Synthesis and Structural Characterization of Amidine, Amide, Urea and Isocyanate Derivatives of the Amino-closo-dodecaborate Anion [B12H11NH3]−


 and
and
Abstract
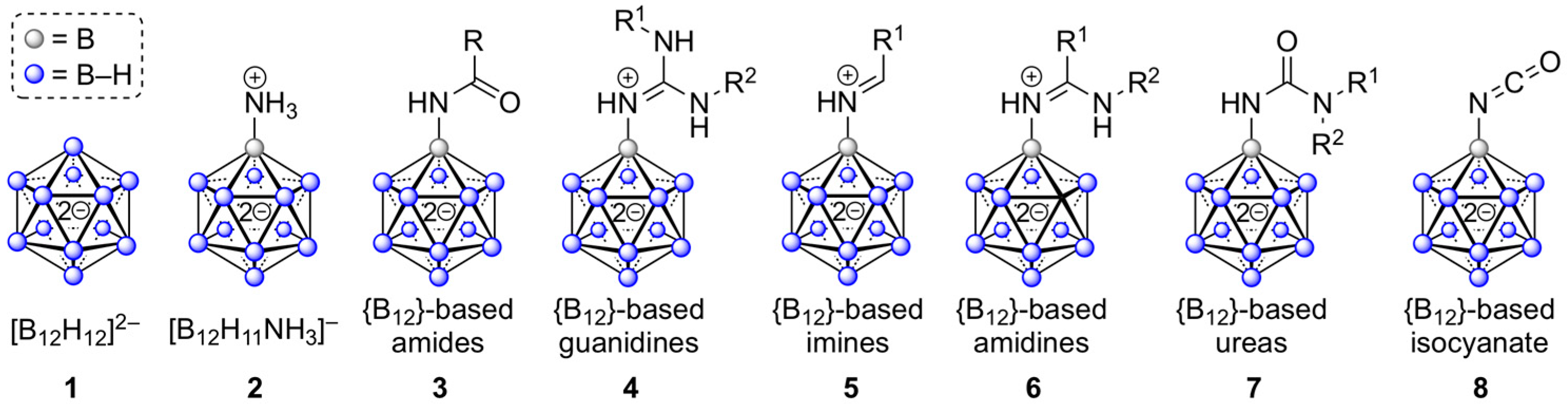
:1. Introduction
2. Results and Discussion
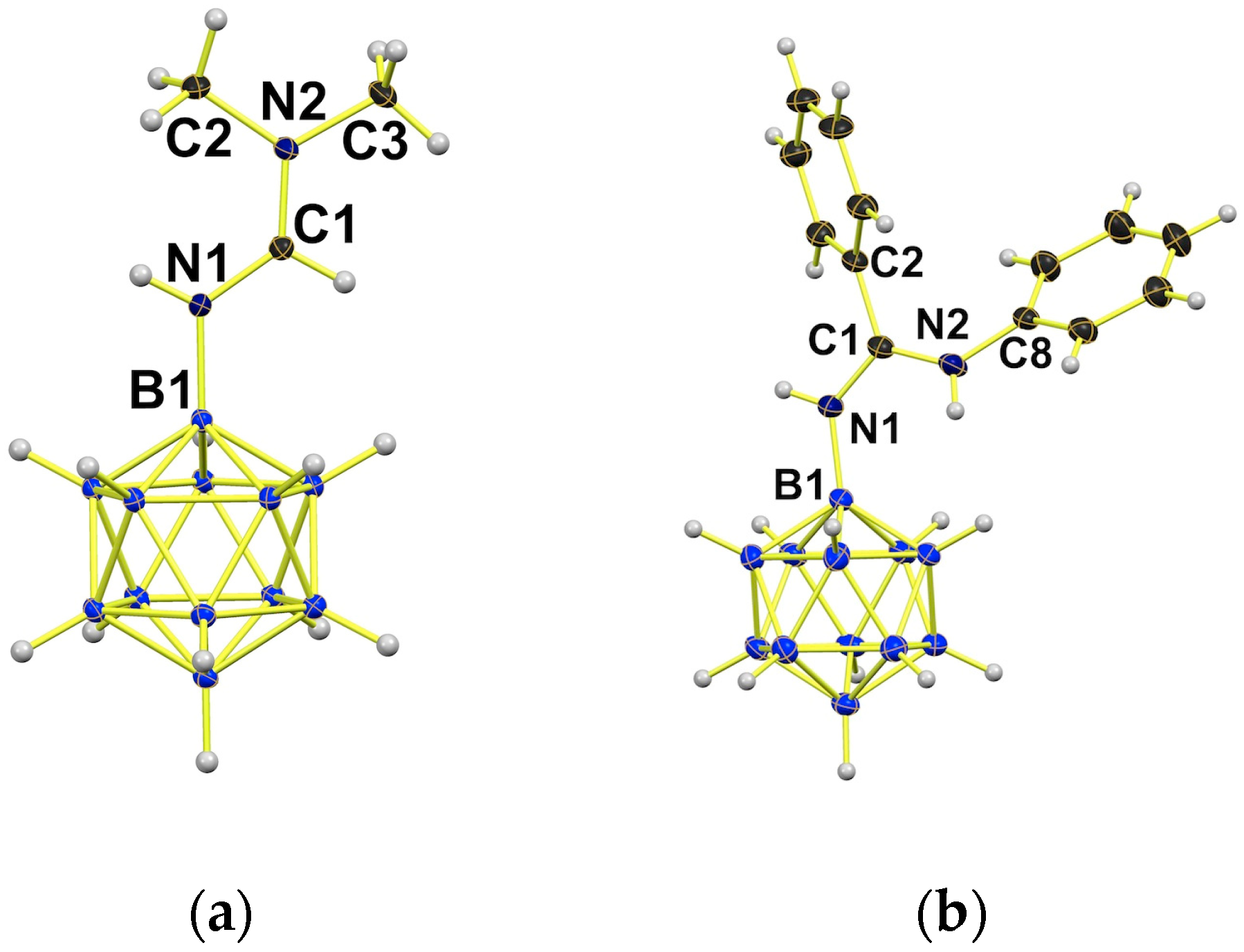
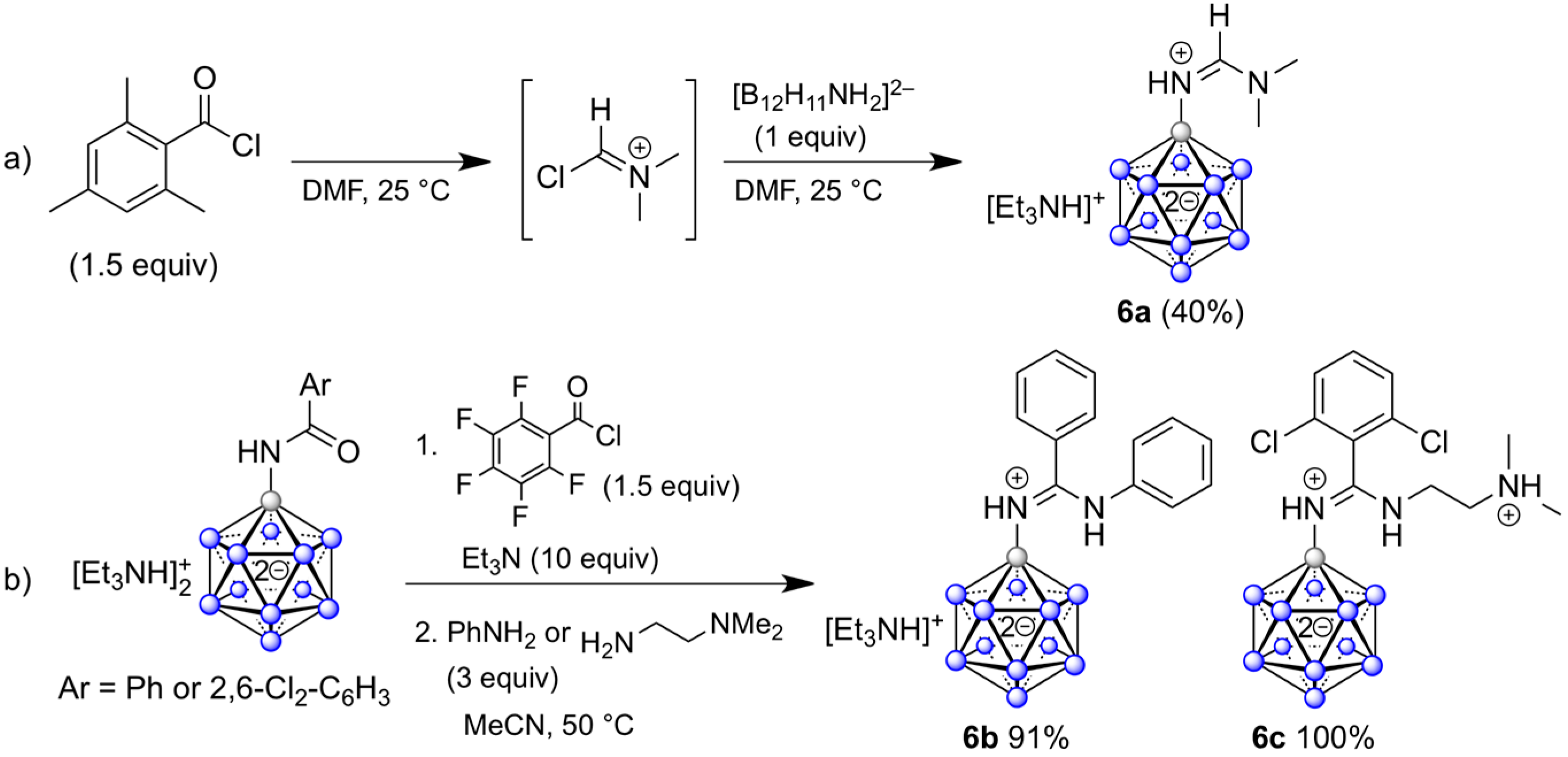
2.1. Synthesis of {B12}-based Amidinium Ions
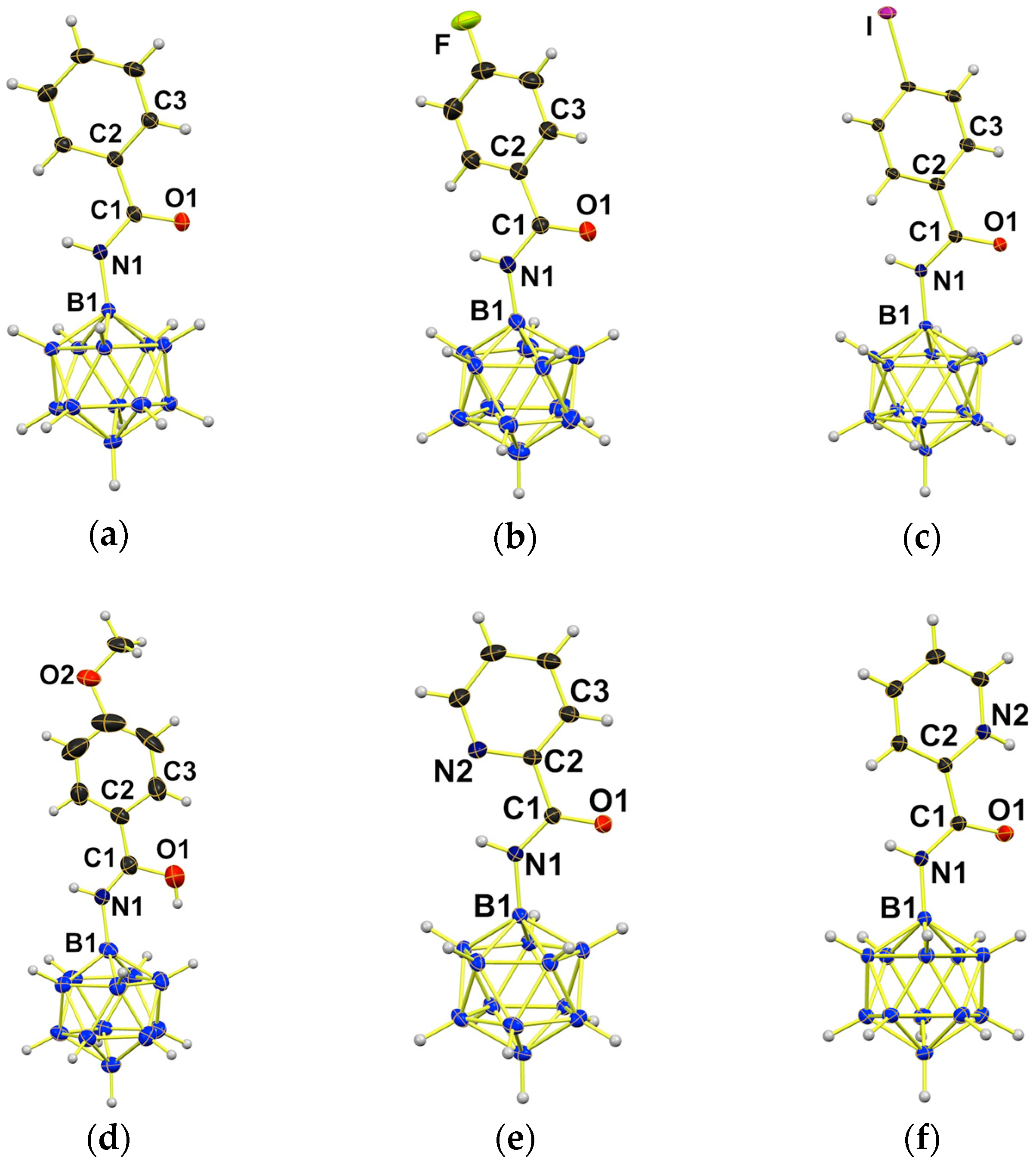
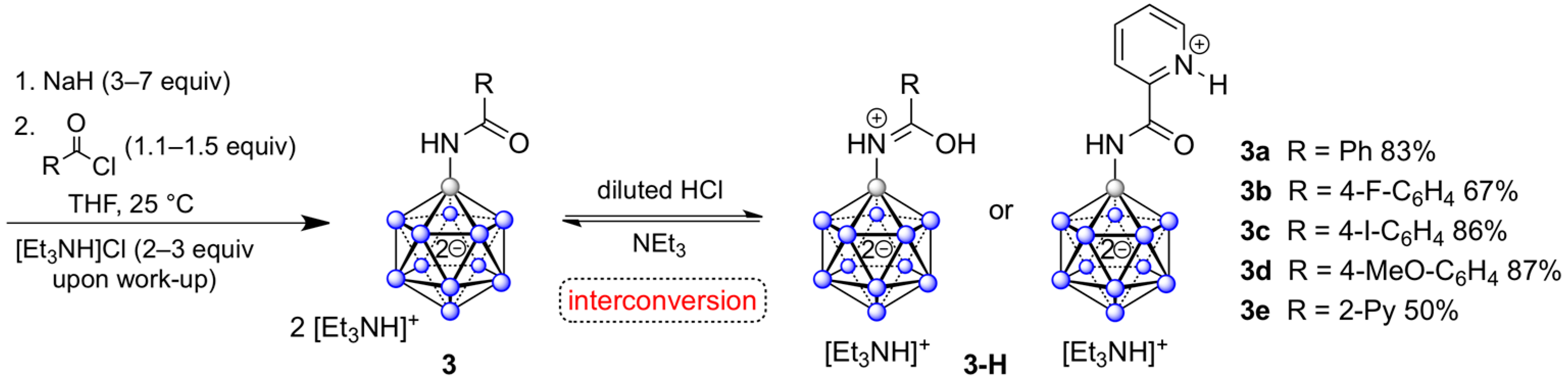
2.2. Synthesis of {B12}-based Amides
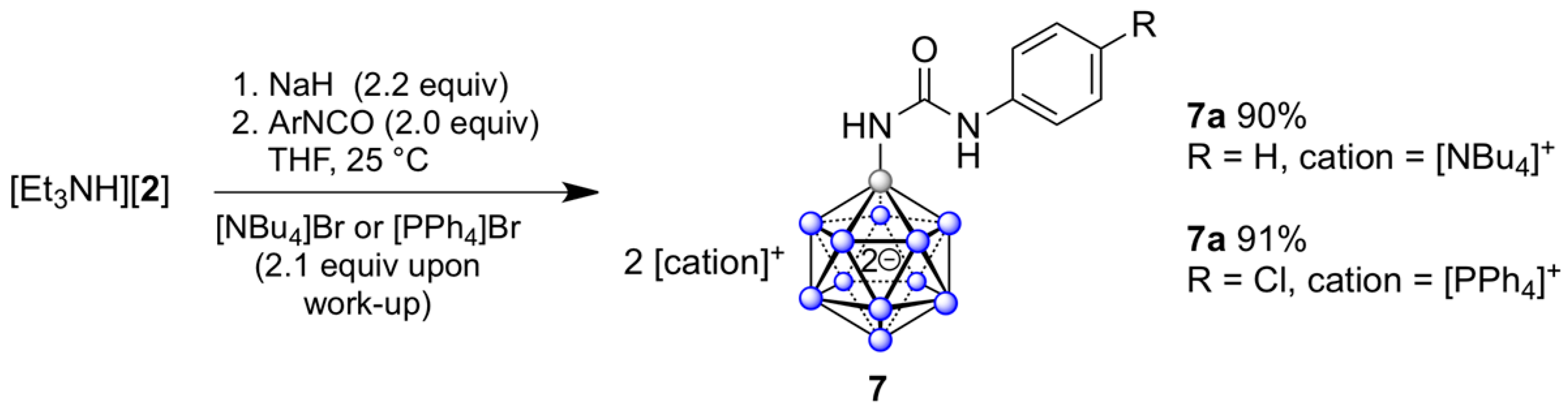
2.3. Synthesis of {B12}-based Ureas
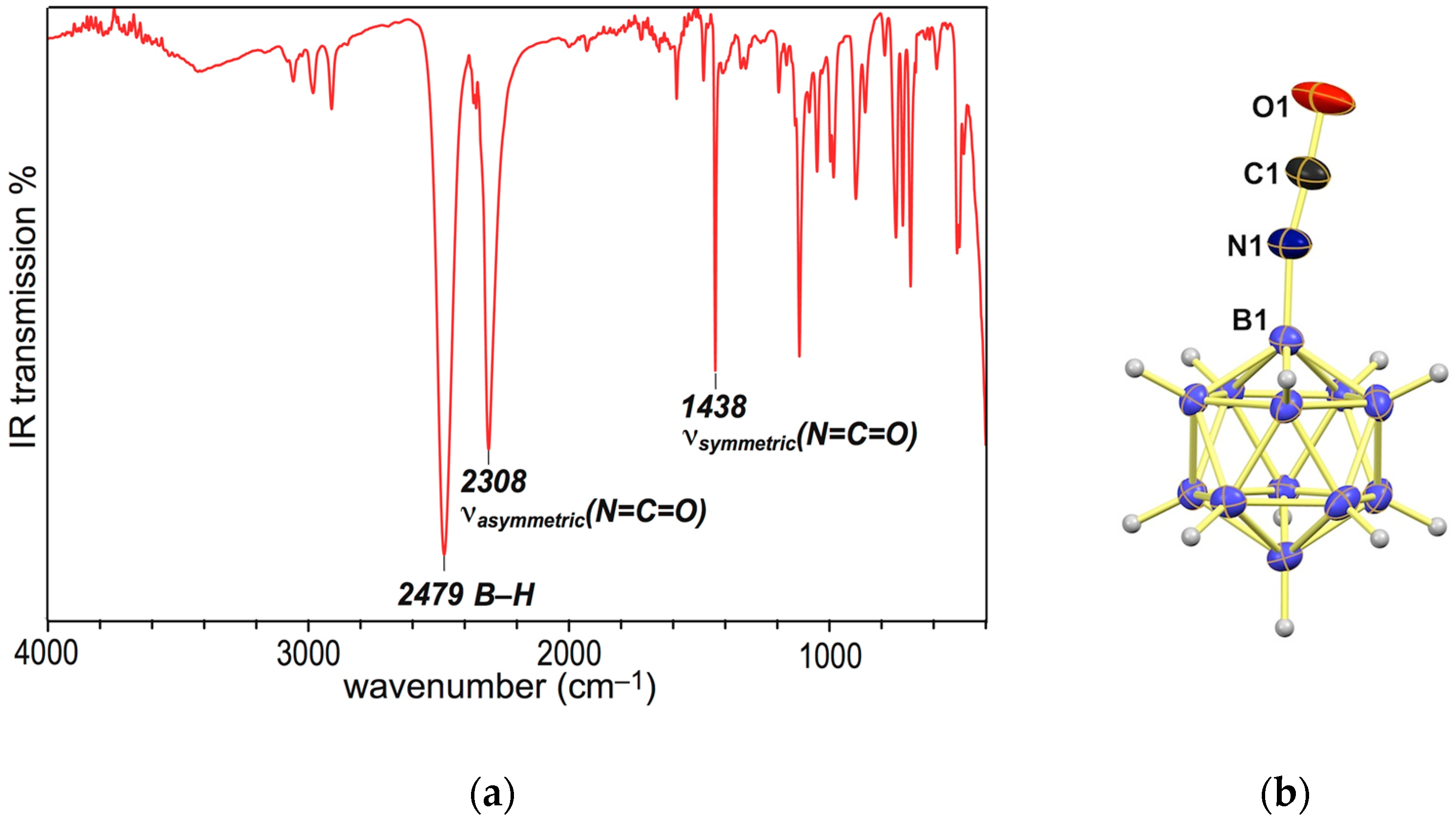
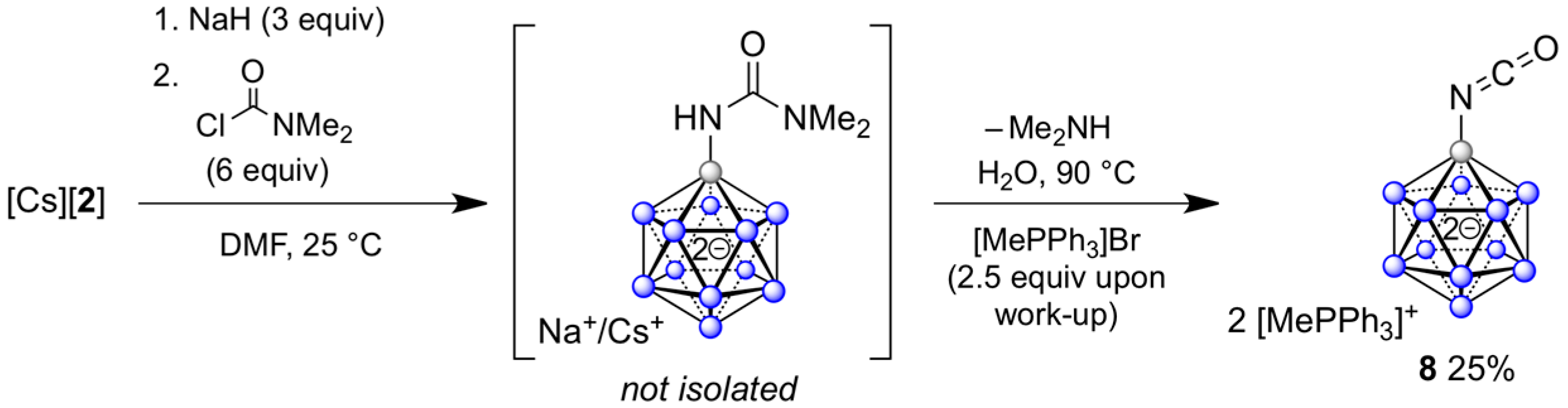
2.4. Synthesis of Dodecaboranyl Isocyanate
3. Materials and Methods
3.1. General
3.2. Experimental Section
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Hosmane, N.S. Boron Science: New Technologies and Applications, 1st ed.; CRC Press, Taylor & Francis Group: Boca Raton, FL, USA, 2012. [Google Scholar]
- Sivaev, I.B.; Bregadze, V.I.; Sjöberg, S. Chemistry of closo-dodecaborate anion [B12H12]2−: A Review. Collect. Czech. Chem. Commun. 2002, 67, 679–727. [Google Scholar] [CrossRef]
- Axtell, J.C.; Saleh, L.M.A.; Qian, E.A.; Wixtrom, A.I.; Spokoyny, A.M. Synthesis and Applications of Perfunctionalized Boron Clusters. Inorg. Chem. 2018, 57, 2333–2350. [Google Scholar] [CrossRef] [PubMed]
- Poater, J.; Solà, M.; Viñas, C.; Teixidor, F. Hückel’s Rule of Aromaticity Categorizes Aromatic closo Boron Hydride Clusters. Chem. Eur. J. 2016, 22, 7437–7443. [Google Scholar] [CrossRef] [PubMed]
- Knapp, C. Weakly coordinating anions: halogenated borates and dodecaborates. In Comprehensive Inorganic Chemistry II; Elsevier: Amsterdam, The Netherlands, 2013; pp. 651–679. [Google Scholar]
- Bolli, C.; Derendorf, J.; Jenne, C.; Scherer, H.; Sindlinger, C.P.; Wegener, B. Synthesis and Properties of the Weakly Coordinating Anion [Me3NB12Cl11]−. Chem. Eur. J. 2014, 20, 13783–13792. [Google Scholar] [CrossRef] [PubMed]
- Kessler, M.; Knapp, C.; Sagawe, V.; Scherer, H.; Uzun, R. Synthesis, Characterization, and Crystal Structures of Silylium Compounds of the Weakly Coordinating Dianion [B12Cl12]2−. Inorg. Chem. 2010, 49, 5223–5230. [Google Scholar] [CrossRef] [PubMed]
- Bolli, C.; Derendorf, J.; Keßler, M.; Knapp, C.; Scherer, H.; Schulz, C.; Warneke, J. Synthesis, Crystal Structure, and Reactivity of the Strong Methylating Agent Me2B12Cl12. Angew. Chem. Int. Ed. 2010, 49, 3536–3538. [Google Scholar] [CrossRef] [PubMed]
- Avelar, A.; Tham, F.S.; Reed, C.A. Superacidity of Boron Acids H2(B12X12) (X=Cl, Br). Angew. Chem. Int. Ed. 2009, 48, 3491–3493. [Google Scholar] [CrossRef] [PubMed]
- Geis, V.; Guttsche, K.; Knapp, C.; Scherer, H.; Uzun, R. Synthesis and characterization of synthetically useful salts of the weakly-coordinating dianion [B12Cl12]2−. Dalton Trans. 2009, 2687–2694. [Google Scholar] [CrossRef] [PubMed]
- Peryshkov, D.V.; Strauss, S.H. Exceptional Structural Compliance of the B12F122− Superweak Anion. Inorg. Chem. 2017, 56, 4072–4083. [Google Scholar] [CrossRef] [PubMed]
- Malischewski, M.; Peryshkov, D.V.; Bukovsky, E.V.; Seppelt, K.; Strauss, S.H. Structures of M2(SO2)6B12F12 (M = Ag or K) and Ag2(H2O)4B12F12: Comparison of the Coordination of SO2 versus H2O and of B12F122− versus Other Weakly Coordinating Anions to Metal Ions in the Solid State. Inorg. Chem. 2016, 55, 12254–12262. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, S.V.; Miller, S.M.; Anderson, O.P.; Solntsev, K.A.; Strauss, S.H. Synthesis and Stability of Reactive Salts of Dodecafluoro-closo-dodecaborate(2−). J. Am. Chem. Soc. 2003, 125, 4694–4695. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, S.V.; Davis, J.A.; Miller, S.M.; Anderson, O.P.; Strauss, S.H. Synthesis and Characterization of Ammonioundecafluoro-closo-dodecaborates(1−). New Superweak Anions. Inorg. Chem. 2003, 42, 4489–4491. [Google Scholar] [CrossRef] [PubMed]
- Leśnikowski, Z.J. Challenges and Opportunities for the Application of Boron Clusters in Drug Design. J. Med. Chem. 2016, 59, 7738–7758. [Google Scholar] [CrossRef] [PubMed]
- Gabel, D. Boron clusters in medicinal chemistry: perspectives and problems. Pure Appl. Chem. 2015, 87, 173–179. [Google Scholar] [CrossRef]
- Scholz, M.; Hey-Hawkins, E. Carbaboranes as Pharmacophores: Properties, Synthesis, and Application Strategies. Chem. Rev. 2011, 111, 7035–7062. [Google Scholar] [CrossRef] [PubMed]
- Wegener, M.; Huber, F.; Bolli, C.; Jenne, C.; Kirsch, S.F. Silver-Free Activation of Ligated Gold(I) Chlorides: The Use of [Me3NB12Cl11]− as a Weakly Coordinating Anion in Homogeneous Gold Catalysis. Chem. Eur. J. 2015, 21, 1328–1336. [Google Scholar] [CrossRef] [PubMed]
- Messina, M.S.; Axtel, J.C.; Wang, Y.; Chong, P.; Wixtrom, A.I.; Kirlikovali, K.O.; Upton, B.M.; Hunter, B.M.; Shafaat, O.S.; Khan, S.I.; et al. Visible-Light-Induced Olefin Activation Using 3D Aromatic Boron-Rich Cluster Photooxidants. J. Am. Chem. Soc. 2016, 138, 6952–6955. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, J.; Duttwyler, S. Synthesis and Structural Characterization of Ammonio/Hydroxo Undecachloro-closo-dodecaborates [B12Cl11(NH3)]−/[B12Cl11(OH)]2− and Their Derivatives. Eur. J. Inorg. Chem. 2015, 5158–5162. [Google Scholar] [CrossRef]
- Kirchmann, M.; Wesemann, L. Amino-closo-dodecaborate—A new ligand in coordination chemistry. Dalton Trans. 2008. [Google Scholar] [CrossRef] [PubMed]
- Kirchmann, M.; Wesemann, L. η1 and η2 Coordination of 1-amino-closo-dodecaborate. Dalton Trans. 2008. [Google Scholar] [CrossRef] [PubMed]
- Warneke, J.; Jenne, C.; Bernarding, J.; Azov, V.A.; Plaumann, M. Evidence for an intrinsic binding force between dodecaborate dianions and receptors with hydrophobic binding pockets. Chem. Commun. 2016, 52, 6300–6303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Assaf, K.I.; Gabel, D.; Zimmermann, W.; Nau, W.M. High-affinity host–guest chemistry of large-ring cyclodextrins. Org. Biomol. Chem. 2016, 14, 7702–7706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Wang, X.; Cao, J.; Liu, J.; Qi, B.; Zhou, X.; Zhang, S.; Gabel, D.; Nau, W.M.; Assaf, K.I.; et al. The chaotropic effect as an orthogonal assembly motif for multi-responsive dodecaborate-cucurbituril supramolecular networks. Chem. Commun. 2018, 54, 2098–2101. [Google Scholar] [CrossRef] [PubMed]
- Assaf, K.I.; Suckova, O.; Danaf, N.A.; von Glasenapp, V.; Gabel, D.; Nau, W.M. Dodecaborate-Functionalized Anchor Dyes for Cyclodextrin-Based Indicator Displacement Applications. Org. Lett. 2016, 18, 932–935. [Google Scholar] [CrossRef] [PubMed]
- Assaf, K.I.; Ural, M.S.; Pan, F.; Georgiev, T.; Simova, S.; Rissanen, K.; Gabel, D.; Nau, W.M. Water Structure Recovery in Chaotropic Anion Recognition: High-Affinity Binding of Dodecaborate Clusters to γ-Cyclodextrin. Angew. Chem. Int. Ed. 2015, 54, 6852–6856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pitochelli, A.R.; Hawthorne, F.M. The isolation of the icosahedral B12H122− Ion. J. Am. Chem. Soc. 1960, 82, 3228–3229. [Google Scholar] [CrossRef]
- Olid, D.; Núñez, R.; Viñas, C.; Teixidor, F. Methods to produce B–C, B–P, B–N and B–S bonds in boron clusters. Chem. Soc. Rev. 2013, 42, 3318–3336. [Google Scholar] [CrossRef] [PubMed]
- Grüner, B.; Bonnetot, B.; Mongeot, H. Synthesis of N-and B-substituted derivatives of closo-amino-undecahydro-dodecaborate(1–) anion. Collect. Czech. Chem. Commun. 1997, 62, 1185–1204. [Google Scholar] [CrossRef]
- Hertler, W.R.; Raasch, M.S. Chemistry of boranes. XIV. Amination of B10H102− and B12H122− with hydroxylamine-O-sulfonic acid. J. Am. Chem. Soc. 1964, 86, 3661–3668. [Google Scholar] [CrossRef]
- Dudenkov, I.V.; Zhizhin, K.Yu.; Chernyavskii, A.S.; Katser, S.B.; Goeva, L.V.; Sergienko, V.S.; Solntsev, K.A.; Kuznetsov, N.T. Synthesis and Crystal Structure of 1,7-(NH3)2B12H10·0.5 H2O. Russ. J. Inorg. Chem. 2000, 45, 1864–1867. [Google Scholar]
- Genady, A.R.; El-Zaria, M.E.; Gabel, D. Non-covalent assemblies of negatively charged boronated porphyrins with different cationic moieties. J. Organomet. Chem. 2004, 689, 3242–3250. [Google Scholar] [CrossRef]
- Hoffmann, S.; Justus, E.; Ratajski, M.; Lork, E.; Gabel, D. B12H11-containing guanidinium derivatives by reaction of carbodiimides with H3N-B12H11(1−). A new method for connecting boron clusters to organic compounds. J. Organomet. Chem. 2005, 690, 2757–2760. [Google Scholar] [CrossRef]
- Koo, M.-S.; Ozawa, T.; Santos, R.A.; Lamborn, K.R.; Bollen, A.W.; Deen, D.F.; Kahl, S.B. Synthesis and Comparative Toxicology of a Series of Polyhedral Borane Anion-Substituted Tetraphenyl Porphyrins. J. Med. Chem. 2007, 50, 820–827. [Google Scholar] [CrossRef] [PubMed]
- El-Zaria, M.E.; Ban, H.S.; Nakamura, H. Boron-Containing Protoporphyrin IX Derivatives and Their Modification for boron Neutron Capture Therapy: Synthesis, Characterization, and Comparative In Vitro Toxicity Evaluation. Chem. Eur. J. 2010, 16, 1543–1552. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Zhang, J.; Zhang, Y.; Liu, J.; van der Veen, S.; Duttwyler, S. The closo-Dodecaborate Dianion Fused with Oxazoles Provides 3D Diboraheterocycles with Selective Antimicrobial Activity. Chem. Eur. J. 2018, 24, 10364–10371. [Google Scholar] [CrossRef] [PubMed]
- Sivaev, I.B.; Bruskin, A.B.; Nesterov, V.V.; Antipin, M.Y.; Bregadze, V.I.; Sjöberg, S.S. Synthesis of Schiff Bases Derived from the Ammoniaundecahydro-closo-dodecaborate (1−) Anion, [B12H11NH=CHR]−, and Their Reduction into Monosubstituted Amines [B12H11NH2CH2R]−: A New Route to Water Soluble Agents for BNCT. Inorg. Chem. 1999, 38, 5887–5893. [Google Scholar] [CrossRef]
- Zhang, Y.; Sun, Y.; Lin, F.; Liu, J.; Duttwyler, S. Rhodium (III)-Catalyzed Alkenylation–Annulation of closo-Dodecaborate Anions through Double B−H Activation at Room Temperature. Angew. Chem. Int. Ed. 2016, 50, 15609–15614. [Google Scholar] [CrossRef] [PubMed]
- Alam, F.; Soloway, A.H.; Barth, R.F.; Mafune, N.; Adams, D.M.; Knoth, W.H. Boron Neutron Capture Therapy: Linkage of a Boronated Macromolecule to Monoclonal Antibodies Directed Against Tumor-Associated Antigens. J. Med. Chem. 1989, 32, 2326–2330. [Google Scholar] [CrossRef] [PubMed]
- Ozaki, S. Recent Advances in Isocyanate Chemistry. Chem. Rev. 1972, 72, 457–496. [Google Scholar] [CrossRef]
- Nyquist, R.A. Interpreting Infrared, Raman, and Nuclear Magnetic Resonance Spectra, 1st ed.; Academic Press: Cambridge, MA, USA, 2001; p. 46. [Google Scholar]
Sample Availability: Not available. |









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Distance [Å]/Angle [°] | 3a 1 | 3a-H 2 | 3b | 3c | 3d-H | 3e | 3e-H |
|---|---|---|---|---|---|---|---|
| B1–N1 | 1.520(6) | 1.534(4) | 1.516(4) | 1.512(5) | 1.577(7) | 1.516(3) | 1.524(3) |
| N1–C1 | 1.324(6) | 1.295(7) | 1.320(4) | 1.327(4) | 1.263(7) | 1.306(2) | 1.316(3) |
| C1–O1 | 1.250(5) | 1.314(2) | 1.238(3) | 1.227(4) | 1.343(8) | 1.234(2) | 1.232(3) |
| C1–C2 | 1.496(6) | 1.471(2) | 1.500(4) | 1.500(5) | 1.466(17) | 1.502(3) | 1.512(3) |
| Σ(C1) | 359.9 | 359.9 | 360.0 | 360.0 | 359.9 | 360.0 | 360.0 |
| O1-C1-C2-C3 3 | –17.8(3) | –9.95(13) | –15.6(4) | 32.0(3) | –4.9(4) | 5.77(2) | 7.4(3) |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.; Sun, Y.; Wang, T.; Liu, J.; Spingler, B.; Duttwyler, S. Synthesis and Structural Characterization of Amidine, Amide, Urea and Isocyanate Derivatives of the Amino-closo-dodecaborate Anion [B12H11NH3]−. Molecules 2018, 23, 3137. https://doi.org/10.3390/molecules23123137
Zhang Y, Sun Y, Wang T, Liu J, Spingler B, Duttwyler S. Synthesis and Structural Characterization of Amidine, Amide, Urea and Isocyanate Derivatives of the Amino-closo-dodecaborate Anion [B12H11NH3]−. Molecules. 2018; 23(12):3137. https://doi.org/10.3390/molecules23123137
Chicago/Turabian StyleZhang, Yuanbin, Yuji Sun, Tao Wang, Jiyong Liu, Bernhard Spingler, and Simon Duttwyler. 2018. "Synthesis and Structural Characterization of Amidine, Amide, Urea and Isocyanate Derivatives of the Amino-closo-dodecaborate Anion [B12H11NH3]−" Molecules 23, no. 12: 3137. https://doi.org/10.3390/molecules23123137