
Electrophilic Bromination in Flow: A Safe and Sustainable Alternative to the Use of Molecular Bromine in Batch

 and
and
Abstract
:
1. Introduction
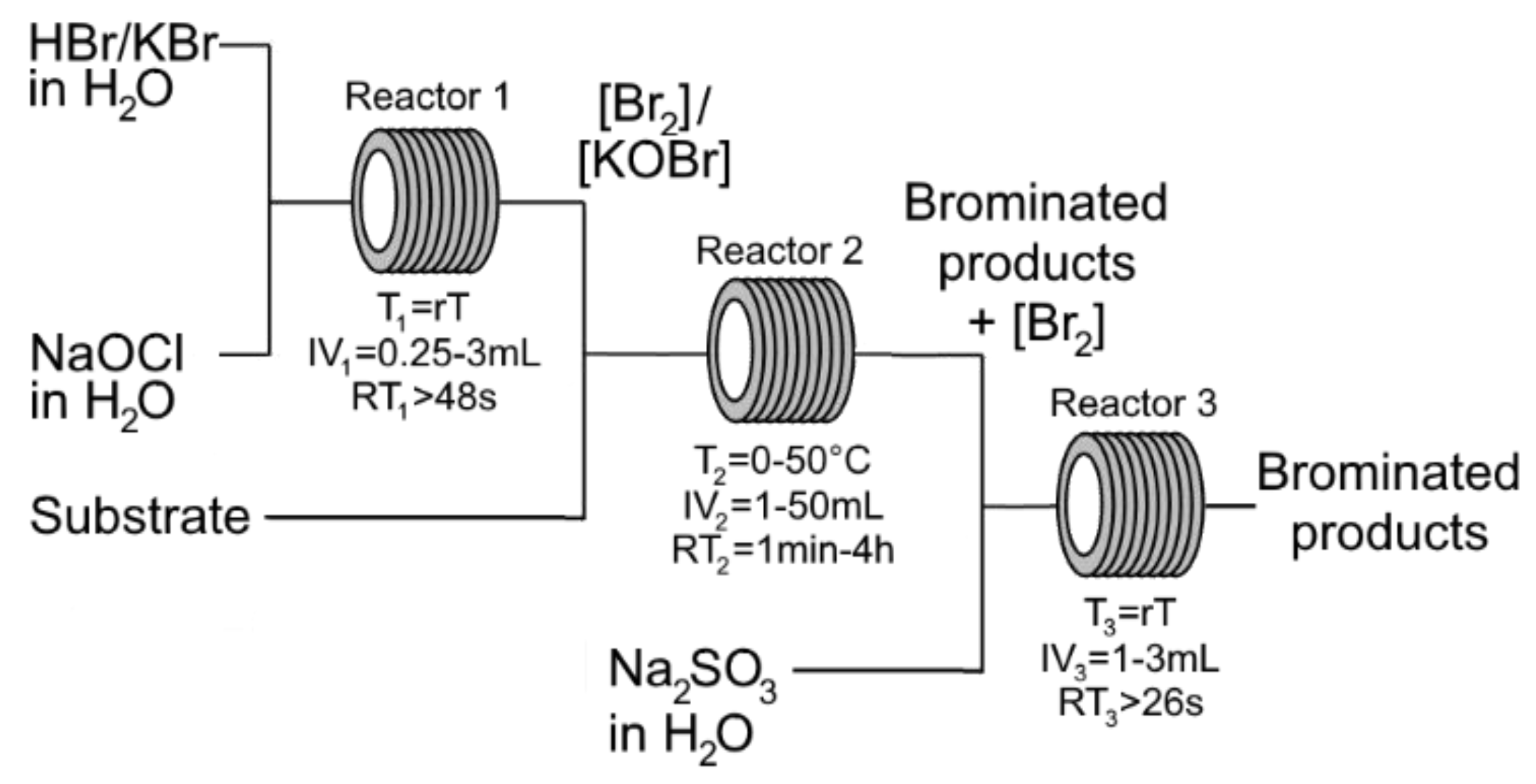
2. Results and Discussion

3. Materials and Methods
3.1. General Procedure
3.2. 2,4,6-Tribromophenol (Table 1, Entry 1)
3.3. 2,2′,6,6′-Tetrabromobisphenol A (Table 1, Entry 2)
3.4. 1,2,5,6,9,10-Hexabromocyclododecane (Table 1, Entry 3)
3.5. Eosin Y (Table 1, entry 4)
3.6. Synthesis of Trioctyl-(3-sulfopropyl)ammonium Perchlorate 4
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Labinger, J.A.; Bercaw, J.E. Understanding and exploiting C–H bond activation. Nature 2002, 417, 507. [Google Scholar] [CrossRef] [PubMed]
- Miyaura, N.; Yamada, K.; Suzuki, A. A new stereospecific cross-coupling by the palladium-catalyzed reaction of 1-alkenylboranes with 1-alkenyl or 1-alkynyl halides. Tetrahedron Lett. 1979, 20, 3437–3440. [Google Scholar] [CrossRef] [Green Version]
- Heck, R.F.; Nolley, J.P. Palladium-catalyzed vinylic hydrogen substitution reactions with aryl, benzyl, and styryl halides. J. Org. Chem. 1972, 37, 2320–2322. [Google Scholar] [CrossRef]
- King, A.O.; Okukado, N.; Negishi, E.-i. Highly general stereo-, regio-, and chemo-selective synthesis of terminal and internal conjugated enynes by the Pd-catalysed reaction of alkynylzinc reagents with alkenyl halides. J. Chem. Soc. Chem. Comm. 1977, 683–684. [Google Scholar] [CrossRef]
- NIOSH Pocket Guide to Chemical Hazards: Bromine. Available online: https://www.cdc.gov/niosh/npg/npgd0064.html (accessed on 14 November 2018).
- Urben, P. Bretherick’s Handbook of Reactive Chemical Hazards; Elsevier: Amsterdam, The Netherlands, 2017. [Google Scholar]
- Ng, W.H.; Shing, T.K.; Yeung, Y.-Y. Mild and Efficient Vicinal Dibromination of Olefins Mediated by Aqueous Ammonium Fluoride. Synlett 2018, 29, 419–424. [Google Scholar]
- De Almeida, L.S.; Esteves, P.M.; de Mattos, M.C. Tribromoisocyanuric acid: A new reagent for regioselective cobromination of alkenes. Synlett 2006, 2006, 1515–1518. [Google Scholar] [CrossRef]
- Kitching, M.O.; Dixon, O.E.; Baumann, M.; Baxendale, I.R. Flow-Assisted Synthesis: A Key Fragment of SR 142948A. Eur. J. Org. Chem. 2017, 2017, 6540–6553. [Google Scholar] [CrossRef]
- Satkar, Y.; Ramadoss, V.; Nahide, P.D.; García-Medina, E.; Juárez-Ornelas, K.A.; Alonso-Castro, A.J.; Chávez-Rivera, R.; Jiménez-Halla, J.O.C.; Solorio-Alvarado, C.R. Practical, mild and efficient electrophilic bromination of phenols by a new I (iii)-based reagent: The PIDA–AlBr 3 system. RSC Adv. 2018, 8, 17806–17812. [Google Scholar] [CrossRef]
- Khusnutdinov, R.; Shchadneva, N.; Mayakova, Y.Y.; Yulamanova, A.; Khazipova, A.; Kutepov, B. Halogenation of Diamantane by Halomethanes Under the Action of Zeolites. Russ. J. Gen. Chem. 2018, 88, 869–873. [Google Scholar] [CrossRef]
- Chaudhuri, M.K.; Khan, A.T.; Patel, B.K.; Dey, D.; Kharmawophlang, W.; Lakshmiprabha, T.; Mandal, G.C. An environmentally benign synthesis of organic ammonium tribromides (OATB) and bromination of selected organic substrates by tetrabutylammonium tribromide (TBATB). Tetrahedron Lett. 1998, 39, 8163–8166. [Google Scholar] [CrossRef] [Green Version]
- Kaushik, M.; Polshettiwar, V. N-octylquinolinium tribromide: A task specific quinoline based ionic liquid as a new brominating agent. Indian. J. Chem. 2006, 45B, 2542–2545. [Google Scholar] [CrossRef]
- Attanasi, O.A.; Berretta, S.; Favi, G.; Filippone, P.; Mele, G.; Moscatelli, G.; Saladino, R. Tetrabromo hydrogenated cardanol: Efficient and renewable brominating agent. Org. Lett. 2006, 8, 4291–4293. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Li, Z.; Yang, C.; Lin, R.; Xia, W. Visible-light photoredox catalysis enabled bromination of phenols and alkenes. Beilstein J. Org. Chem. 2014, 10, 622–627. [Google Scholar] [CrossRef] [PubMed]
- Barhate, N.B.; Gajare, A.S.; Wakharkar, R.D.; Bedekar, A.V. Simple and practical halogenation of arenes, alkenes and alkynes with hydrohalic acid/H2O2 (or TBHP). Tetrahedron 1999, 55, 11127–11142. [Google Scholar] [CrossRef]
- Podgoršek, A.; Stavber, S.; Zupan, M.; Iskra, J. Environmentally benign electrophilic and radical bromination ‘on water’: H2O2–HBr system versus N-bromosuccinimide. Tetrahedron 2009, 65, 4429–4439. [Google Scholar] [CrossRef]
- Semwal, R.; Ravi, C.; Kumar, R.; Meena, R.; Adimurthy, S. Sodium Salts (NaI/NaBr/NaCl) for the Halogenation of Imidazo-Fused Heterocycles. J. Org. Chem. 2019, 84, 792–805. [Google Scholar] [CrossRef] [PubMed]
- Olsen, K.L.; Jensen, M.R.; MacKay, J.A. A mild halogenation of pyrazoles using sodium halide salts and Oxone. Tetrahedron Lett. 2017, 58, 4111–4114. [Google Scholar] [CrossRef]
- Adimurthy, S.; Ghosh, S.; Patoliya, P.U.; Ramachandraiah, G.; Agrawal, M.; Gandhi, M.R.; Upadhyay, S.C.; Ghosh, P.K.; Ranu, B.C. An alternative method for the regio-and stereoselective bromination of alkenes, alkynes, toluene derivatives and ketones using a bromide/bromate couple. Green. Chem. 2008, 10, 232–237. [Google Scholar] [CrossRef]
- Adimurthy, S.; Ramachandraiah, G.; Bedekar, A.V.; Ghosh, S.; Ranu, B.C.; Ghosh, P.K. Eco-friendly and versatile brominating reagent prepared from a liquid bromine precursor. Green. Chem. 2006, 8, 916–922. [Google Scholar] [CrossRef]
- Ghorpade, A.K.; Huddar, S.N.; Akamanchi, K.G. Aq HBr–NaNO2–KI/air: A new catalytic system for α-monobromination of ketones. Tetrahedron Lett. 2016, 57, 4918–4921. [Google Scholar] [CrossRef]
- de la Vega, F.; Sasson, Y.; Huddersman, K. Selectivity in the liquid-phase bromination of aromatics catalyzed by zeolites. Zeolites 1991, 11, 617–621. [Google Scholar] [CrossRef]
- Movsisyan, M.; Delbeke, E.; Berton, J.; Battilocchio, C.; Ley, S.; Stevens, C. Taming hazardous chemistry by continuous flow technology. Chem. Soc. Rev. 2016, 45, 4892–4928. [Google Scholar] [CrossRef]
- Van Waes, F.E.; Seghers, S.; Dermaut, W.; Cappuyns, B.; Stevens, C.V. Efficient continuous-flow bromination of methylsulfones and methanesulfonates and continuous synthesis of hypobromite. J. Flow. Chem. 2014, 4, 118–124. [Google Scholar] [CrossRef]
- Cantillo, D.; Kappe, C.O. Halogenation of organic compounds using continuous flow and microreactor technology. React. Chem. Eng. 2017, 2, 7–19. [Google Scholar] [CrossRef]
- Battilocchio, C.; Bosica, F.; Rowe, S.M.; Abreu, B.L.; Godineau, E.; Lehmann, M.; Ley, S.V. Continuous Preparation and Use of Dibromoformaldoxime as a Reactive Intermediate for the Synthesis of 3-Bromoisoxazolines. Org. Process. Res. Dev. 2017, 21, 1588–1594. [Google Scholar] [CrossRef]
- Godineau, E.; Battilocchio, C.; Lehmann, M.; Ley, S.V.; Labes, R.; Birnoschi, L.; Subramanian, S.; Prasanna, C.S.; Gorde, A.; Kalbagh, M.; et al. A Convergent Continuous Multistep Process for the Preparation of C4-Oxime-Substituted Thiazoles. Org. Process. Res. Dev. 2018, 22, 955–962. [Google Scholar] [CrossRef]
- Yu, W.-b.; Yu, D.-p.; Zheng, M.-m.; Shan, S.-t.; Li, Y.-j.; Gao, J.-r. Catalyst and solvent-free bromination of toluene derivatives by HBr-H2O2 with visible-light photocatalysis using a continuous-flow micro reactor. J. Chem. Res. 2012, 36, 258. [Google Scholar] [CrossRef]
- Howe, P.; Dobson, S.; Malcolm, H. 2, 4, 6-Tribromophenol and Other Simple Brominated Phenols; World Health Organization: Geneva, Switzerland, 2005. [Google Scholar]
- Alaee, M.; Arias, P.; Sjödin, A.; Bergman, Å. An overview of commercially used brominated flame retardants, their applications, their use patterns in different countries/regions and possible modes of release. Environ. Int. 2003, 29, 683–689. [Google Scholar] [CrossRef]
- Ali, N.; Dirtu, A.C.; Eede, N.V.d.; Goosey, E.; Harrad, S.; Neels, H.; Mannetje, A.; Coakley, J.; Douwes, J.; Covaci, A. Occurrence of alternative flame retardants in indoor dust from New Zealand: Indoor sources and human exposure assessment. Chemosphere 2012, 88, 1276–1282. [Google Scholar] [CrossRef]
- Loschen, C.; Hellweg, A.; Klamt, A. COSMOquick, Version 1.7; COSMOlogic GmbH & Co. KG: Leverkusen, Germany, 2018.
- Hornig, M.; Klamt, A. COSMOfrag: A Novel Tool for High-Throughput ADME Property Prediction and Similarity Screening Based on Quantum Chemistry. J. Chem. Inf. Model. 2005, 45, 1169–1177. [Google Scholar] [CrossRef]
- Loschen, C.; Klamt, A. COSMOquick: A novel interface for fast σ-profile composition and its application to COSMO-RS solvent screening using multiple reference solvents. Ind. Eng. Chem. Res. 2012, 51, 14303–14308. [Google Scholar] [CrossRef]
- Ahmed, B.; Barrow, D.; Wirth, T. Enhancement of reaction rates by segmented fluid flow in capillary scale reactors. Adv. Synth. Catal. 2006, 348, 1043–1048. [Google Scholar] [CrossRef]
- Dupont, D.; Raiguel, S.; Binnemans, K. Sulfonic acid functionalized ionic liquids for dissolution of metal oxides and solvent extraction of metal ions. Chem. Commun. 2015, 51, 9006–9009. [Google Scholar] [CrossRef]
- Naert, P.; Rabaey, K.; Stevens, C.V. Ionic liquid ion exchange: Exclusion from strong interactions condemns cations to the most weakly interacting anions and dictates reaction equilibrium. Green Chem. 2018, 20, 4277–4286. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compound trioctyl-(3-sulfopropyl)ammonium perchlorate 4 are available from the authors. |




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Substrate | Product | Solvent | Eq. NaOCl | Eq. Br− | RT2 | T2 | Conversion | Yield |
|---|---|---|---|---|---|---|---|---|---|
| 1 |  |  | CHCl3 | 6 | 9 | 25 min | 50 °C | 100% | 95% |
| 2 |  |  | Et2O | 5 | 7.5 | 150 s | rT | 100% | 83% |
| 3 |  |  | Cyclohexane/DCM | 9 | 22.5 | 1 min | 0 °C | 100% | 97% |
| 4 |  |  | 2-MeTHF/4 | 9 | 22.5 | 3 h | rT | 100% | 78% |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Van Kerrebroeck, R.; Naert, P.; Heugebaert, T.S.A.; D’hooghe, M.; Stevens, C.V. Electrophilic Bromination in Flow: A Safe and Sustainable Alternative to the Use of Molecular Bromine in Batch. Molecules 2019, 24, 2116. https://doi.org/10.3390/molecules24112116
Van Kerrebroeck R, Naert P, Heugebaert TSA, D’hooghe M, Stevens CV. Electrophilic Bromination in Flow: A Safe and Sustainable Alternative to the Use of Molecular Bromine in Batch. Molecules. 2019; 24(11):2116. https://doi.org/10.3390/molecules24112116
Chicago/Turabian StyleVan Kerrebroeck, Reinout, Pieter Naert, Thomas S. A. Heugebaert, Matthias D’hooghe, and Christian V. Stevens. 2019. "Electrophilic Bromination in Flow: A Safe and Sustainable Alternative to the Use of Molecular Bromine in Batch" Molecules 24, no. 11: 2116. https://doi.org/10.3390/molecules24112116