
Selective Targeting of the Interconversion between Glucosylceramide and Ceramide by Scaffold Tailoring of Iminosugar Inhibitors

 ,
,
Abstract
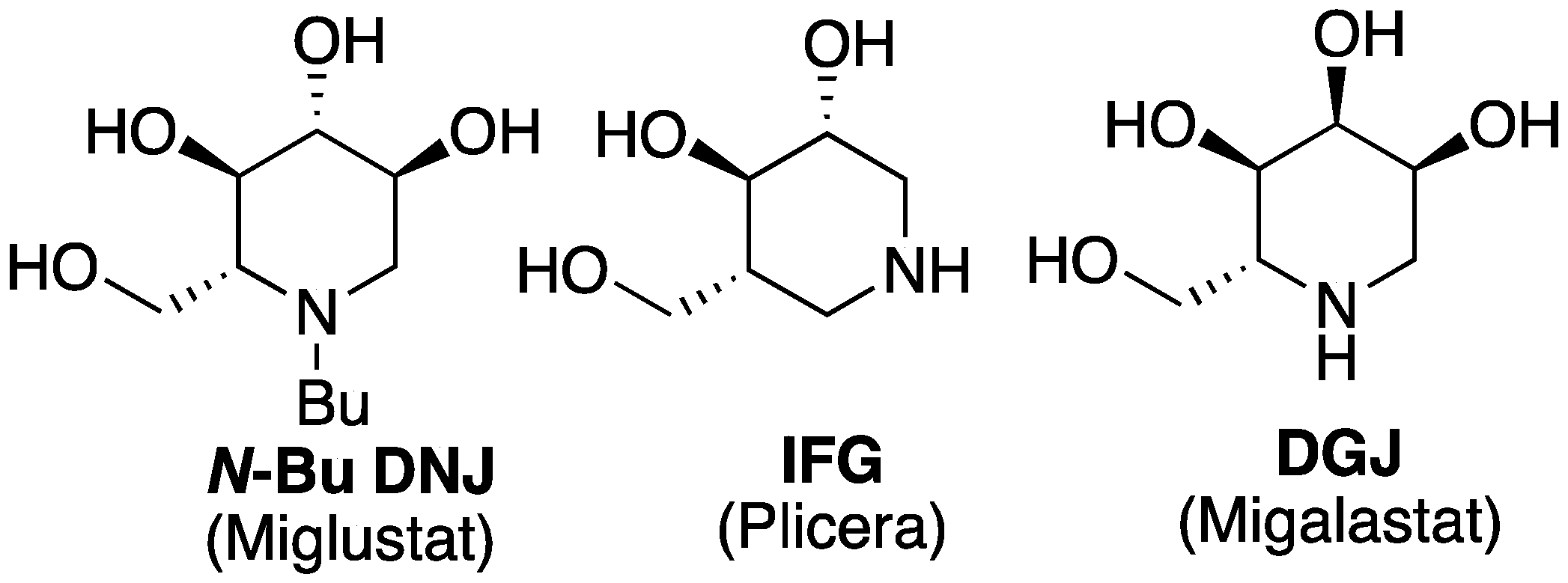
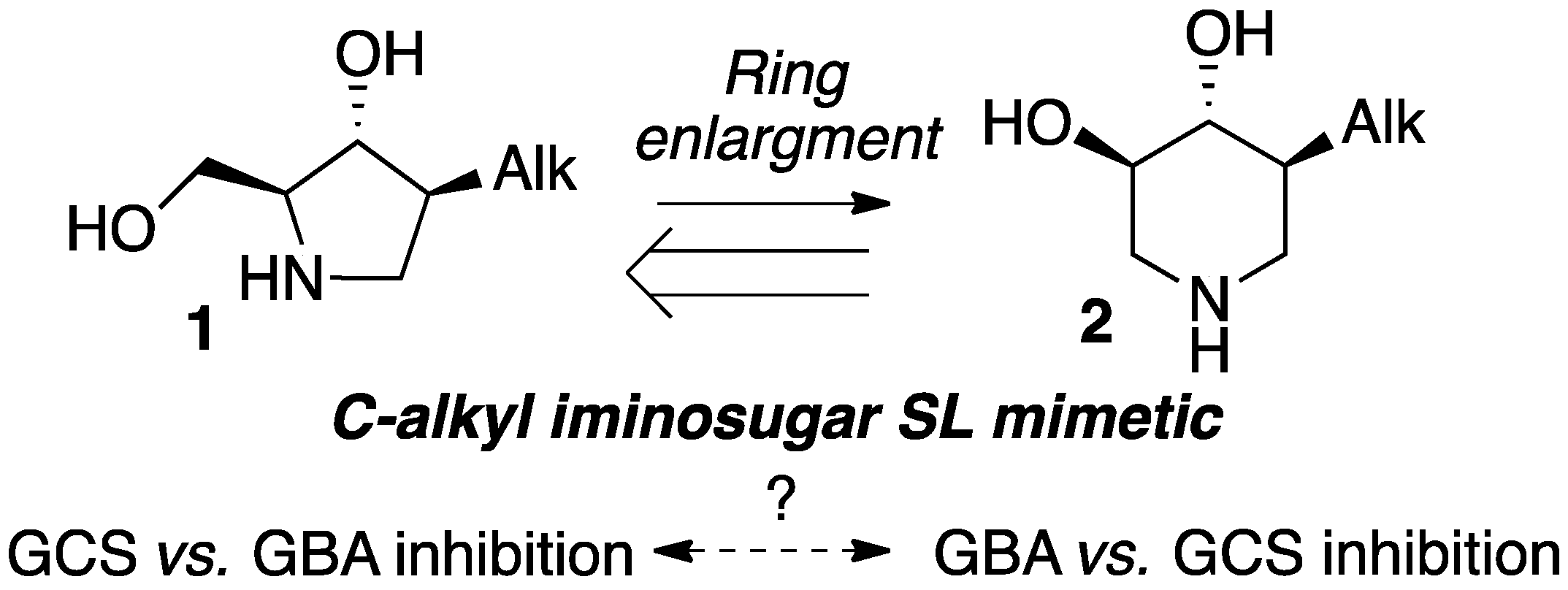
:1. Introduction
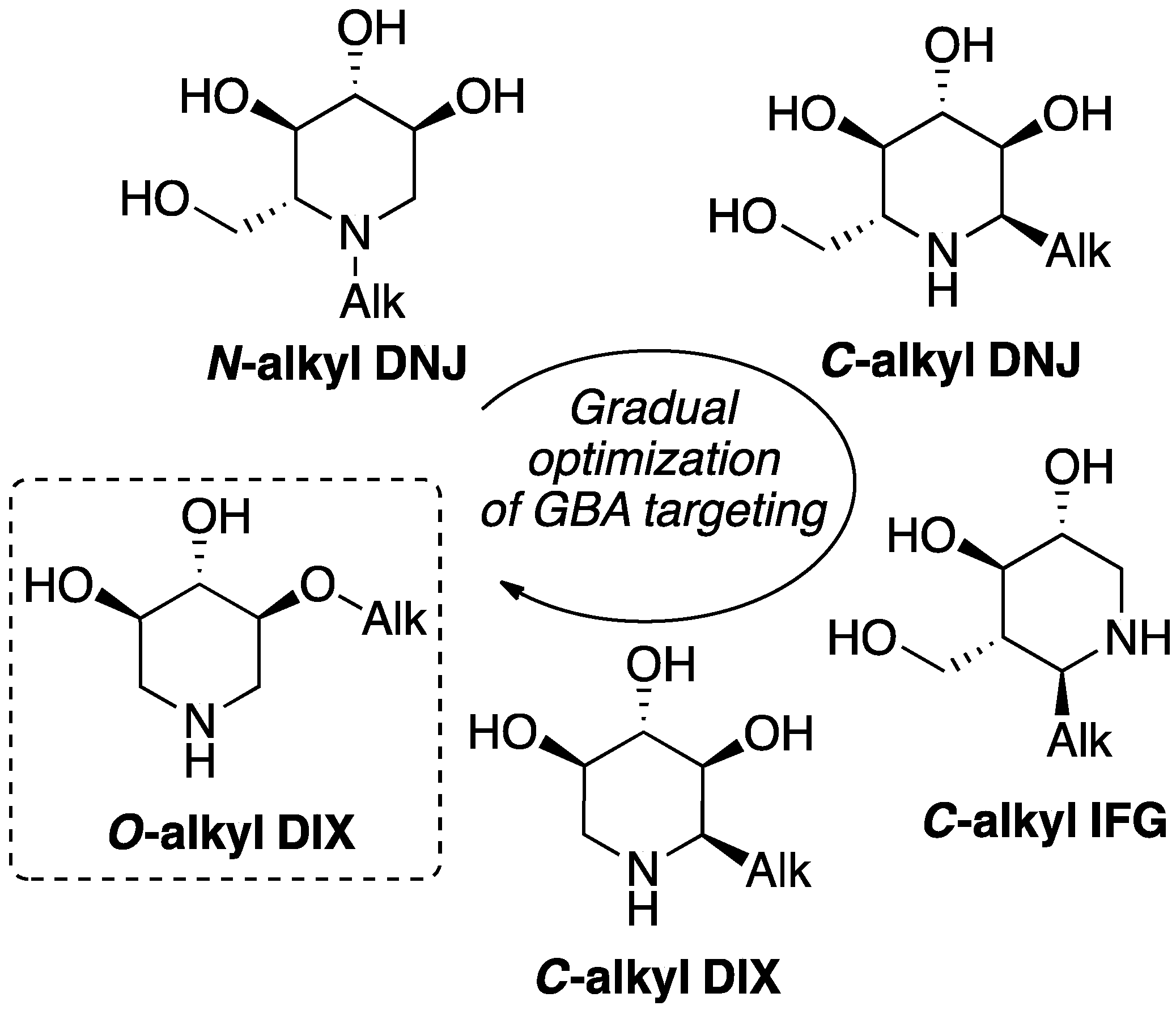
2. Results and Discussion
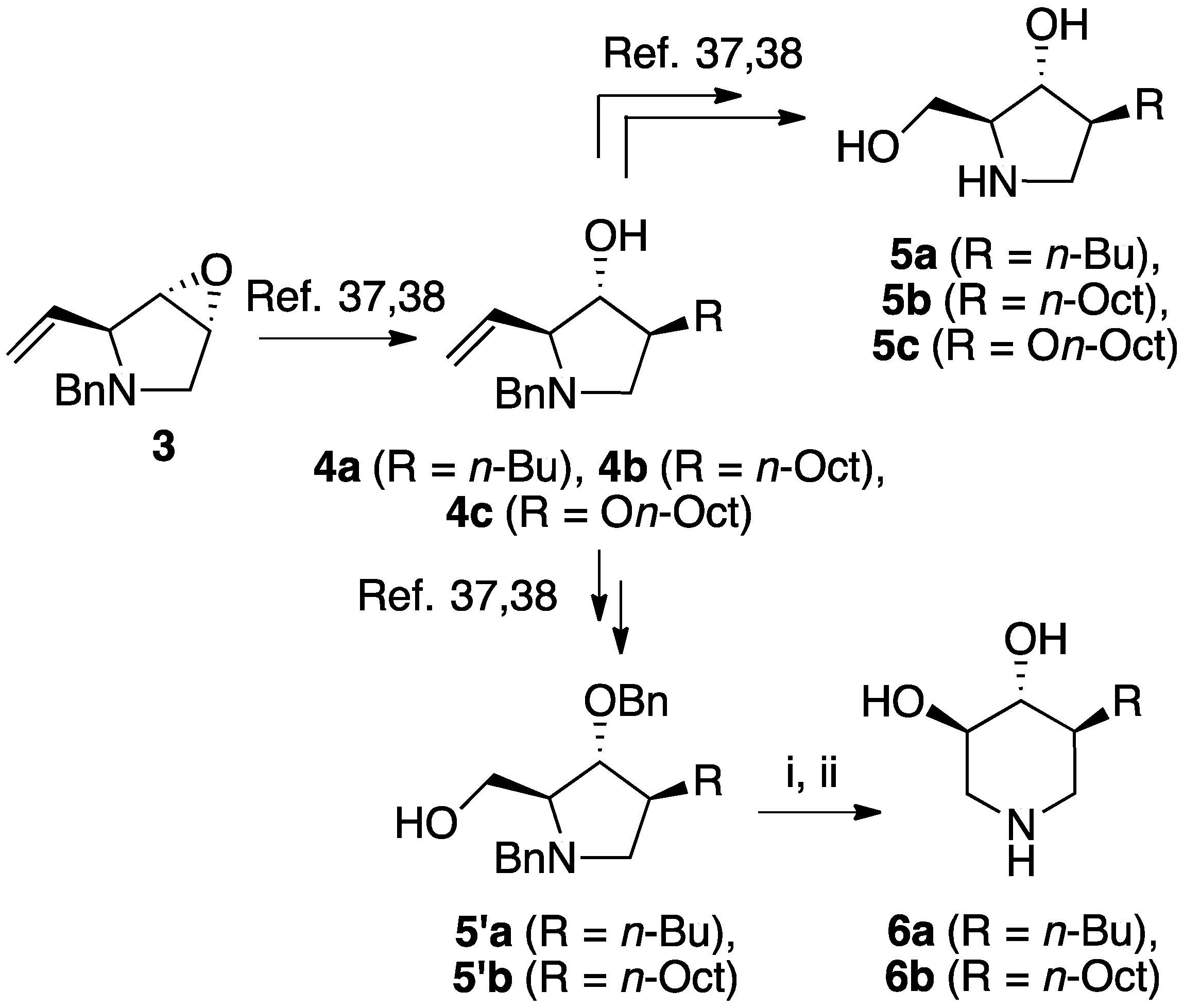
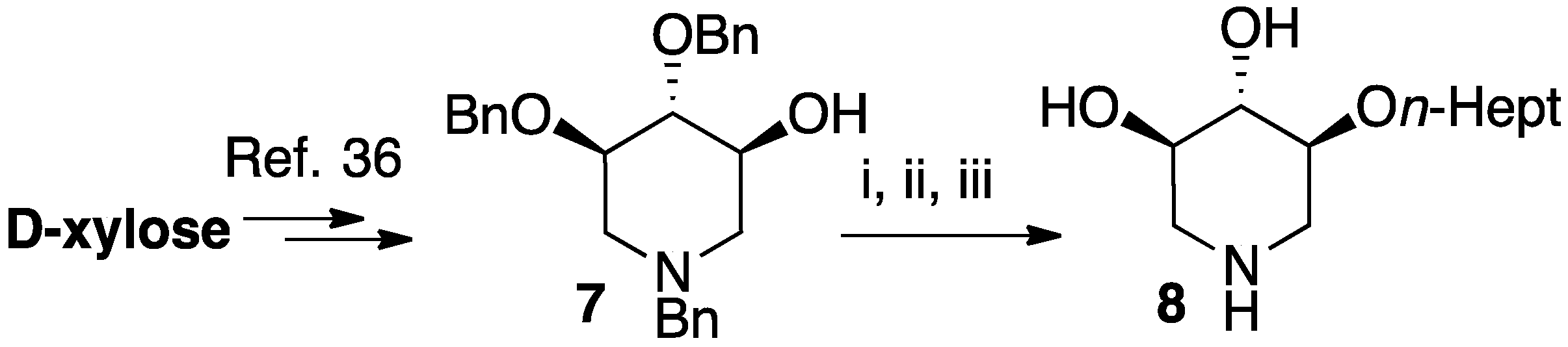
2.1. Chemical Synthesis
2.2. Effect of Iminosugars on Cell Viability
2.3. Effect of Iminosugars on GCS Inhibition
2.4. Effect of Iminosugars on GBA Inhibition
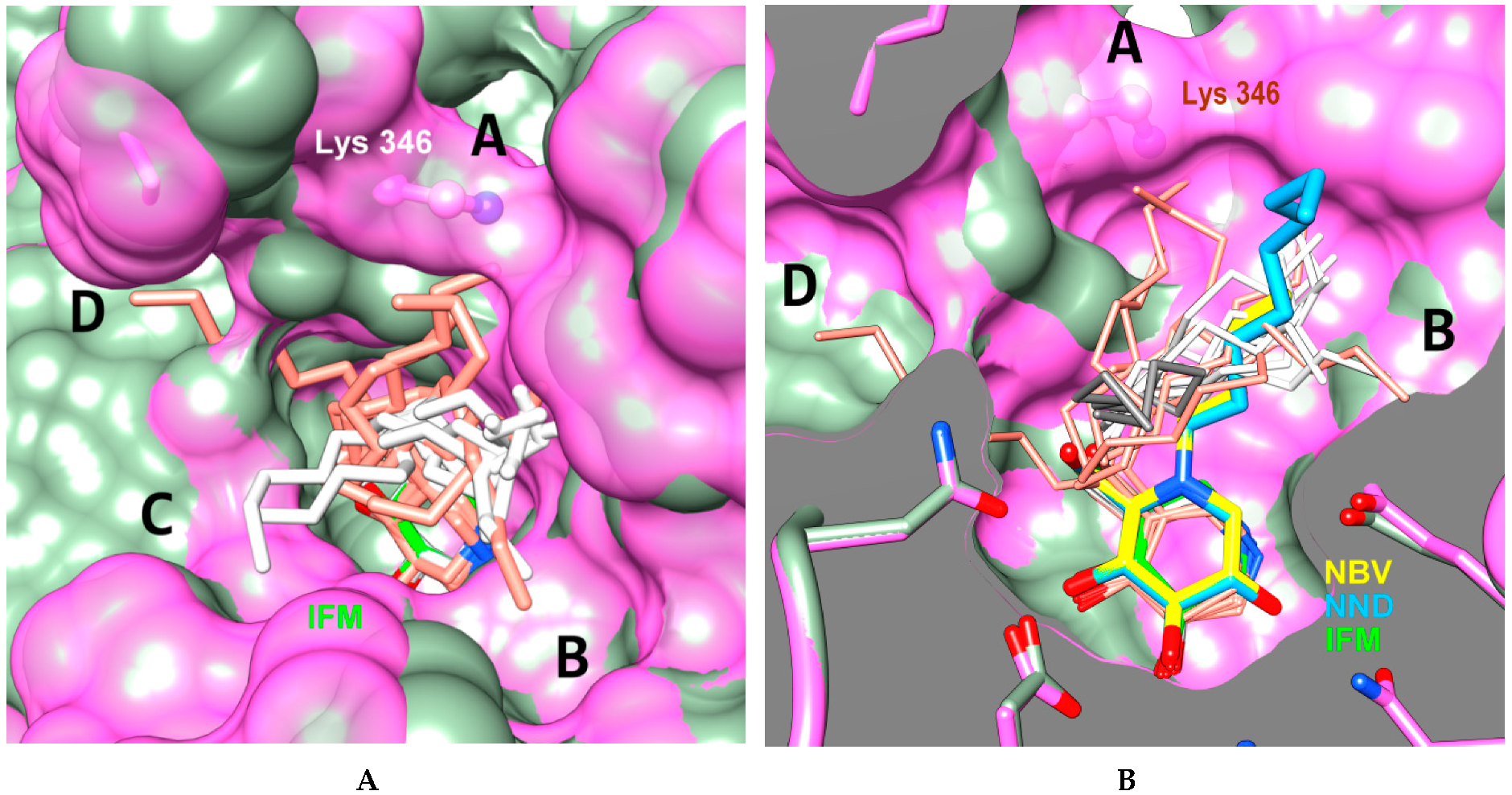
2.5. Molecular Docking
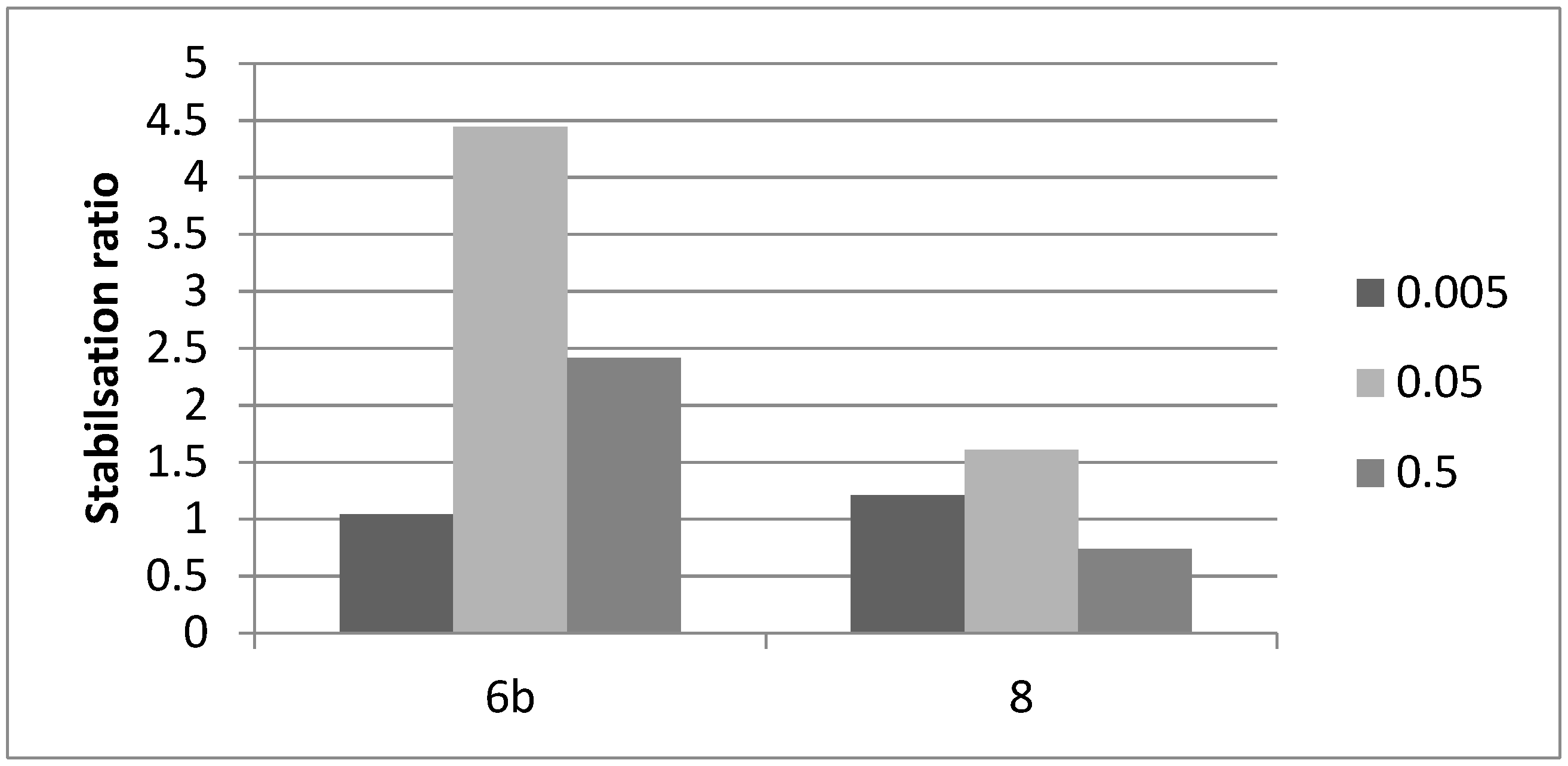
2.6. GBA Pharmacological Chaperone Activity
2.6.1. Preliminary In Vitro Studies
2.6.2. Enhancement of GBA Activity in GD Patient Cells
3. Discussion
4. Materials and Methods
4.1. General Methods
4.2. Synthesis of Compounds 6b and 8
4.3. Cellular Inhibition Assays against Glucosylceramide Synthase
4.4. Inhibition Assays against Recombinant β-glucocerebrosidase.
4.5. Molecular Docking
4.6. Thermal Stabilization Assay Using Recombinant β-Glucocerebrosidase
4.7. Culture and Cell Viability Assays
4.8. Assay of β-glucocerebrosidase Activity
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References and Note
- Merrill, A.H. Sphingolipid and Glycosphingolipid Metabolic Pathways in the Era of Sphingolipidomics. Chem. Rev. 2011, 111, 6387–6422. [Google Scholar] [CrossRef] [PubMed]
- Hannun, Y.A.; Obeid, L.M. Principles of bioactive lipid signalling: Lessons from sphingolipids. Nat. Rev. Mol. Cell Biol. 2008, 9, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Morad, S.A.F.; Cabot, M.C. Ceramide-orchestrated signalling in cancer cells. Nat. Rev. Cancer 2013, 13, 51–65. [Google Scholar] [CrossRef] [PubMed]
- Platt, F.M. Emptying the stores: Lysosomal diseases and therapeutic strategies. Nat. Rev. Drug Discov. 2018, 17, 133–150. [Google Scholar] [CrossRef]
- Siebert, M.; Sidransky, E.; Westbroek, W. Glucocerebrosidase is shaking up the synucleinopathies. Brain 2014, 137, 1304–1322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schapira, A.H.V.; Chiasserini, D.; Beccari, T.; Parnetti, L. Glucocerebrosidase in Parkinson’s disease: Insights into Pathogenesis and Prospects for Treatment. Mov. Disord. 2016, 31, 830–835. [Google Scholar] [CrossRef] [PubMed]
- McMahon, B.; Aflaki, E.; Sidransky, E. Chaperoning glucocerebrosidase: A therapeutic strategy for both Gaucher disease and Parkinsonism. Neural Regen. Res. 2016, 11, 1760–1761. [Google Scholar]
- Jung, O.; Patnaik, S.; Marugan, J.; Sidransky, E.; Westbroek, W. Progress and potential of non-inhibitory small molecule chaperones for the treatment of Gaucher disease and its implications for Parkinson disease. Expert Rev. Proteomic 2016, 13, 471–479. [Google Scholar] [CrossRef]
- Yandim, M.K.; Apohan, E.; Baran, Y. Therapeutic potential of targeting ceramide/glucosylceramide pathway in cancer. Cancer Chemother. Pharmacol. 2013, 71, 13–20. [Google Scholar] [CrossRef]
- Giussani, P.; Tringali, C.; Riboni, L.; Viani, P.; Venerando, B. Sphingolipids: Key Regulators of Apoptosis and Pivotal Players in Cancer Drug Resistance. Int. J. Mol. Sci. 2014, 15, 4356–4392. [Google Scholar] [CrossRef] [Green Version]
- Coutinho, M.F.; Santos, J.I.; Alves, S. Less Is More: Substrate Reduction Therapy for Lysosomal Storage Disorders. Int. J. Mol. Sci. 2016, 17, 1065. [Google Scholar] [CrossRef] [PubMed]
- Boyd, R.E.; Lee, G.; Rybczynski, P.; Benjamin, E.R.; Khanna, R.; Wustman, B.A.; Valenzano, K.J. Pharmacological Chaperones as Therapeutics for Lysosomal Storage Diseases. J. Med. Chem. 2013, 56, 2705–2725. [Google Scholar] [CrossRef]
- Sanchez-Fernandez, E.M.; Fernandez, J.M.G.; Mellet, C.O. Glycomimetic-based pharmacological chaperones for lysosomal storage disorders: Lessons from Gaucher, G(M1)-gangliosidosis and Fabry diseases. Chem. Commun. 2016, 52, 5497–5515. [Google Scholar] [CrossRef] [PubMed]
- Convertino, M.; Das, J.; Dokholyan, N.V. Pharmacological Chaperones: Design and Development of New Therapeutic Strategies for the Treatment of Conformational Diseases. ACS Chem. Biol. 2016, 11, 1471–1489. [Google Scholar] [CrossRef] [PubMed]
- Parenti, G. Treating lysosomal storage diseases with pharmacological chaperones: From concept to clinics. EMBO Mol. Med. 2009, 1, 268–279. [Google Scholar] [CrossRef] [PubMed]
- Parenti, G.; Andria, G.; Valenzano, K.J. Pharmacological Chaperone Therapy: Preclinical Development, Clinical Translation, and Prospects for the Treatment of Lysosomal Storage Disorders. Mol. Ther. 2015, 23, 1138–1148. [Google Scholar] [CrossRef] [PubMed]
- Home, G.; Wilson, F.X.; Tinsley, J.; Williams, D.H.; Storer, R. Iminosugars past, present and future: Medicines for tomorrow. Drug Discov. Today 2011, 16, 107–118. [Google Scholar]
- Lachmann, R.H. Miglustat: Substrate reduction therapy for glycosphingolipid lysosomal storage disorders. Drug Today 2006, 42, 29–38. [Google Scholar] [CrossRef]
- Lyseng-Williamson, K.A. Miglustat: A Review of Its Use in Niemann-Pick Disease Type C. Drugs 2014, 74, 61–74. [Google Scholar] [CrossRef]
- Pineda, M.; Walterfang, M.; Patterson, M.C. Miglustat in Niemann-Pick disease type C patients: A review. Orphanet J. Rare Dis. 2018, 13, 140. [Google Scholar] [CrossRef]
- Okumiya, T.; Kroos, M.A.; Van Wet, L.; Takeuchi, H.; Van der Ploeg, A.T.; Reuser, A.J.J. Chemical chaperones improve transport and enhance stability of mutant alpha-glucosidases in glycogen storage disease type II. Mol. Genet. Metab. 2007, 90, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Parenti, G.; Zuppaldi, A.; Pittis, M.G.; Tuzzi, M.R.; Annunziata, I.; Meroni, G.; Porto, C.; Donaudy, F.; Rossi, B.; Rossi, M.; et al. Pharmacological enhancement of mutated alpha-glucosidase activity in fibroblasts from patients with Pompe disease. Mol. Ther. 2007, 15, 508–514. [Google Scholar] [CrossRef] [PubMed]
- Sawkar, A.R.; Cheng, W.C.; Beutler, E.; Wong, C.H.; Balch, W.E.; Kelly, J.W. Chemical chaperones increase the cellular activity of N370S beta-glucosidase: A therapeutic strategy for Gaucher disease. Proc. Natl. Acad. Sci. USA 2002, 99, 15428–15433. [Google Scholar] [CrossRef] [PubMed]
- Diot, J.D.; Moreno, I.G.; Twigg, G.; Mellet, C.O.; Haupt, K.; Butters, T.D.; Kovensky, J.; Gouin, S.G. Amphiphilic 1-Deoxynojirimycin Derivatives through Click Strategies for Chemical Chaperoning in N370S Gaucher Cells. J. Org. Chem. 2011, 76, 7757–7768. [Google Scholar] [CrossRef] [PubMed]
- Steet, R.A.; Chung, S.; Wustman, B.; Powe, A.; Do, H.; Kornfeld, S.A. The iminosugar isofagomine increases the activity of N370S mutant acid beta-glucosidase in Gaucher fibroblasts by several mechanisms. Proc. Natl. Acad. Sci. USA 2006, 103, 13813–13818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Z.Q.; Sawkar, A.R.; Whalen, L.J.; Wong, C.H.; Kelly, J.W. Isofagomine- and 2,5-anhydro-2,5-imino-D-glucitol-based glucocerebrosidase pharmacological chaperones for Gaucher disease intervention. J. Med. Chem. 2007, 50, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Markham, A. Migalastat: First Global Approval. Drugs 2016, 76, 1147–1152. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Ikeda, K.; Kato, A.; Adachi, I.; Godin, G.; Compain, P.; Martin, O.; Asano, N. alpha-1-C-octyl-1-deoxynojirimycin as a pharmacological chaperone for Gaucher disease. Bioorg. Med. Chem. 2006, 14, 7736–7744. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.X.; Sheth, K.A.; Li, S.H.; Chang, H.H.; Fan, J.Q. Rational design and synthesis of highly potent beta-glucocerebrosidase inhibitors. Angew. Chem. Int. Ed. 2005, 44, 7450–7453. [Google Scholar] [CrossRef]
- Hill, T.; Tropak, M.B.; Mahuran, D.; Withers, S.G. Synthesis, Kinetic Evaluation and Cell-Based Analysis of C-Alkylated Isofagomines as Chaperones of beta-Glucocerebrosidase. ChemBioChem 2011, 12, 2151–2154. [Google Scholar] [CrossRef]
- Compain, P.; Martin, O.R.; Boucheron, C.; Godin, G.; Yu, L.; Ikeda, K.; Asano, N. Design and synthesis of highly potent and selective pharmacological chaperones for the treatment of Gaucher’s disease. ChemBioChem 2006, 7, 1356–1359. [Google Scholar] [CrossRef] [PubMed]
- Wennekes, T.; van den Berg, R.J.B.H.N.; Boltje, T.J.; Donker-Koopman, W.E.; Kuijper, B.; van der Marel, G.A.; Strijland, A.; Verhagen, C.P.; Aerts, J.M.F.G.; Overkleeft, H.S. Synthesis and Evaluation of Lipophilic Aza-C-glycosides as Inhibitors of Glucosylceramide Metabolism. Eur. J. Org. Chem. 2010, 2010, 1258–1283. [Google Scholar] [CrossRef]
- Goddard-Borger, E.D.; Tropak, M.B.; Yonekawa, S.; Tysoe, C.; Mahuran, D.J.; Withers, S.G. Rapid Assembly of a Library of Lipophilic Iminosugars via the Thiol-Ene Reaction Yields Promising Pharmacological Chaperones for the Treatment of Gaucher Disease. J. Med. Chem. 2012, 55, 2737–2745. [Google Scholar] [CrossRef] [PubMed]
- Schonemann, W.; Gallienne, E.; Ikeda-Obatake, K.; Asano, N.; Nakagawa, S.; Kato, A.; Adachi, I.; Gorecki, M.; Frelek, J.; Martin, O.R. Glucosylceramide Mimics: Highly Potent GCase Inhibitors and Selective Pharmacological Chaperones for Mutations Associated with Types 1 and 2 Gaucher Disease. ChemMedChem 2013, 8, 1805–1817. [Google Scholar] [CrossRef] [PubMed]
- Serra-Vinardell, J.; Diaz, L.; Casas, J.; Grinberg, D.; Vilageliu, L.; Michelakakis, H.; Mavridou, I.; Aerts, J.M.F.G.; Decroocq, C.; Compain, P.; et al. Glucocerebrosidase Enhancers for Selected Gaucher Disease Genotypes by Modification of alpha-1-C-Substituted Imino-D-xylitols (DIXs) by Click Chemistry. ChemMedChem 2014, 9, 1744–1754. [Google Scholar] [PubMed]
- Oulaidi, F.; Front-Deschamps, S.; Gallienne, E.; Lesellier, E.; Ikeda, K.; Asano, N.; Compain, P.; Martin, O.R. Second-Generation Iminoxylitol-Based Pharmacological Chaperones for the Treatment of Gaucher Disease. ChemMedChem 2011, 6, 353–361. [Google Scholar] [CrossRef] [PubMed]
- Faugeroux, V.; Genisson, Y.; Andrieu-Abadie, N.; Colie, S.; Levade, T.; Baltas, M. C-Alkyl 5-membered ring imino sugars as new potent cytotoxic glucosylceramide synthase inhibitors. Org. Biomol. Chem. 2006, 4, 4437–4439. [Google Scholar] [CrossRef]
- Rives, A.; Genisson, Y.; Faugeroux, V.; Zedde, C.; Lepetit, C.; Chauvin, R.; Saffon, N.; Andrieu-Abadie, N.; Colie, S.; Levade, T.; et al. Highly Regioselective Oxirane Ring-Opening of a Versatile Epoxypyrrolidine Precursor of New Imino-Sugar-Based Sphingolipid Mimics. Eur. J. Org. Chem. 2009, 2009, 2474–2489. [Google Scholar] [CrossRef]
- Rives, A.; Genisson, Y.; Faugeroux, V.; Saffon, N.; Baltas, M. Enantioselective Access to All-trans 5-Alkylpiperidine-3,4-diols: Application to the Asymmetric Synthesis of the 1-N-Iminosugar (+)-Isofagomine. Synthesis 2009, 2009, 3251–3258. [Google Scholar]
- Pardo, D.G.; Cossy, J. Access to Optically Active 3-Substituted Piperidines by Ring Expansion of Prolinols and Derivatives. Chem. Eur. J. 2014, 20, 4516–4525. [Google Scholar] [CrossRef]
- Kato, A.; Nakagome, I.; Sato, K.; Yamamoto, A.; Adachi, I.; Nash, R.J.; Fleet, G.W.J.; Natori, Y.; Watanabe, Y.; Imahori, T.; et al. Docking study and biological evaluation of pyrrolidine-based iminosugars as pharmacological chaperones for Gaucher disease. Org. Biomol. Chem. 2016, 14, 1039–1048. [Google Scholar] [CrossRef] [PubMed]
- Nakagome, I.; Kato, A.; Yamaotsu, N.; Yoshida, T.; Ozawa, S.-I.; Adachi, I.; Hirono, S. Design of a New α-1-C-Alkyl-DAB Derivative Acting as a Pharmacological Chaperone for β-Glucocerebrosidase Using Ligand Docking and Molecular Dynamics Simulation. Molecules 2018, 23, 2683. [Google Scholar] [CrossRef] [PubMed]
- Kato, A.; Miyauchi, S.; Kato, N.; Nash, R.J.; Yoshimura, Y.; Nakagome, I.; Hirono, S.; Takahata, H.; Adachi, I. Docking and SAR Studies of D- and L-Isofagomine Isomers as Human β-Glucocerebrosidase Inhibitors. Bioorg. Med. Chem. 2011, 19, 3558–3568. [Google Scholar] [CrossRef] [PubMed]
- Brumshtein, B.; Greenblatt, H.M.; Butters, T.D.; Shaaltiel, Y.; Aviezer, D.; Silman, I.; Futerman, A.H.; Sussman, J.L. Crystal Structures of Complexes of N-Butyl- and N-Nonyl-Deoxynojirimycin Bound to Acid Beta-Glucosidase: Insights into the Mechanism of Chemical Chaperone Action in Gaucher Disease. J. Biol. Chem. 2007, 282, 29052. [Google Scholar] [CrossRef] [PubMed]
- For a selected example, see: Trapero, A.; Gonzalez-Bulnes, P.; Butters, T.D.; Llebaria, A. Potent Aminocyclitol Glucocerebrosidase Inhibitors are Subnanomolar Pharmacological Chaperones for Treating Gaucher Disease. J. Med. Chem. 2012, 55, 4479–4488. [Google Scholar] and references cited therein.[Green Version]
- Trapero, A.; Alfonso, I.; Butters, T.D.; Llebaria, A. Polyhydroxylated bicyclic isoureas and guanidines are potent glucocerebrosidase inhibitors and nanomolar enzyme activity enhancers in Gaucher cells. J. Am. Chem. Soc. 2011, 133, 5474–5484. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera: A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dvir, H.; Harel, M.; McCarthy, A.A.; Toker, L.; Silman, I.; Futerman, A.H.; Sussman, J.L. X-ray structure of human acid-β-glucosidase, the defective enzyme in Gaucher disease. EMBO Rep. 2003, 4, 704–709. [Google Scholar] [CrossRef] [Green Version]
- Meng, E.C.; Pettersen, E.F.; Couch, G.S.; Huang, C.C.; Ferrin, T.E. Tools for integrated sequence-structure analysis with UCSF Chimera. BMC Bioinf. 2006, 7, 339. [Google Scholar]
- Sousa, S.F.; Ribeiro, A.J.M.; Coimbra, J.T.S.; Neves, R.P.P.; Martins, S.A.; Moorthy, N.S.H.N.; Fernandes, P.A.; Ramos, M.J. Protein-Ligand Docking in the New Millennium—A Retrospective of 10 Years in the Field. Curr. Med. Chem. 2013, 20, 2296–2314. [Google Scholar] [CrossRef]
- Lieberman, R.L.; Wustman, B.A.; Huertas, P.; Powe, A.C.; Pine, C.W.; Khanna, R.; Schlossmacher, M.G.; Ringe, D.; Petsko, G.A. Structure of acid β-glucosidase with pharmacological chaperone provides insight into Gaucher disease. Nat. Chem. Biol. 2007, 3, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Brumshtein, B.; Aguilar-Moncayo, M.; García-Moreno, M.I.; Ortiz Mellet, C.; García Fernández, J.M.; Silman, I.; Shaaltiel, Y.; Aviezer, D.; Sussman, J.L.; Futerman, A.H. 6-Amino-6-deoxy-5,6-di-N-(N′-octyliminomethylidene)nojirimycin: Synthesis, Biological Evaluation, and Crystal Structure in Complex with Acid β-Glucosidase. ChemBioChem 2009, 10, 1480–1485. [Google Scholar] [CrossRef] [PubMed]
- Thomsen, R.; Christensen, M.H. MolDock: A New Technique for High-Accuracy Molecular Docking. J. Med. Chem. 2006, 49, 3315–3321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wenger, D.A.; Williams, C. Techniques in Diagnostic Human Biochemical Genetics: A Laboratory Manual; Hommes, F.A., Ed.; Wiley-Liss Inc.: New York, NY, USA, 1991; pp. 587–617. [Google Scholar]
Sample Availability: Samples of the compounds are not available from the authors. |











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
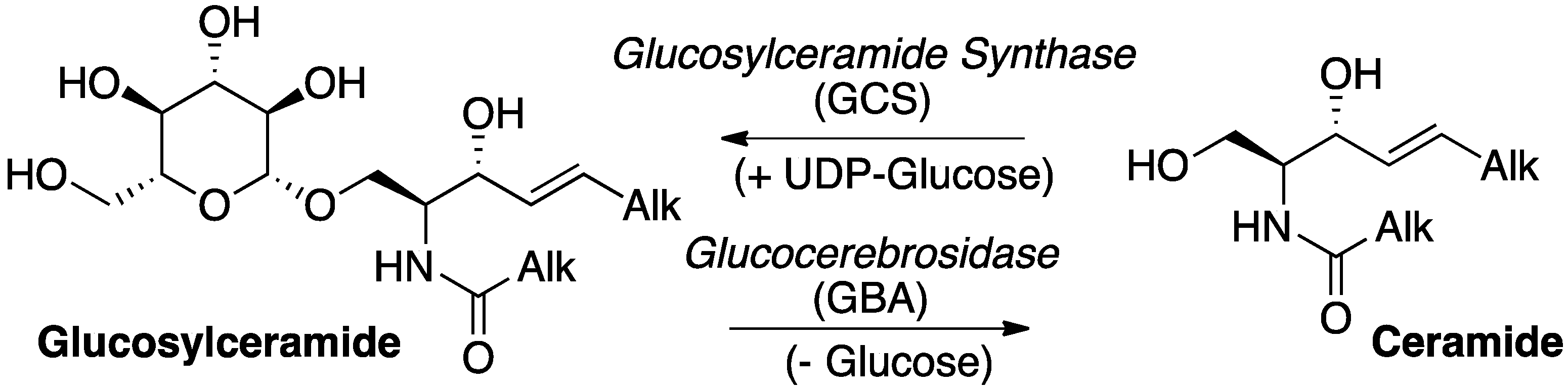{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Viability 1 | Murine Melanoma 2 | Human Fibroblast 3 | Gaucher Fibroblast 4 |
|---|---|---|---|
 | 85% | ND 5 | ND |
 | 14% | 12.5% | 14% |
 | 16% | 13.7% | 14% |
 | 85% | 91% | 84% |
 | ND | 99% | 94% |
| Enzyme Inhibition | GCS 1 | GBA 2 |
|---|---|---|
 | 15% | >8000 nM |
 | 47% | 4200 nM |
 | 20% | 6000 nM |
 | NI 3 | 108 nM |
 | NI | 11 nM |
 | ND 4 | 4 nM |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baudoin-Dehoux, C.; Castellan, T.; Rodriguez, F.; Rives, A.; Stauffert, F.; Garcia, V.; Levade, T.; Compain, P.; Génisson, Y. Selective Targeting of the Interconversion between Glucosylceramide and Ceramide by Scaffold Tailoring of Iminosugar Inhibitors. Molecules 2019, 24, 354. https://doi.org/10.3390/molecules24020354
Baudoin-Dehoux C, Castellan T, Rodriguez F, Rives A, Stauffert F, Garcia V, Levade T, Compain P, Génisson Y. Selective Targeting of the Interconversion between Glucosylceramide and Ceramide by Scaffold Tailoring of Iminosugar Inhibitors. Molecules. 2019; 24(2):354. https://doi.org/10.3390/molecules24020354
Chicago/Turabian StyleBaudoin-Dehoux, Cécile, Tessa Castellan, Frédéric Rodriguez, Arnaud Rives, Fabien Stauffert, Virginie Garcia, Thierry Levade, Philippe Compain, and Yves Génisson. 2019. "Selective Targeting of the Interconversion between Glucosylceramide and Ceramide by Scaffold Tailoring of Iminosugar Inhibitors" Molecules 24, no. 2: 354. https://doi.org/10.3390/molecules24020354