieghe, P.; Lisowski, V.; Martinez, J.; Khrestchatisky, M. Synthetic therapeutic peptides: Science and market. Drug Discov. Today 2010, 15, 40–56. [Google Scholar] [CrossRef] [PubMed]
- Sawyer, T.K.; Partridge, A.W.; Kaan, H.Y.K.; Juang, Y.C.; Lim, S.; Johannes, C.; Yuen, Y.T.; Verma, C.; Kannan, S.; Aronica, P.; et al. Macrocyclic alpha helical peptide therapeutic modality: A perspective of learnings and challenges. Bioorg. Med. Chem. 2018, 26, 2807–2815. [Google Scholar] [CrossRef] [PubMed]
- Carvajal, L.A.; Neriah, D.B.; Senecal, A.; Benard, L.; Thiruthuvanathan, V.; Yatsenko, T.; Narayanagari, S.R.; Wheat, J.C.; Todorova, T.I.; Mitchell, K.; et al. Dual inhibition of MDMX and MDM2 as a therapeutic strategy in leukemia. Sci. Transl. Med. 2018, 10, eaao3003. [Google Scholar] [CrossRef] [PubMed]
- Schafmeister, C.E.; Po, J.; Verdine, G.L. An All-Hydrocarbon Cross-Linking System for Enhancing the Helicity and Metabolic Stability of Peptides. J. Am. Chem. Soc. 2000, 122, 5891–5892. [Google Scholar] [CrossRef]
- Blackwell, H.E.; Sadowsky, J.D.; Howard, R.J.; Sampson, J.N.; Chao, J.A.; Steinmetz, W.E.; O’Leary, D.J.; Grubbs, R.H. Ring-closing metathesis of olefinic peptides: Design, synthesis, and structural characterization of macrocyclic helical peptides. J. Org. Chem. 2001, 66, 5291–5302. [Google Scholar] [CrossRef] [PubMed]
- Blackwell, H.E.; Grubbs, R.H. Highly Efficient Synthesis of Covalently Cross-Linked Peptide Helices by Ring-Closing Metathesis. Angew. Chem. Int. Ed. Engl. 1998, 37, 3281–3284. [Google Scholar] [CrossRef]
- Chang, Y.S.; Graves, B.; Guerlavais, V.; Tovar, C.; Packman, K.; To, K.H.; Olson, K.A.; Kesavan, K.; Gangurde, P.; Mukherjee, A.; et al. Stapled alpha-helical peptide drug development: A potent dual inhibitor of MDM2 and MDMX for p53-dependent cancer therapy. Proc. Natl. Acad. Sci. USA 2013, 110, E3445–E3454. [Google Scholar] [CrossRef]
- Bernal, F.; Tyler, A.F.; Korsmeyer, S.J.; Walensky, L.D.; Verdine, G.L. Reactivation of the p53 tumor suppressor pathway by a stapled p53 peptide. J. Am. Chem. Soc. 2007, 129, 2456–2457. [Google Scholar] [CrossRef]
- Hu, B.; Gilkes, D.M.; Chen, J. Efficient p53 activation and apoptosis by simultaneous disruption of binding to MDM2 and MDMX. Cancer Res. 2007, 67, 8810–8817. [Google Scholar] [CrossRef] [PubMed]
- Pazgier, M.; Liu, M.; Zou, G.; Yuan, W.; Li, C.; Li, C.; Li, J.; Monbo, J.; Zella, D.; Tarasov, S.G.; et al. Structural basis for high-affinity peptide inhibition of p53 interactions with MDM2 and MDMX. Proc. Natl. Acad. Sci. USA 2009, 106, 4665–4670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernal, F.; Wade, M.; Godes, M.; Davis, T.N.; Whitehead, D.G.; Kung, A.L.; Wahl, G.M.; Walensky, L.D. A stapled p53 helix overcomes HDMX-mediated suppression of p53. Cancer Cell 2010, 18, 411–422. [Google Scholar]
- Brown, C.J.; Quah, S.T.; Jong, J.; Goh, A.M.; Chiam, P.C.; Khoo, K.H.; Choong, M.L.; Lee, M.A.; Yurlova, L.; Zolghadr, K.; et al. Stapled Peptides with Improved Potency and Specificity That Activate p53. ACS Chem. Biol. 2013, 8, 506–512. [Google Scholar] [CrossRef] [PubMed]
- Kabsch, W. XDS. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Winn, M.D.; Ballard, C.C.; Cowtan, K.D.; Dodson, E.J.; Emsley, P.; Evans, P.R.; Keegan, R.M.; Krissinel, E.B.; Leslie, A.G.W.; McCoy, A.; et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. Sect. D Biol. Crystallogr. 2011, 67, 235–242. [Google Scholar] [CrossRef] [PubMed]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. [Google Scholar] [CrossRef] [PubMed]
- Moriarty, N.A.-O.; Liebschner, D.; Klei, H.E.; Echols, N.; Afonine, P.V.; Headd, J.J.; Poon, B.K.; Adams, P.D. Interactive comparison and remediation of collections of macromolecular structures. Protein Sci. 2018, 27, 182–194. [Google Scholar] [CrossRef]
- Murshudov, G.N.; Vagin, A.A.; Dodson, E.J. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. Sect. D Biol. Crystallogr. 1997, 53, 240–255. [Google Scholar] [CrossRef] [PubMed]
- Lebedev, A.A.; Young, P.; Isupov, M.N.; Isupov, M.N.; Moroz, O.V.; Murshudov, G.N. JLigand: A graphical tool for the CCP4 template-restraint library. Acta Crystallogr. Sect. D Biol. Crystallogr. 2012, D64, 431–440. [Google Scholar] [CrossRef]
- Prisant, M.G.; Richardson, J.S.; Richardson, D.C. Structure validation by Calpha geometry: Phi,psi and Cbeta deviation. Proteins 2003, 50, 437–450. [Google Scholar]
- Pearlman, D.A.; Case, D.A.; Caldwell, J.W.; Ross, W.S.; Cheatham, T.E.; DeBolt, S.; Ferguson, D.; George, S.; Kollman, P. AMBER, a package of computer programs for applying molecular mechanics, normal mode analysis, molecular dynamics and free energy calculations to simulate the structural and energetic properties of molecules. Comput. Phys. Commun. 1995, 91, 1–41. [Google Scholar] [CrossRef]
- Tan, Y.S.; Reeks, J.; Brown, C.J.; Thean, D.; Ferrer Gago, F.J.; Yuen, T.Y.; Goh, E.T.L.; Lee, X.E.C.; Jennings, C.E.; Joseph, T.L.; et al. Benzene Probes in Molecular Dynamics Simulations Reveal Novel Binding Sites for Ligand Design. J. Phys. Chem. Lett. 2016, 7, 3452–3457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ostermeir, K.; Zacharias, M. Hamiltonian replica-exchange simulations with adaptive biasing of peptide backbone and side chain dihedral angles. J. Comput. Chem. 2014, 35, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Delano, W.L. The PyMOL Molecular Graphics System. Available online: https://www.researchgate.net/publication/225159692_The_PyMOL_Molecular_Graphics_System_2002_DeLano_Scientific_Palo_Alto_CA_USA_httpwwwpymolorg (accessed on 17 June 2019).
- Cruz, J.; Mihailescu, M.; Wiedman, G.; Herman, K.; Searson, P.C.; Searson, P.C.; Wimley, W.C.; Hristova, K. A membrane-translocating peptide penetrates into bilayers without significant bilayer perturbations. Biophys. J. 2013, 104, 2419–2428. [Google Scholar] [CrossRef]
- Valko, K.; Bevan, C.; Reynolds, D. Chromatographic Hydrophobicity Index by Fast-Gradient RP-HPLC: A High-Throughput Alternative to log P/log D. Analy. Chem. 1997, 69, 2022–2029. [Google Scholar] [CrossRef] [PubMed]
- Ghose, A.K.; Viswanadhan, V.N.; Wendoloski, J.J. Prediction of Hydrophobic (Lipophilic) Properties of Small Organic Molecules Using Fragmental Methods: An Analysis of ALOGP and CLOGP Methods. J. Phys. Chem. A 1998, 102, 3762–3772. [Google Scholar] [CrossRef]
- Ghose, A.K.; Crippen, G.M. Atomic physicochemical parameters for three-dimensional-structure-directed quantitative structure-activity relationships. 2. Modeling dispersive and hydrophobic interactions. J. Chem. Inform. Comput. Sci. 1987, 27, 21–35. [Google Scholar] [CrossRef]
- Furukawa, A.; Townsend, C.E.; Schwochert, J.; Pye, C.R.; Bednarek, M.A.; Lokey, R.S. Passive Membrane Permeability in Cyclic Peptomer Scaffolds Is Robust to Extensive Variation in Side Chain Functionality and Backbone Geometry. J. Med. Chem. 2016, 59, 9503–9512. [Google Scholar] [CrossRef]
- Bird, G.H.; Mazzola, E.; Opoku-Nsiah, K.; Lammert, M.A.; Godes, M.; Neuberg, D.S.; Walensky, L.D. Biophysical determinants for cellular uptake of hydrocarbon-stapled peptide helices. Nat. Chem. Biol. 2016, 12, 845. [Google Scholar] [CrossRef] [PubMed]
- Sakagami, K.; Masuda, T.; Kawano, K.; Futaki, S. Importance of Net Hydrophobicity in the Cellular Uptake of All-Hydrocarbon Stapled Peptides. Mol. Pharm. 2018, 15, 1332–1340. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.; Juang, Y.C.; Kaan, H.Y.K.; Sadruddinb, A.; Yuen, T.Z.; Ferrer, F.J.; Lee, X.C.; Xi, L.; Johannes, C.W.; Brown, C.J.; et al. De-risking drug discovery of intracellular targeting macrocyclic peptides: Screening strategies to eliminate false-positive hits. BioRxiv 2019, 636563. [Google Scholar]
- Rich, R.L.; Myszka, D.G. Survey of the 2009 commercial optical biosensor literature. J. Mol. Recognit. 2011, 24, 892–914. [Google Scholar] [CrossRef] [PubMed]
- Myszka, D.G. Survey of the 1998 optical biosensor literature. J. Mol. Recognit. 1999, 12, 390–408. [Google Scholar] [CrossRef]
- Li, X.; Liu, C.; Chen, S.; Hu, H.; Su, J.; Zou, Y. d-Amino acid mutation of PMI as potent dual peptide inhibitors of p53-MDM2/MDMX interactions. Bioorg. Med. Chem. Lett. 2017, 27, 4678–4681. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds used in this study may be available from the authors pending sufficient stocks and execution of material transfer agreements. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Table section | Peptide | N-term | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | C-term | Helicity (%) | Cellular (µM) | Mdm2 (nM) | Ratio |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A | MP-292 | Ac- | L | T | F | R8 | E | Y | W | Q | L | Cba | S5 | -SAA-amide | 32.8 | 0.54 | 18.6 | 29 |
| L1A | Ac- | A | T | F | R8 | E | Y | W | Q | L | Cba | S5 | -SAA-amide | 53.3 | 0.50 | 17.6 | 28 | |
| T2A | Ac- | L | A | F | R8 | E | Y | W | Q | L | Cba | S5 | -SAA-amide | 36.2 | 1.0 | 21.3 | 47 | |
| F3A | Ac- | L | T | A | R8 | E | Y | W | Q | L | Cba | S5 | -SAA-amide | 48.4 | 37 | 3600 | 10 | |
| E5A | Ac- | L | T | F | R8 | A | Y | W | Q | L | Cba | S5 | -SAA-amide | 27.6 | 0.18 | 29.8 | 6 | |
| Y6A | Ac- | L | T | F | R8 | E | A | W | Q | L | Cba | S5 | -SAA-amide | 50.3 | 0.55 | 38.9 | 14 | |
| W7A | Ac- | L | T | F | R8 | E | Y | A | Q | L | Cba | S5 | -SAA-amide | ND | 8.8 | 5093 | 2 | |
| Q8A | Ac- | L | T | F | R8 | E | Y | W | A | L | Cba | S5 | -SAA-amide | 33.4 | 0.10 | 26.5 | 4 | |
| L9A | Ac- | L | T | F | R8 | E | Y | W | Q | A | Cba | S5 | -SAA-amide | 48.7 | 0.16 | 12.0 | 13 | |
| Cba10A | Ac- | L | T | F | R8 | E | Y | W | Q | L | A | S5 | -SAA-amide | ND | 11 | 176 | 61 | |
| S12A | Ac- | L | T | F | R8 | E | Y | W | Q | L | Cba | S5 | -AAA-amide | 36.5 | 0.35 | 20.4 | 17 | |
| B | L1(D-Leu) | Ac- | D-Leu | T | F | R8 | E | Y | W | Q | L | Cba | S5 | -SAA-amide | 44.1 | 0.6 | 31 | 18 |
| T2(D-Thr) | Ac- | L | D-Thr | F | R8 | E | Y | W | Q | L | Cba | S5 | -SAA-amide | 40.7 | 3.4 | 136 | 25 | |
| F3(D-Phe) | Ac- | L | T | D-Phe | R8 | E | Y | W | Q | L | Cba | S5 | -SAA-amide | 51.8 | >50 | 6485 | >8 | |
| E5(D-Glu) | Ac- | L | T | F | R8 | D-Glu | Y | W | Q | L | Cba | S5 | -SAA-amide | 38.2 | 1.0 | 32 | 30 | |
| Y6 (D-Tyr) | Ac- | L | T | F | R8 | E | D-Tyr | W | Q | L | Cba | S5 | -SAA-amide | 43.2 | 3.2 | 286 | 11 | |
| W6(D-Trp) (MP-384) | Ac- | L | T | F | R8 | E | Y | D-Trp | Q | L | Cba | S5 | -SAA-amide | 41.0 | 0.6 | 30 | 20 | |
| Q8(D-Gln) | Ac- | L | T | F | R8 | E | Y | W | D-Gln | L | Cba | S5 | -SAA-amide | 27.1 | 2.1 | 22 | 95 | |
| L9(D-Leu) | Ac- | L | T | F | R8 | E | Y | W | Q | D-Leu | Cba | S5 | -SAA-amide | 39.0 | 1.1 | 30 | 37 | |
| Cba10(D-Cba) | Ac- | L | T | F | R8 | E | Y | W | Q | L | D-Cba | S5 | -SAA-amide | ND | 8.1 | 1650 | 5 | |
| S12(D-Ser) | Ac- | L | T | F | R8 | E | Y | W | Q | L | Cba | S5 | -(D-Ser)-AA-amide | 41.2 | 1.2 | 8.6 | 140 | |
| A13(D-Ala) | Ac- | L | T | F | R8 | E | Y | W | Q | L | Cba | S5 | -S-(D-Ala)-A-amide | 41.8 | 0.6 | 17 | 35 | |
| A14(D-Ala) | Ac- | L | T | F | R8 | E | Y | W | Q | L | Cba | S5 | -SA-(D-Ala)-amide | 34.6 | 0.5 | 18 | 28 | |
| C | Linear Parent (MP-189) | Ac- | L | T | F | Aib | E | Y | W | Q | L | Cba | Aib | -SAA-amide | 35.3 | 33.5 | 43.1 | 777 |
| Linear L1(D-Leu) | Ac- | D-Leu | T | F | Aib | E | Y | W | Q | L | Cba | Aib | -SAA-amide | 37.2 | >50 | 277 | >180 | |
| Linear E5(D-Glu) | Ac- | L | T | F | Aib | D-Glu | Y | W | Q | L | Cba | Aib | -SAA-amide | 21.4 | >50 | 135 | >370 | |
| Linear Q8(D-Gln) | Ac- | L | T | F | Aib | E | Y | W | D-Gln | L | Cba | Aib | -SAA-amide | 21.8 | >50 | 119 | >370 | |
| Linear L9(D-Leu) | Ac- | L | T | F | Aib | E | Y | W | Q | D-Leu | Cba | Aib | -SAA-amide | 19.7 | >50 | 88 | >568 |
| Peptide | N-term | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | C-term | Helicity (%) | Cellular (µM) | Mdm2 (nM) | Ratio |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MP-081 | Ac-K(N3)-(βA)- | L | T | F | R8 | E | Y | W | A | Q | Cba | S5 | -SAA-amide | 49.6 | 0.42 | 19.0 | 22 |
| L1(Aib) | Ac-K(N3)-(βA)- | Aib | T | F | R8 | E | Y | W | A | Q | Cba | S5 | -SAA-amide | 42.7 | 0.49 | 29.5 | 17 |
| T2(Aib) | Ac-K(N3)-(βA)- | L | Aib | F | R8 | E | Y | W | A | Q | Cba | S5 | -SAA-amide | 38.3 | 2.9 | 1706 | 2 |
| A8(Aib) | Ac-K(N3)-(βA)- | L | T | F | R8 | E | Y | W | Aib | Q | Cba | S5 | -SAA-amide | 37.5 | 1.5 | 119.4 | 12 |
| Q9(Aib) | Ac-K(N3)-(βA)- | L | T | F | R8 | E | Y | W | A | Aib | Cba | S5 | -SAA-amide | 28 | 0.25 | 33.1 | 8 |
| S12(Aib) | Ac-K(N3)-(βA)- | L | T | F | R8 | E | Y | W | A | Q | Cba | S5 | -(Aib)-AA-amide | 37.3 | 0.25 | 24.4 | 10 |
| A13(Aib) | Ac-K(N3)-(βA)- | L | T | F | R8 | E | Y | W | A | Q | Cba | S5 | -A-(Aib)-A-amide | 34.4 | 0.59 | 25.7 | 23 |
| L1(N-methyl L-Leu) | Ac-K(N3)-(βA)- | NMe-L | T | F | R8 | E | Y | W | A | Q | Cba | S5 | -SAA-amide | 32.6 | 0.46 | 23.5 | 20 |
| L1(N-methyl L-Ala) | Ac-K(N3)-(βA)- | NMe-A | T | F | R8 | E | Y | W | A | Q | Cba | S5 | -SAA-amide | 47.2 | 1.49 | 15.4 | 97 |
| T2(N-methyl L-Thr) | Ac-K(N3)-(βA)- | L | NMe-T | F | R8 | E | Y | W | A | Q | Cba | S5 | -SAA-amide | 38.2 | 7.7 | 352.2 | 22 |
| A8(N-methyl L-Ala) | Ac-K(N3)-(βA)- | L | T | F | R8 | E | Y | W | NMe-A | Q | Cba | S5 | -SAA-amide | 16.9 | >50 | 671.3 | >75 |
| Q9(N-methyl L-Gln) | Ac-K(N3)-(βA)- | L | T | F | R8 | E | Y | W | A | NMe-Q | Cba | S5 | -SAA-amide | 20.2 | >50 | 211.2 | >237 |
| A13(N-methyl L-Ala) | Ac-K(N3)-(βA)- | L | T | F | R8 | E | Y | W | A | Q | Cba | S5 | -S-(Nme-A)-A-amide | 38.6 | 1.6 | 10.0 | 158 |
| A14(N-methyl L-Ala) | Ac-K(N3)-(βA)- | L | T | F | R8 | E | Y | W | A | Q | Cba | S5 | -SA-(Nme-A)-amide | 47.5 | 0.71 | 5.9 | 120 |
| L1P | Ac-K(N3)-(βA)- | P | T | F | R8 | E | Y | W | A | Q | Cba | S5 | -SAA-amide | 41.6 | 0.75 | 21.9 | 34 |
| Q9P | Ac-K(N3)-(βA)- | L | T | F | R8 | E | Y | W | A | P | Cba | S5 | -SAA-amide | 27.4 | 3.1 | 77.1 | 41 |
| S12P | Ac-K(N3)-(βA)- | L | T | F | R8 | E | Y | W | A | Q | Cba | S5 | -PAA-amide | 30.8 | 3.7 | 45.7 | 80 |
| L1G | Ac-K(N3)-(βA)- | G | T | F | R8 | E | Y | W | A | Q | Cba | S5 | -SAA-amide | 44.6 | 1.6 | 22.1 | 73 |
| E5G | Ac-K(N3)-(βA)- | L | T | F | R8 | G | Y | W | A | Q | Cba | S5 | -SAA-amide | 28.6 | 0.26 | 61.8 | 4 |
| Q9G | Ac-K(N3)-(βA)- | L | T | F | R8 | E | Y | W | A | G | Cba | S5 | -SAA-amide | 38.8 | 0.68 | 42.0 | 16 |
| S12G | Ac-K(N3)-(βA)- | L | T | F | R8 | E | Y | W | A | Q | Cba | S5 | -GAA-amide | 46.5 | 0.65 | 18.9 | 34 |
| L1G, E5G, A8G | Ac-K(N3)-(βA)- | G | T | F | R8 | G | Y | W | G | Q | Cba | S5 | -SAA-amide | 20.4 | 1.7 | 49.8 | 34 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Partridge, A.W.; Kaan, H.Y.K.; Juang, Y.-C.; Sadruddin, A.; Lim, S.; Brown, C.J.; Ng, S.; Thean, D.; Ferrer, F.; Johannes, C.; et al. Incorporation of Putative Helix-Breaking Amino Acids in the Design of Novel Stapled Peptides: Exploring Biophysical and Cellular Permeability Properties. Molecules 2019, 24, 2292. https://doi.org/10.3390/molecules24122292
Partridge AW, Kaan HYK, Juang Y-C, Sadruddin A, Lim S, Brown CJ, Ng S, Thean D, Ferrer F, Johannes C, et al. Incorporation of Putative Helix-Breaking Amino Acids in the Design of Novel Stapled Peptides: Exploring Biophysical and Cellular Permeability Properties. Molecules. 2019; 24(12):2292. https://doi.org/10.3390/molecules24122292
Chicago/Turabian StylePartridge, Anthony W., Hung Yi Kristal Kaan, Yu-Chi Juang, Ahmad Sadruddin, Shuhui Lim, Christopher J. Brown, Simon Ng, Dawn Thean, Fernando Ferrer, Charles Johannes, and et al. 2019. "Incorporation of Putative Helix-Breaking Amino Acids in the Design of Novel Stapled Peptides: Exploring Biophysical and Cellular Permeability Properties" Molecules 24, no. 12: 2292. https://doi.org/10.3390/molecules24122292