Recent Advances in Substrate-Controlled Asymmetric Cyclization for Natural Product Synthesis


1
College of Pharmacy, Pusan National University, Busan 46241, Korea
2
College of Pharmacy, Institute of Pharmaceutical Sciences, Cha University, Pocheon-si 11160, Korea
3
College of Pharmacy, Dankook University, Cheonan 31116, Korea
4
College of Pharmacy, Kyungsung University, Busan 48434, Korea
*
Author to whom correspondence should be addressed.
†
These authors contributed equally to this work.
Molecules 2017, 22(7), 1069; https://doi.org/10.3390/molecules22071069
Submission received: 28 May 2017
/
Accepted: 21 June 2017
/
Published: 26 June 2017
(This article belongs to the Special Issue Asymmetric Synthesis 2017)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Asymmetric synthesis of naturally occurring diverse ring systems is an ongoing and challenging research topic. A large variety of remarkable reactions utilizing chiral substrates, auxiliaries, reagents, and catalysts have been intensively investigated. This review specifically describes recent advances in successful asymmetric cyclization reactions to generate cyclic architectures of various natural products in a substrate-controlled manner.
1. Introduction
Asymmetric construction of structurally diverse ring architectures has always been considered a formidable task in natural product synthesis. Various natural sources have provided an enormous number of enantiomerically enriched carbo- and heterocycles [1,2,3,4,5]. Their ring systems include monocycles, such as small-sized, medium-sized, and large-sized rings and polycycles, such as spiro-, fused-, bridged-, and ansa-rings. These intriguing structures have attracted considerable attention from the organic synthesis communities. Many synthetic chemists have explored fascinating tactics to construct chiral ring frameworks using chiral substrates, auxiliaries, reagents, and catalysts [6,7,8]. In particular the substrate-controlled cyclization strategy, which utilizes the nature of the built-in chiral environment in the starting material, is a very powerful method. In addition, this strategy is more environmentally and economically attractive than using chiral auxiliaries or catalysts [9,10].
The aim of this review is to highlight outstanding achievements in the construction of enantioenriched rings in the field of total synthesis from 2010 to April 2017. Selected original articles in this review contain various substrate-controlled cyclization strategies. The cyclization reactions are categorized by reaction type, such as anionic, cationic, transition metal-mediated, pericyclic, and radical reactions.
2. Anionic Cyclizations
Recently, a wide range of natural product syntheses via anionic cyclization in a substrate-controlled manner have been reported. Carreira et al. accomplished the total synthesis of (−)-dendrobine (8) [11], which was isolated from the ornamental orchid Dendrobium nobile Lindl [12,13]. To construct the core of 8, they utilized a special cascade sequence including enamine conjugate addition and stereoselective protonation as shown in Scheme 1. Highly advanced precursor 2 was readily prepared from ester 1 [14] in twelve steps. Treatment of 2 with N-methylbenzylamine followed by exposure to H2 and Pd/C led to bicycle 7 in 68% yield. High diastereoselectivity was obtained by a substrate-controlled cascade process involving the conjugate addition of enamine 3, enamine formation by proton loss of 4 and the convex protonation of enamine 5. This transformation allowed the asymmetric formations of the important C–C bond and the desired pendant amine.
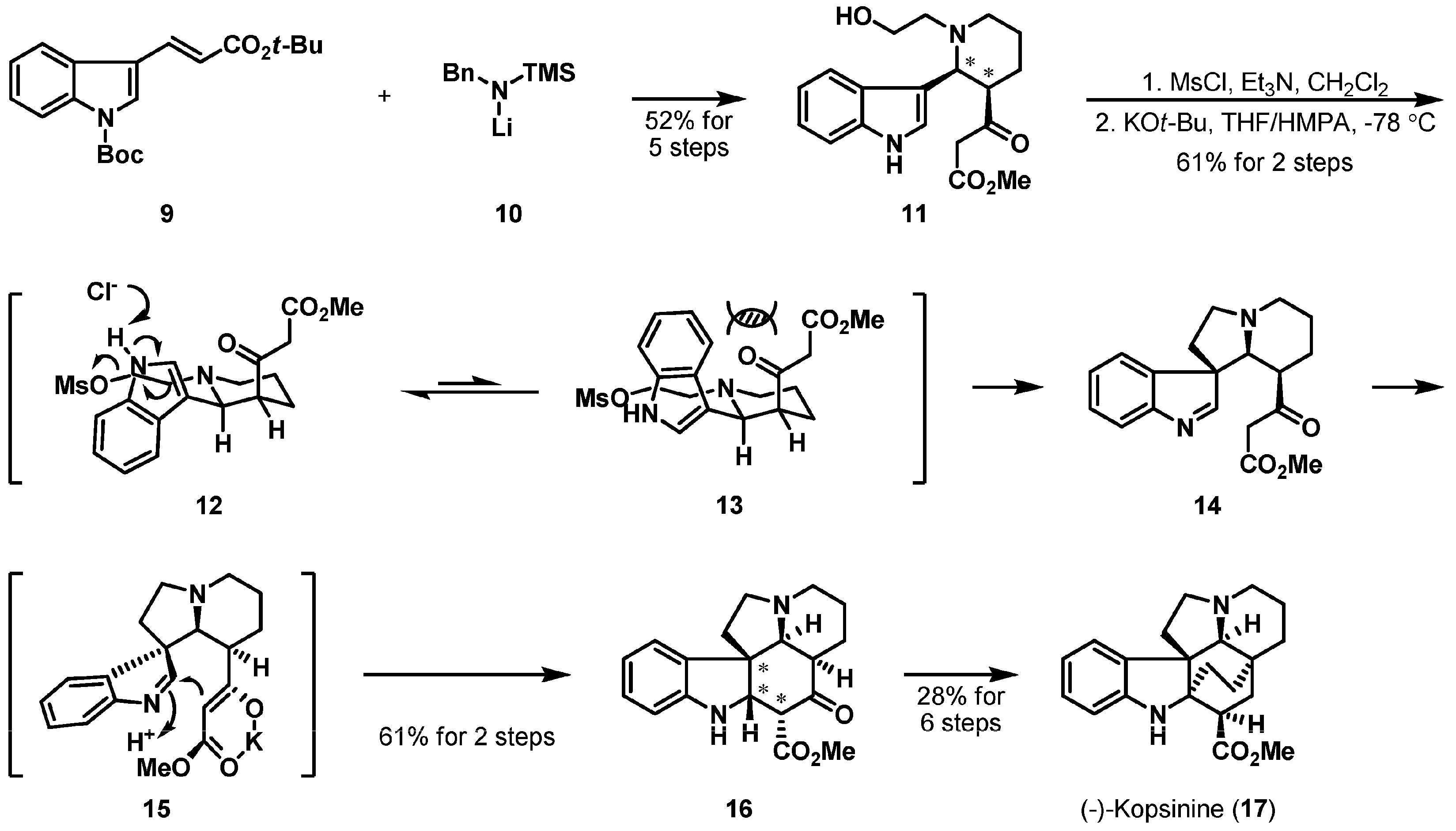
Another substrate-controlled asymmetric cyclization is summarized in Scheme 2 which depicts the total synthesis reported by Tomioka et al. [15] of (−)-kopsinine (17), a Kopsia alkaloid isolated from Kopsia longiflora Merr. in 1955 [16,17]. Key precursor 11 was smoothly formed via a high-yielding process including the asymmetric one-pot [N+2+3] cyclization of tert-butyl N-Boc-indole-3-propenoate (9) and lithium N-benzyltrimethylsilylamide (10). Having successfully inserted two stereocenters onto 11, the three stereocenters of pentacyclic intermediate 16 were generated in a substrate-controlled manner. Mesylation of β-ketoester 11 and subsequent anionic cyclization of the resulting tetracycle 14 gave optically pure 16, presumably through transition states 12 and 15. The bridged ring of 17 was later introduced by Diels-Alder cyclization.
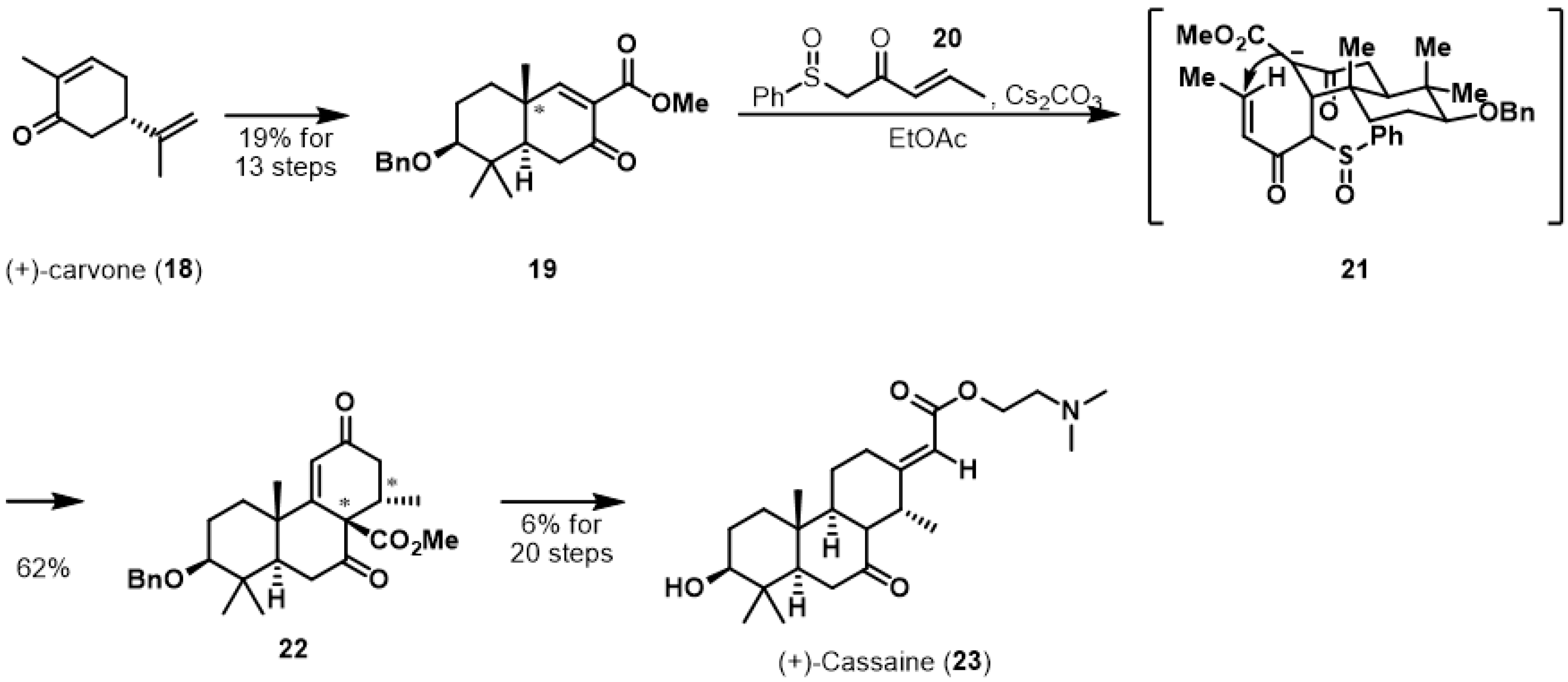
An elegant anionic polycyclization strategy of Deslongchamps et al. led to the total synthesis of (+)-cassaine (23) as shown in Scheme 3 [18]. (+)-Cassaine was isolated from Erythrophleum guineense in 1935 and reported as a nonsteroidal Na+-K+-ATPase inhibitor [19]. (+)-Carvone (18) was selected as the starting building block to synthesize the pharmacologically interesting natural product. The authors successfully prepared cyclization precursor 19 based on their previous synthetic route [20]. With the asymmetric formation of the trans-decalin system of 19, the desired anionic cyclization was performed using Nazarov reagent 20. Thus, treatment with cesium carbonate in EtOAc gave rise to diastereomerically pure tricycle 22 via Michael adduct 21 in 62% yield. The newly created stereocenters originated from the α-face attack of 20 toward 19, which was controlled by the steric repulsion of the angular methyl group in the ring junction of 19. Synthesis of (+)-cassaine (23) was completed through multiple chemical reactions.
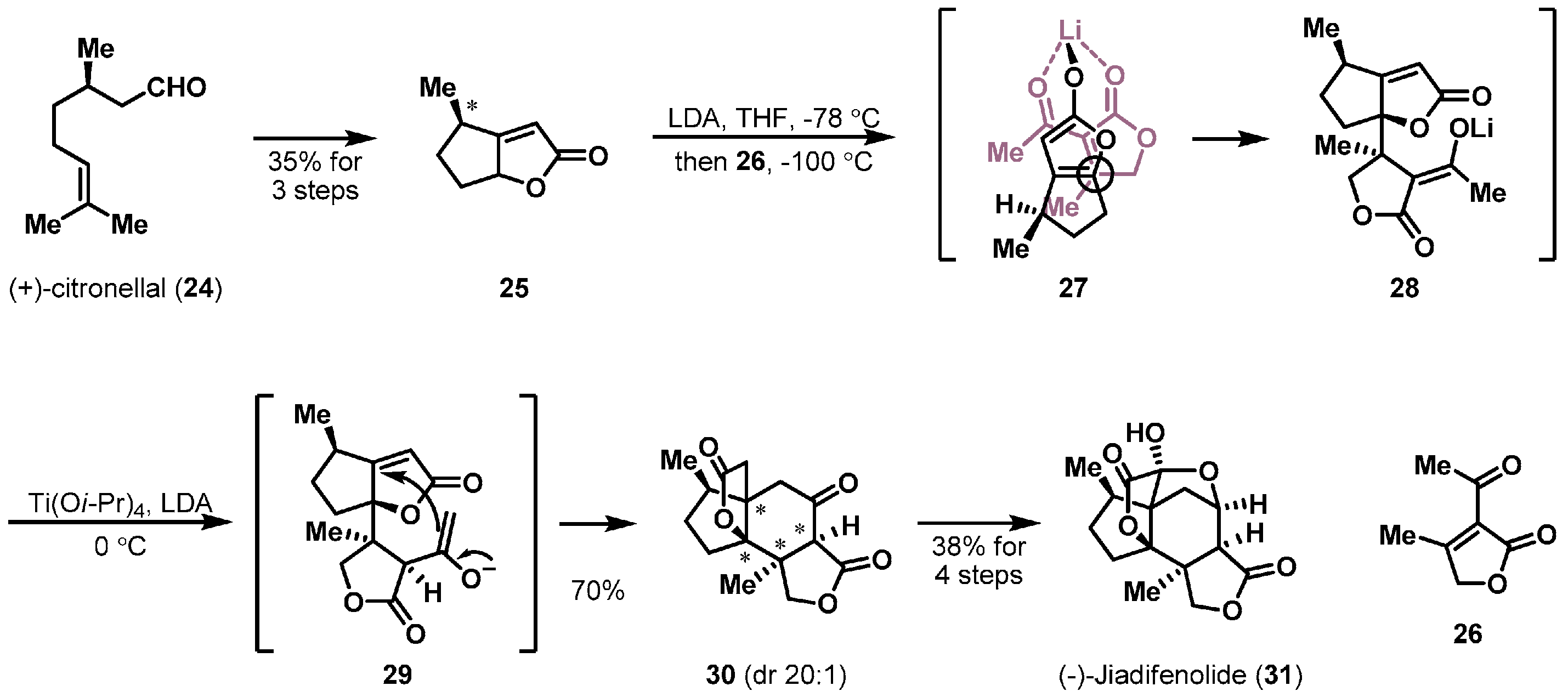
Stereoselective double Michael reactions have also been adopted for the construction of unique ring systems. Shenvi et al. completed the impressive synthesis of (−)-jiadifenolide (31) [21], which is one of the Illicium terpenes [22], as shown in Scheme 4. Key precursors butenolides 25 and 26 were quickly synthesized through three- and two-step routes, respectively. The first intermolecular Michael reaction of chiral butenolide 25 with achiral butenolide 26 in the presence of lithium diisopropylamide (LDA) provided stable intermediate 28. The stereochemistry of the process derived from a chelated transition state 27 and the newly created stereocenters of 28 were controlled by the chiral methyl group of 25. Subsequent exposure of the resulting 28 to titanium(IV) isopropoxide and additional LDA finally furnished ketolactone 30 via enolate 29 in 70% yield. The second intramolecular Michael reaction enabled the construction of the entire skeleton of 31 in a substrate-controlled manner.
3. Cationic Cyclizations
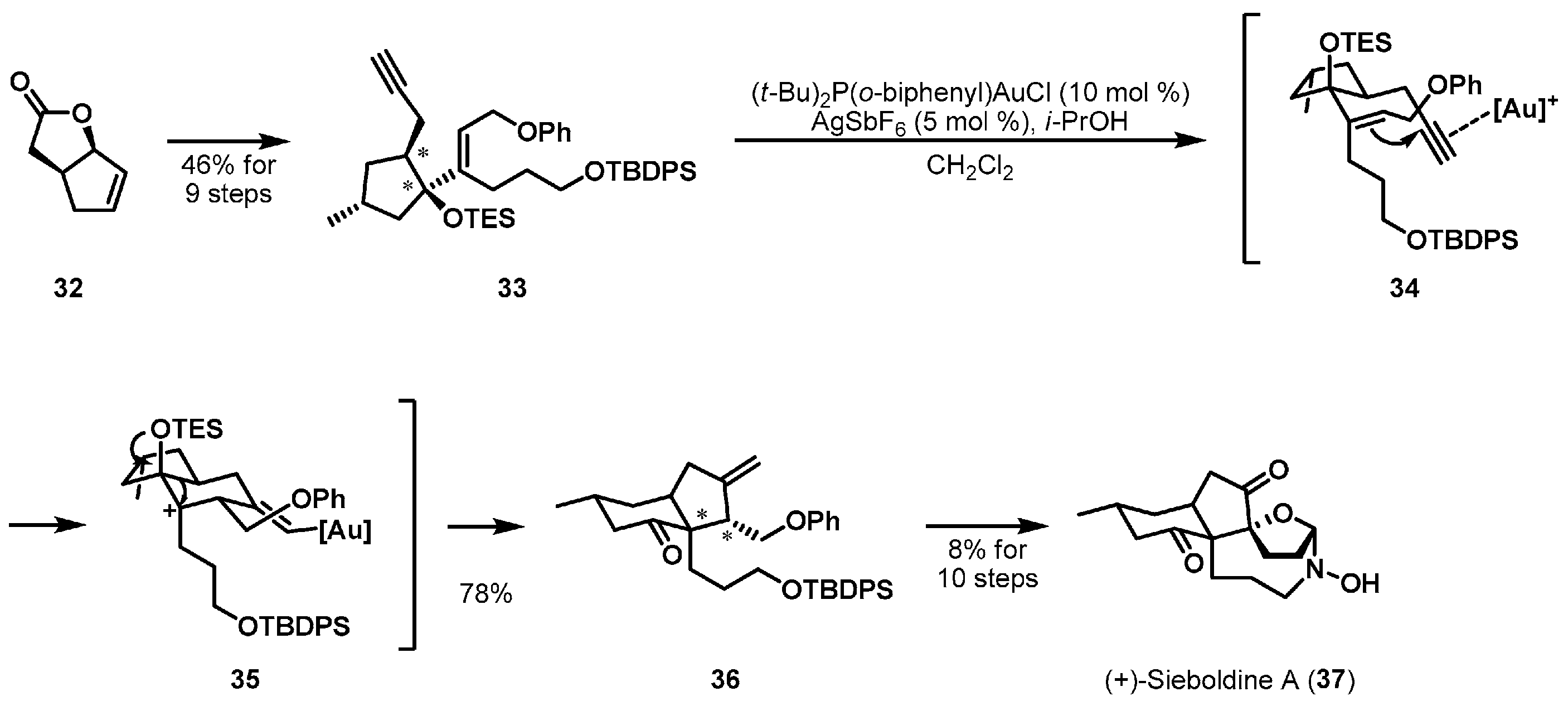
Cation-induced cyclization has been a powerful tool for controlling the stereochemistry of various ring structures in natural product synthesis. The total synthesis of (+)-siebolidine A (37), which is an alkaloid of club moss Lycopodium sieboldii [23], was firstly reported by Overman et al. [24,25]. This landmark synthesis was accomplished with a pinacol-terminated cyclization cascade as depicted in Scheme 5. Cyclization precursor 33 was built from the readily available allylic lactone 32 [26,27] in 9 steps. Initially, an increased cationic environment of gold alkyne species 34 enabled 1,6-enyne cyclization. The subsequent pinacol shift of cyclized cationic intermediate 35 afforded the desired cis-hydrindanone 36 as a single stereoisomer. The stereochemistry of enyne 33 was efficiently reorganized in a substrate-controlled manner.
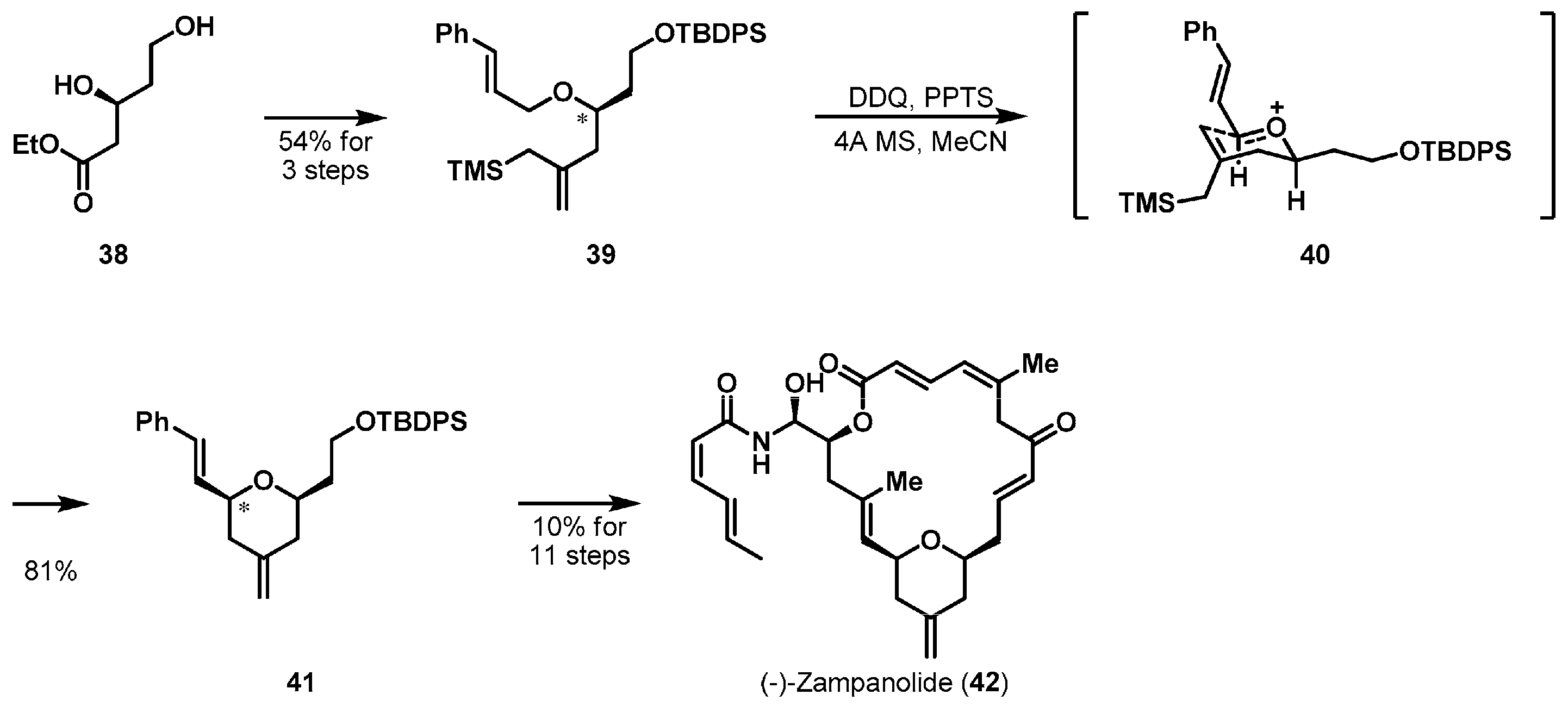
Another interesting example of substrate-controlled cyclization is the total synthesis of (−)-zampanolide (42) published by Ghosh et al. [28,29] (Scheme 6). (−)-Zampanolide was initially separated from the marine sponge Fasciospongia rimosa and exhibited a potent microtubule- stabilizing activity [30,31]. Synthesis of interesting macrolide 42 commenced from the known ester 38, which was effectively prepared by the Noyori hydrogenation procedure [32]. Optically active starting material 38 was converted to allylsilane 39 in three steps. With the desired starting material 39 in hand, the authors carried out an oxidative Sakurai type cyclization using 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) with mild Brønsted acid pyridinium p-toluenesulfonate (PPTS). Cyclized product 41 was obtained diastereoselectively through the Zimmerman-Traxler transition state 40. The cis-stereochemistry of tetrahydropyran ring 41 was caused by the equatorial orientations of all substituents in 40. Having successfully assembled the core ring, macrocycle 42 was successfully constructed via cross and ring-closing metatheses.
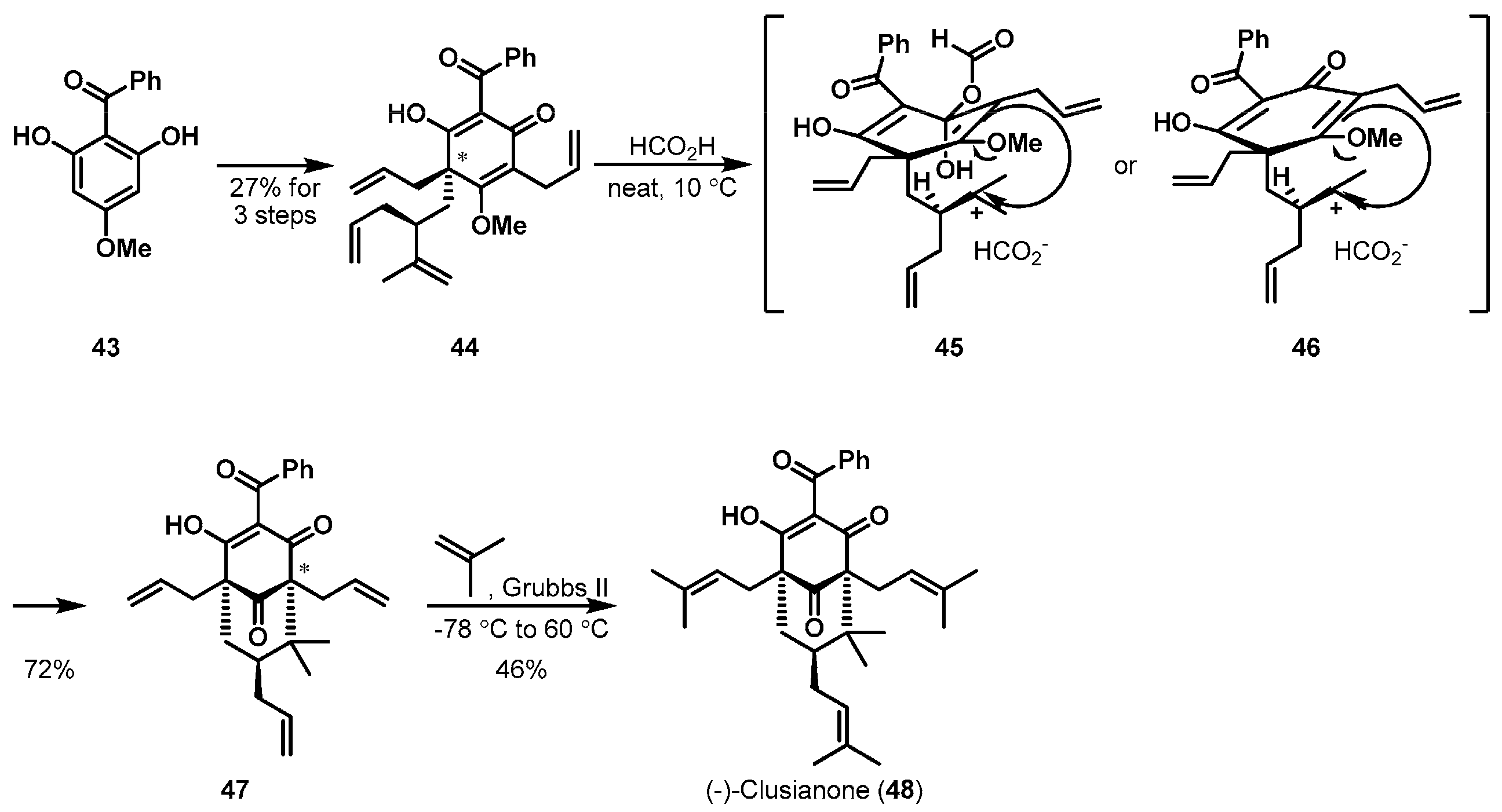
Natural product clusianone is one of the polyprenylated polycyclic acylphloroglucinols (PPAPs) possessing a structurally unique bicyclo[3.3.1]nonane-2,4,9-trione core [33]. A biomimetic cationic cyclization was applied to construct the bicyclic template of (−)-clusianone (48) as shown in Scheme 7 [34]. Porco Jr. et al. were interested in the synthesis and structure-activity relationship of PPAP natural products and derivatives. Cyclization precursor 44 was accessible from starting material 43 via an unprecedented alkylative dearomatization strategy. Thus, when optically active 44 was subjected to neat formic acid, a tertiary carbocation was initially formed, which led to the intramolecular attack of the methyl enol ether to stereoselectively furnish the desired bicycle 47 in 72% yield. The authors proposed the transition states 45 and 46 due to the observation of formate adduct from ultrahigh performance liquid chromatography (UPLC) measurements. A final olefin metathesis produced (−)-clusianone (48).
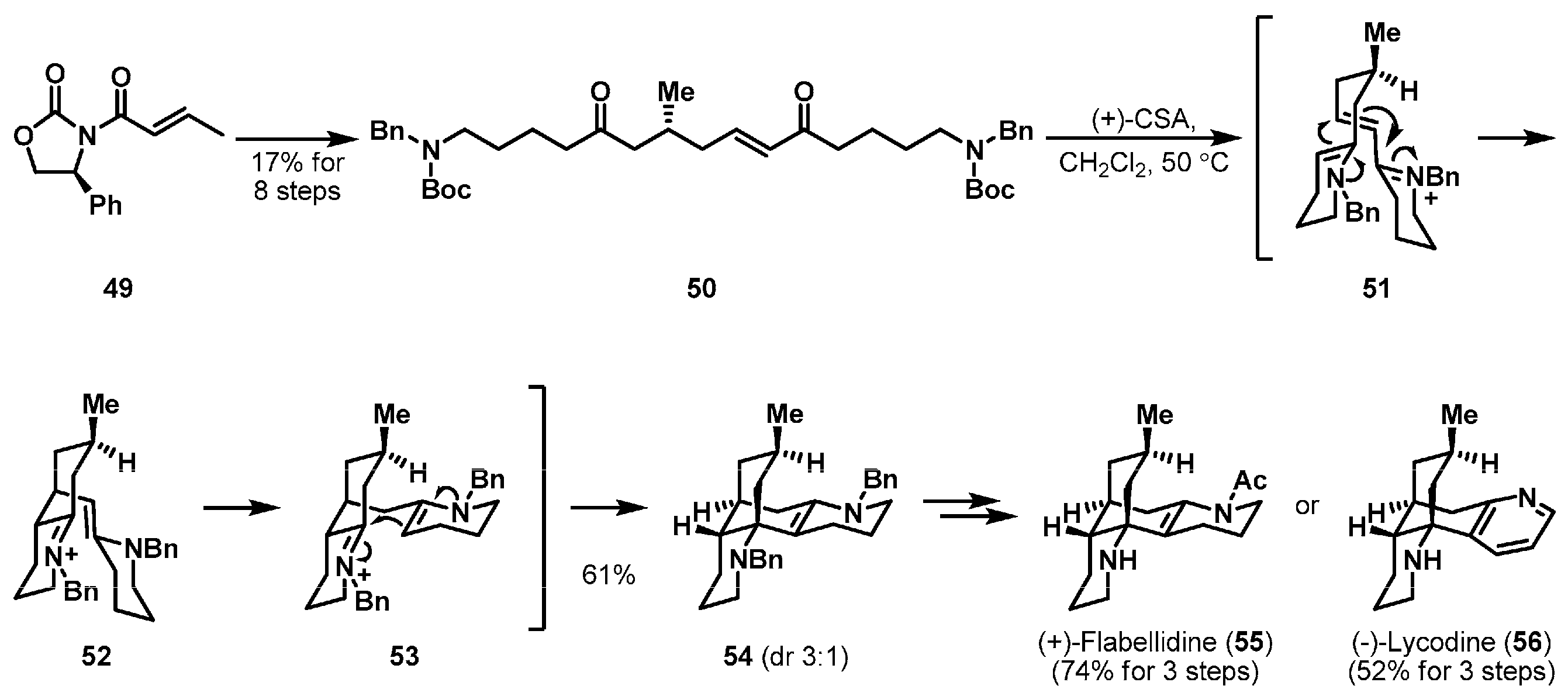
As depicted in Scheme 8, another biomimetic cationic cyclization study was performed by Takayama et al. [35]. Lycopodium alkaloids (+)-flabellidine (55) and (−)-lycodine (56) were reported from Lycopodium complanatum in 1942 and Lycopodium annotinum in 1958, respectively [36,37]. The methyl group of linear precursor 50 was diastereoselectively introduced by Hosomi-Sakurai allylation of commercially available crotonamide 49 [38]. With linear substrate 50 in hand, the authors examined the designed cationic cascade cyclization to form the tetracyclic backbone. Exposure of 50 in CH2Cl2 to an excess amount of (+)-camphorsulfonic acid (CSA) provided tetracycle 54 as the major product, presumably through conjugate addition of ene-iminium intermediate 51 and olefin migration of 52, Mannich-like reaction of 53. The diastereoselectivity was dominantly controlled by the stereocenter of the methyl group. After protecting group manipulation a minor diastereomer of the cationic cascade cyclization could be removed, and syntheses of (+)-flabellidine (55) and (−)-lycodine (56) were completed by a chemoselective acetylation and selective IBX oxidation, respectively.
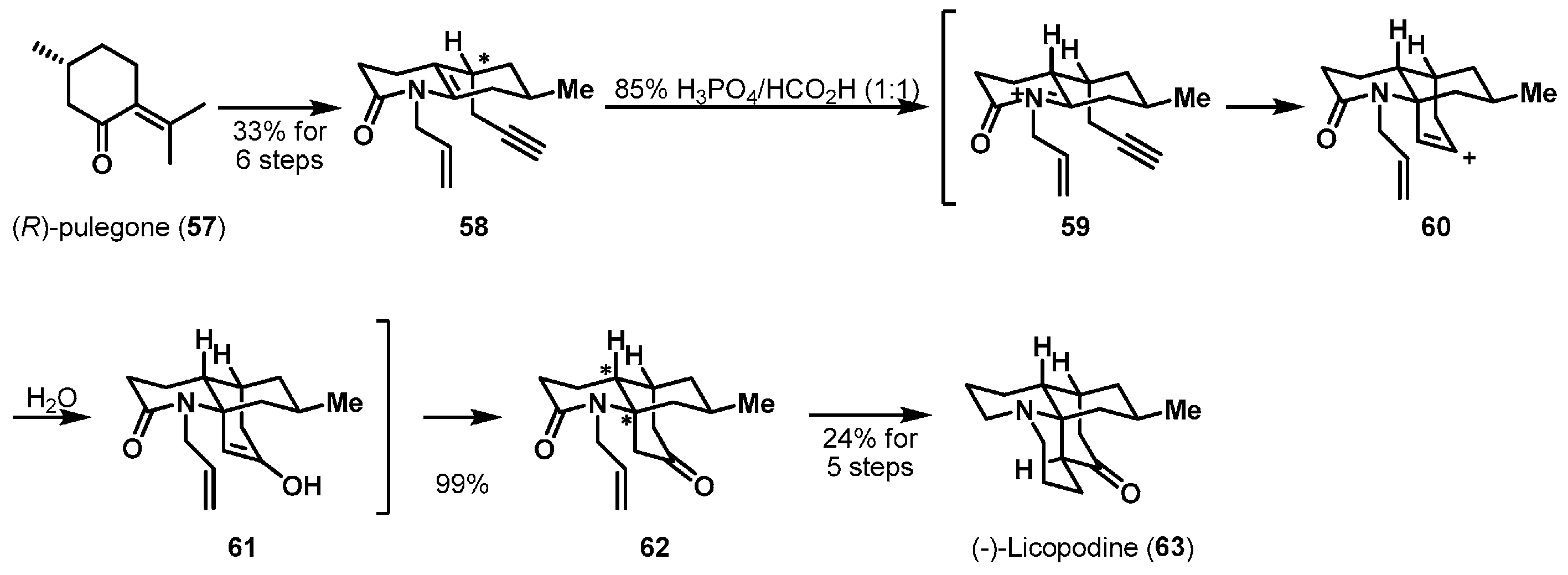
More recently, a protecting-group-free total synthesis of (−)-lycopodine (63) was described (Scheme 9). (−)-Lycopodine is one of the Lycopodium alkaloids and was originally isolated from Lycopodium complanatum in 1881 [39]. To construct its polycyclic architecture, She et al. employed an acid-promoted aza-Prins cyclization [40]. Cyclization substrate 58 was smoothly synthesized from (R)-pulegone (57) via Wade’s enone synthesis, a one-pot amidation, and cyclization [41]. Alkyne-enamide 58 was subjected to a phosphoric acid-promoted cyclization and the desired product 62 was obtained in almost quantitative yield. Initially, enamine 58 was protonated stereoselectively to furnish N-acyliminium 59. Subsequent intramolecular 6-exo-trig cyclization provided unstable carbocation species 60, which was quickly transformed to enol 61 via the capture of water. Further manipulation, including an intramolecular aldol cyclization, led to completion of the synthesis of (−)-licopodine (63).
4. Transition Metal-Mediated Cyclizations
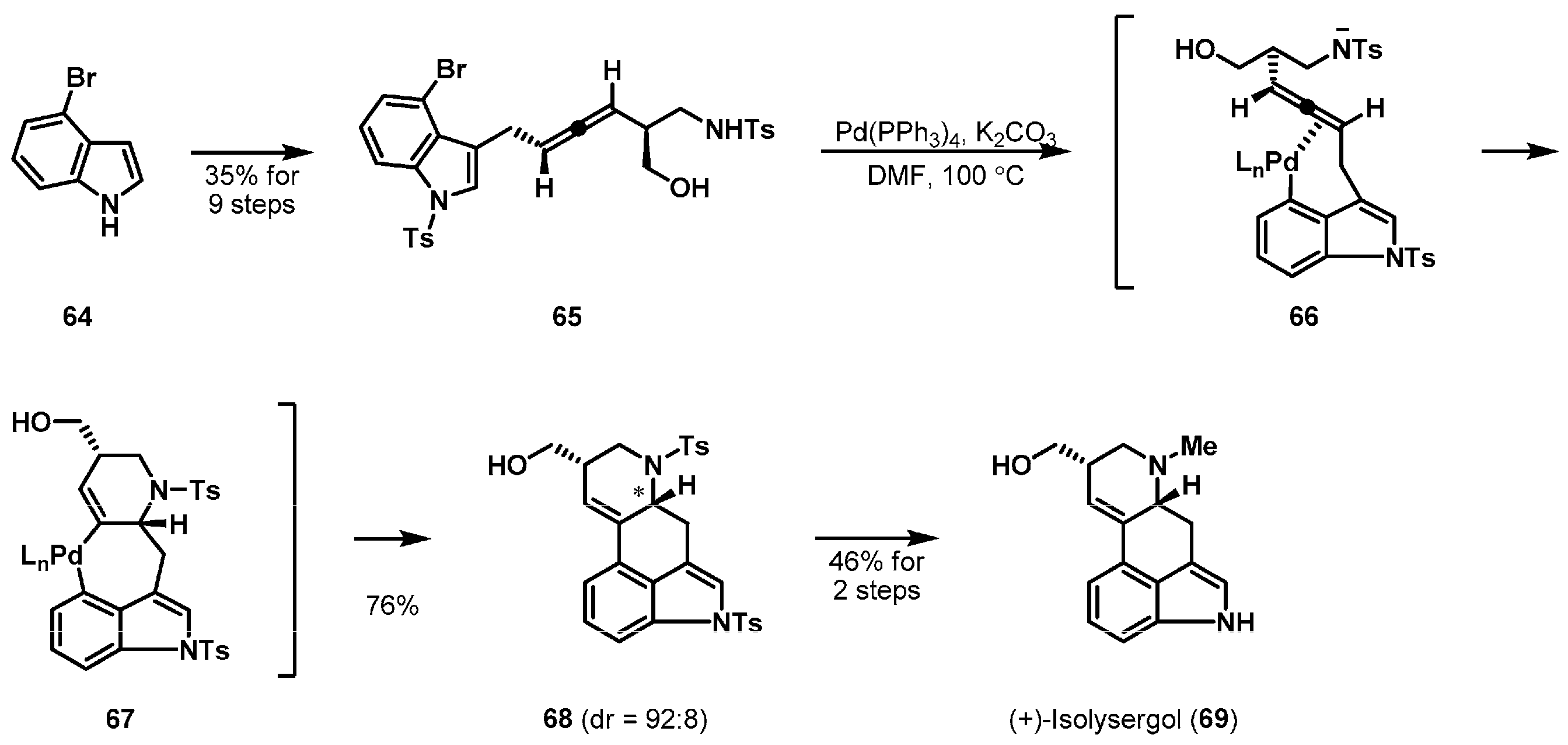
Transition metals have been used as powerful reagents for cyclization in the synthesis of complex natural products. The enantioselective total synthesis of (+)-isolysergol (69) [42], one of the biologically important ergot alkaloids [43], was completed by Fujii and Ohno et al. The authors examined a special palladium-catalyzed domino cyclization to build the ergot alkaloid backbone (Scheme 10). Allenic amide 65, required for the cyclization, was prepared from commercially available 4-bromoindole (64). Successful substrate-controlled cyclization of allene 65 in the presence of 5 mol % of Pd(PPh3)4 and K2CO3 was achieved leading to the desired product 68 in 76% yield with high diastereoselectivity. After oxidative addition, aminopalladation of the indolylpalladium halide proceeded via the conformation 66 to provide alkenylpalladium(II) intermediate 67. The stereochemistry of the ring junction in 68 ultimately derived from chiral allene 65.
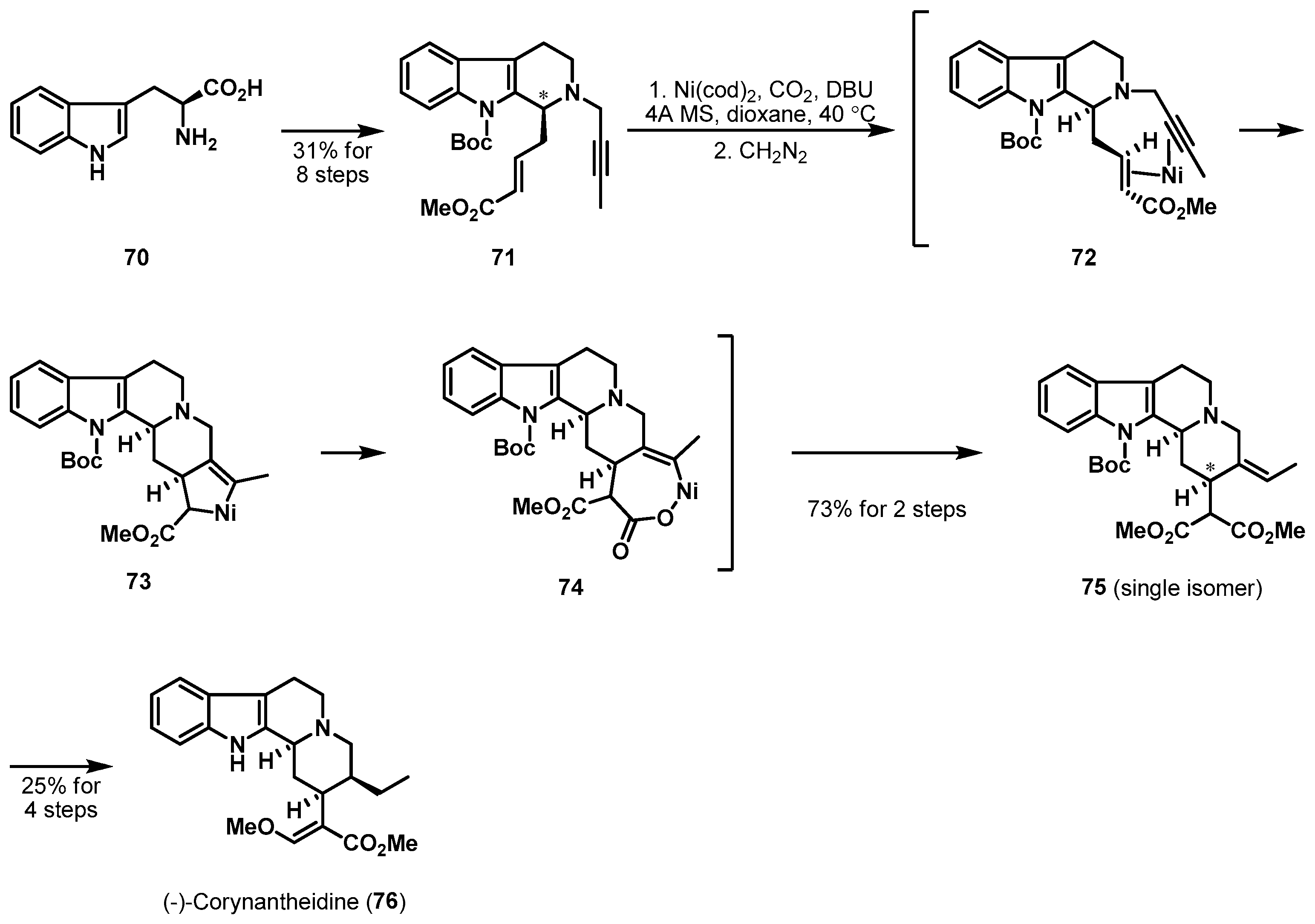
The nickel-mediated carboxylative cyclization of enyne precursor is another example of a transition-metal mediated cyclization as depicted in Scheme 11. Sato et al. described the total synthesis of indole alkaloid (−)-corynantheidine (76) [44], which was first reported in 1944 from the African plant Pseudocinchona africana [45]. Desired enyne 71 was readily accessible from l-tryptophan (70) in an optically active form through a sequence of usual transformations involving a cis-selective Pictet-Spengler reaction [46]. Upon treatment of precursor 71 with a stoichiometric amount of Ni(cod)2 and 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) under an atmosphere of gaseous CO2, the crude materials were obtained. Hydrolysis and methylation of the crude nickelacycle 73 stereoselectively led to the desired tetracycle 75 in 73% yield. The fourth ring was formed through oxidative cycloaddition of 71 and subsequent insertion of CO2 between the Csp3–nickel bond in 73. The new stereogenic center of the ring junction in 75 was created by an asymmetric formation of the nickelacycle in a substrate-controlled manner.
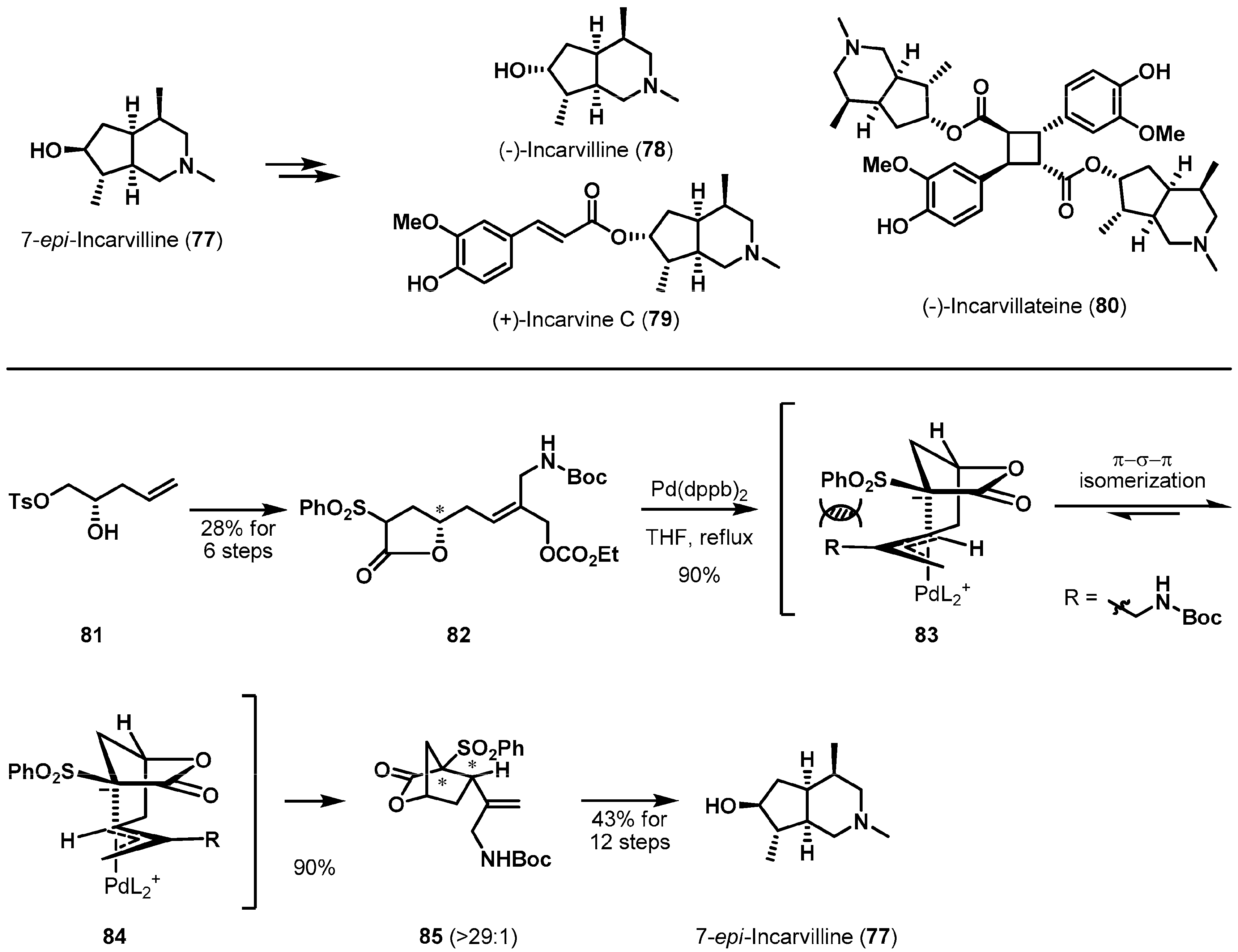
The asymmetric synthesis of incarvillea alkaloids was studied by using palladium(0)-catalyzed cyclization. Suh et al. completed an enantioselective synthesis of 7-epi-incarvilline (77) [47], which is an advanced architecture for the formal syntheses of (−)-incarvilline (78), (+)-incarvine C (79), and (−)-incarvillateine (80) (Scheme 12). Structurally, they consist of a common bicyclic piperidine skeleton including five contiguous stereocenters [48,49,50]. The authors focused on the diastereoselective construction of key intermediate 85, a highly functionalized bicyclic lactone containing three stereocenters. The desired precursor 82 of the reaction was easily obtained from the known chiral tosylate 81 [51]. Exposure of 82 in THF to Pd(dppb)2 resulted in the desired bicyclic lactone 85 in 90% yield and with excellent diastereoselectivity (>29:1). The two transition states, 83 and 84, were controlled by the built-in chirality of lactone 82 and the high diastereoselectivity likely occurred due to the steric repulsion between the benzenesulfonyl group and the R substituent in Pd–π-allyl complex of 83. With the bicyclic lactone 85 in hand, 7-epi-incarvilline (77) was synthesized using further manipulations such as a substrate-controlled catalytic hydrogenation and a 1,4-addition.
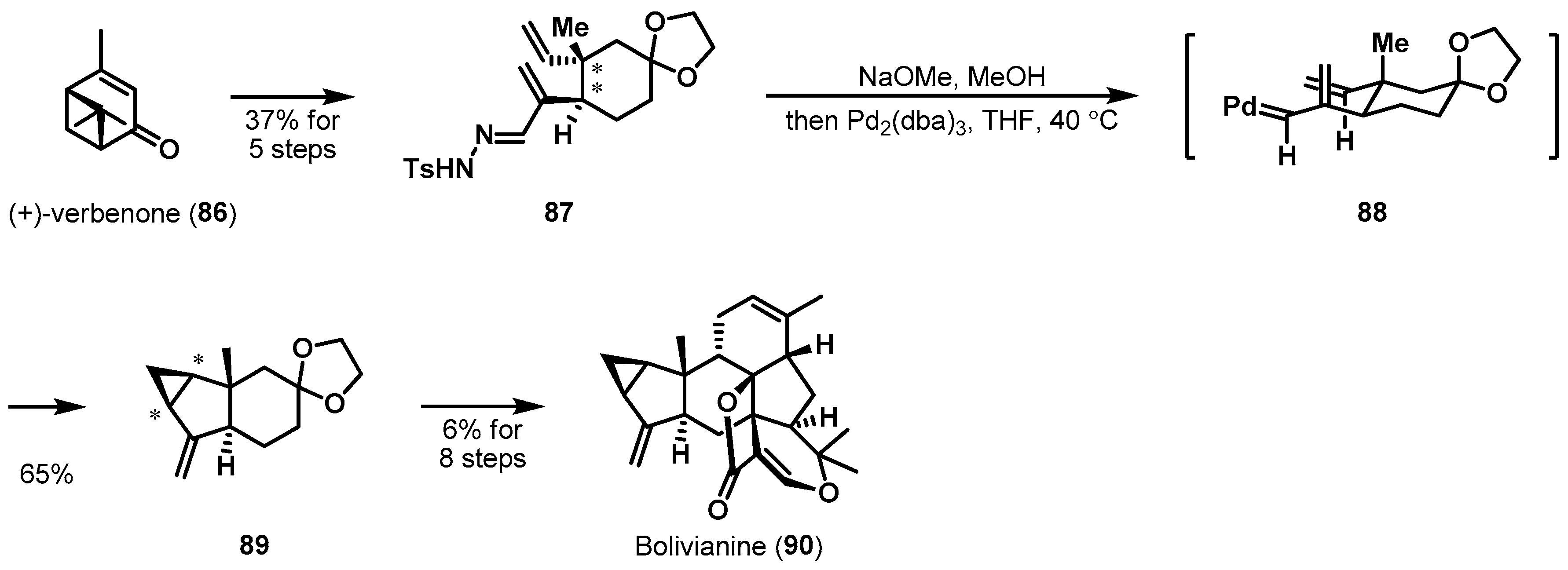
Metal-catalyzed carbenoid transfer is also a useful method for asymmetric cyclizations. Bolivianine was isolated from Hedyosmum angustifolium (Chloranthaceae) in 2007 [52]. The sesterterpenoid natural product consists of a highly complex heptacyclic skeleton and nine stereocenters. Liu et al. reported a bioinspired total synthesis of bolivianine (90), which is summarized in Scheme 13. [53,54]. Precursor tosylhydrazone 87 was well designed for the formation of the chiral cyclopropyl moiety in 89 and produced from commercially available (+)-verbenone (86). The programmed intramolecular cyclopropanation of 87 with a palladium catalyst and a sodium salt afforded the desired product 89 as the sole isolable diastereomer in 65% yield. The stereochemistry of the chiral cyclopropane was substrate-controlled via allylic metal carbene species 88, which its conformation was caused by an equatorial positioning of two alkyl chains in chair-like transition state.
5. Pericyclic Reactions
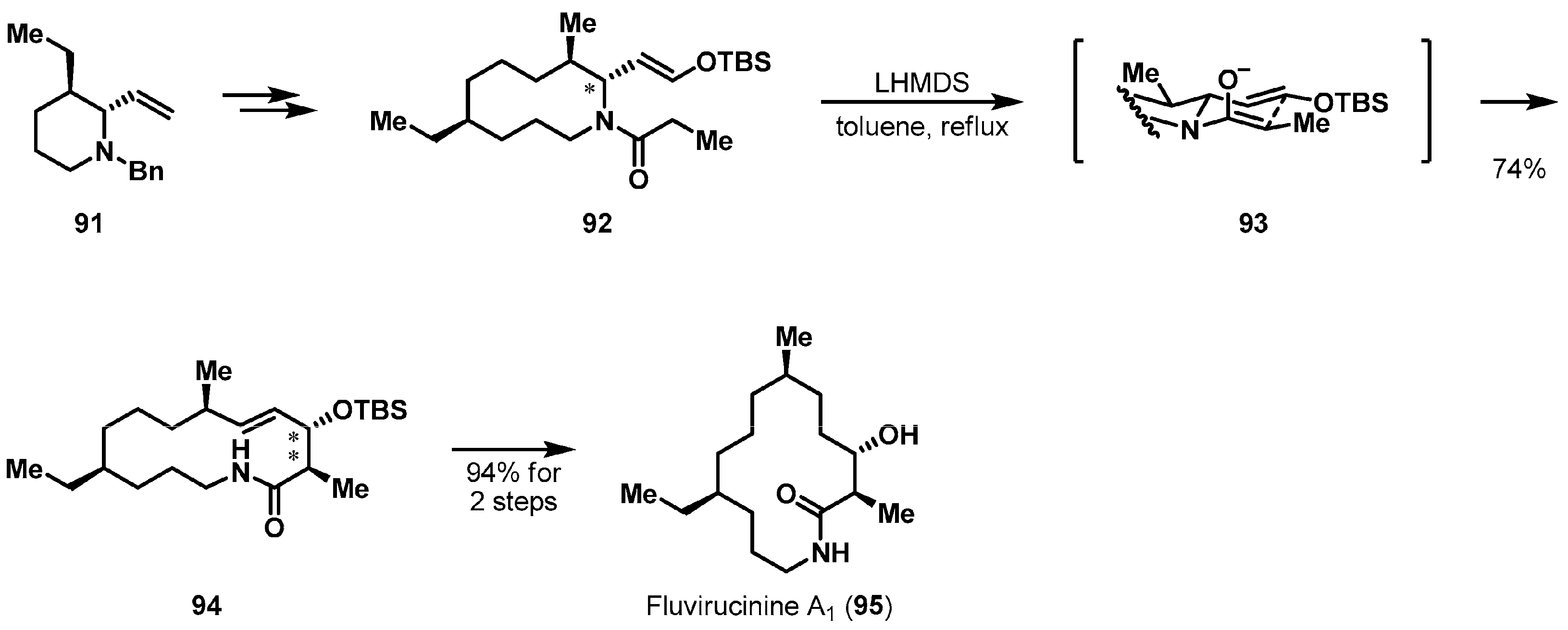
Diverse types of pericyclic reactions have been used as an attractive tactic for substrate-controlled asymmetric cyclizations. Structurally interesting fluvirucins were isolated from actinomycetes by scientists from Bristol-Myers Squibb [55,56,57,58] and Schering-Plough [59,60] independently. Suh et al. employed a stereoselective aza-Claisen rearrangement-promoted ring expansion in the total synthesis of antibiotic macrolactam fluvirucinine A1 (95), an aglycon of fluvirucine A1 (Scheme 14) [61]. The key precursor, 10-membered α-alkoxyvinyl acylazacycle 92, was obtained from piperidine 91 by a diastereoselective α-alkylation of an iminium ion generated from an N,O-acetal TMS ether [62]. With the functionalized precursor 92 in hand, the projected aza-Claisen reorganization was executed in the presence of lithium bis(trimethylsilyl)amide (LHMDS) in refluxing toluene, to affording 14-membered macrolactam 94. The origin of the stereoselectivity could be explained based on a selective (Z)-lactam enolate formation and an equatorial positioning of the alkoxyvinyl substituent in chair-like transition state 93.
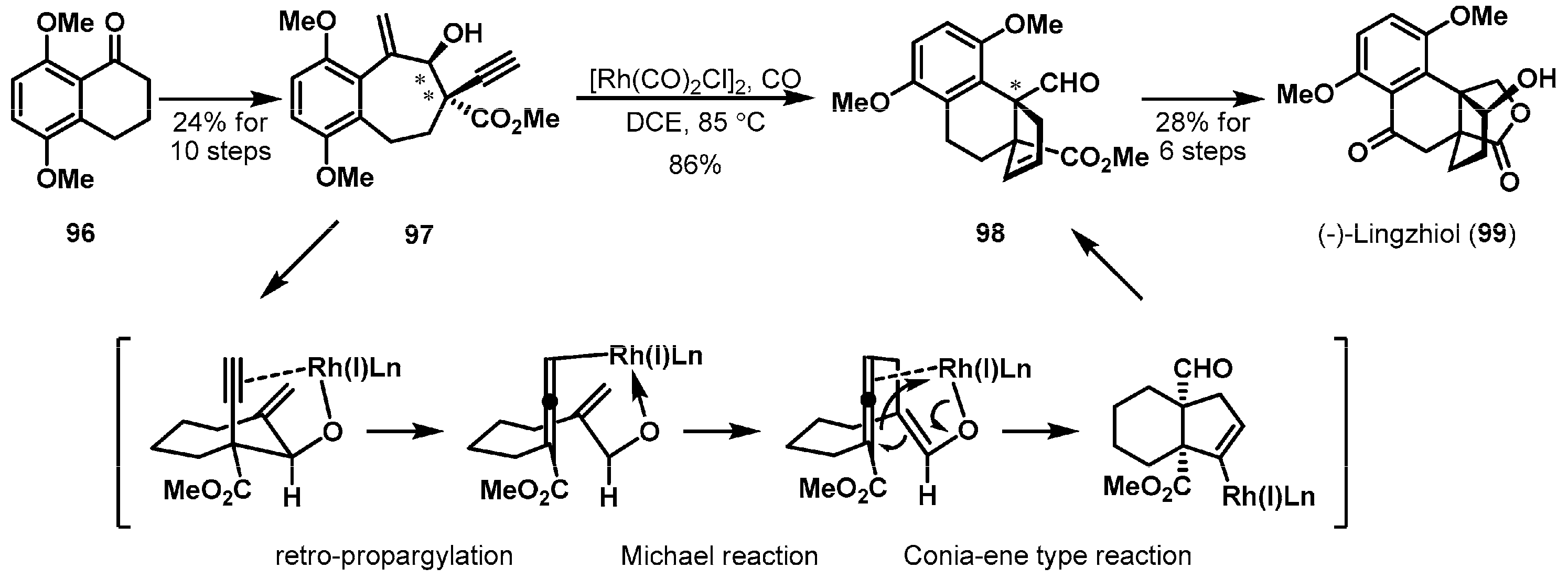
(−)-Lingzhiol and its enantiomer were separated from Ganoderma lucidum in 2013 and found to inhibit selectively p-Smad3 [63]. The architecture of lingzhiol is composed of a carbocyclo[4.3.0]nonane skeleton and two quaternary bridgehead carbons (Scheme 15). Long et al. achieved the total synthesis of (−)-lingzhiol (99) via a rhodium-catalyzed intramolecular [3 + 2] cycloaddition [64]. Important precursor 97 was easily generated from 5,8-dimethoxy-3,4-dihydronaphthalen-1(2H)-one (96) in 10 steps by a ring expansion reaction employing Koser’s reagent [65] and an alkynylation using Waser’s reagent [66]. Treatment of compound 97 with the rhodium catalyst in the presence of CO in DCE gave rise to tricycle 98 in a diastereoselective manner. The catalytic cycle underwent a retro-propargylation, Michael reaction, Conia-ene type reaction, and protonolysis. The stereochemistry of the product was transferred stereospecifically from the chirality of the substrate. Further six steps completed the synthesis of (−)-lingzhiol (99).
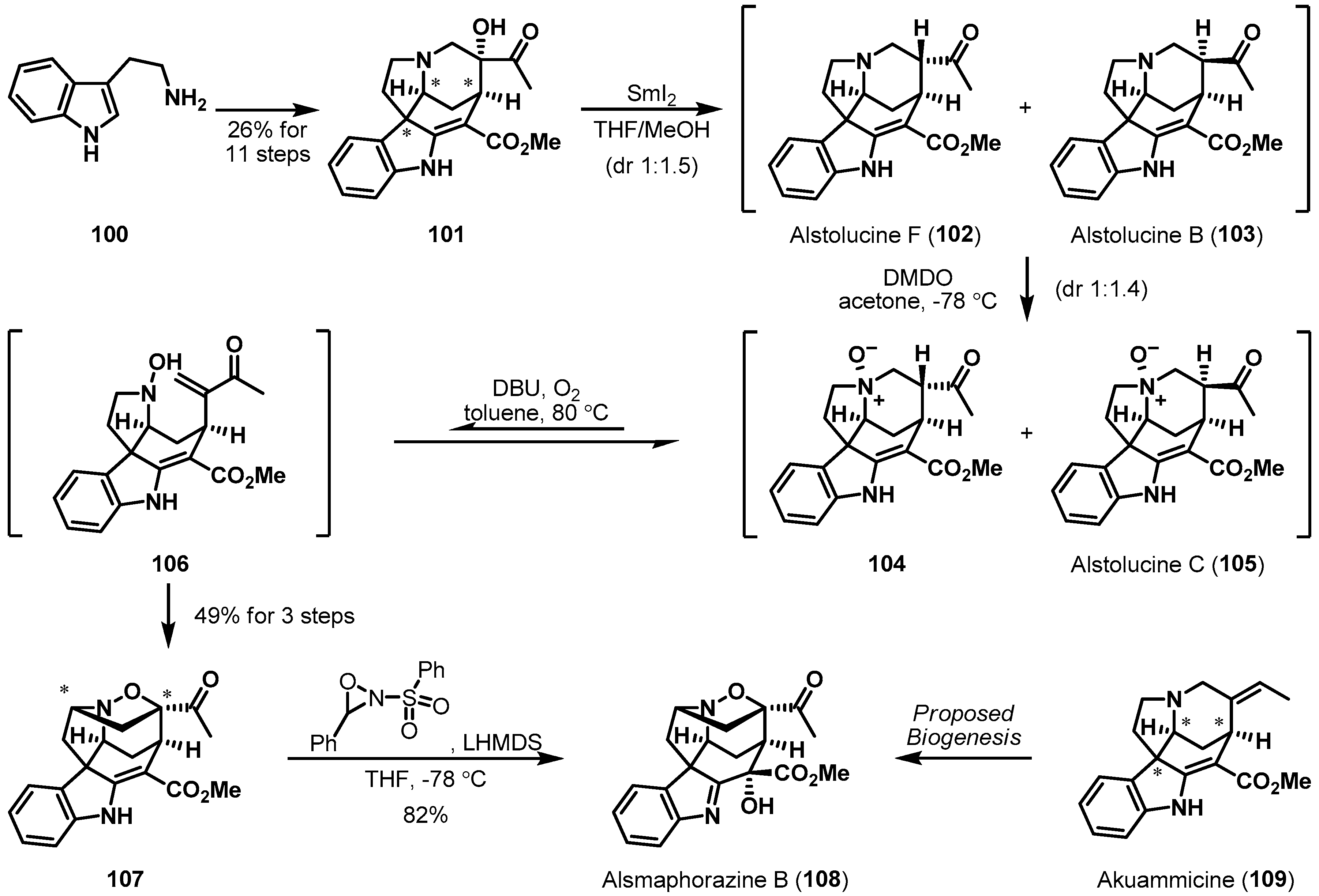
Vanderwal et al. recently described the total synthesis of alsmaphorazine B (108) by an intramolecular nitrone/alkene dipolar cycloaddition (Scheme 16) [67]. Alsmaphorazine B possesses a highly dense, oxidized, and cage-like hexacyclic structure with a rare endo N–O bond [68]. The authors analyzed the structure of 108 and then proposed an alternative biogenetic hypothesis of multiple oxidations from akuammicine (109). Akuammicine-derived pivotal substrate 101 was prepared from tryptamine (100) by applying a scalable process involving a Diels-Alder reaction and a Heck reaction. Firstly, deoxygenation of α-ketol 101 with samarium iodide furnished two diastereomers, 102 and 103, in 1:1.5 ratio. Subsequent DMDO oxidation of the mixture of tertiary amines 102 and 103 readily led to the corresponding N-oxides, 104 and 105. Then, treatment of the obtained N-oxide mixture, 104 and 105, with DBU liberated a hydroxylamine and an enone. Finally, the desired cycloadduct 107 was spontaneously produced in 49% yield for 3 steps. This surprising sequence proceeded stereoselectively in a substrate-controlled manner.
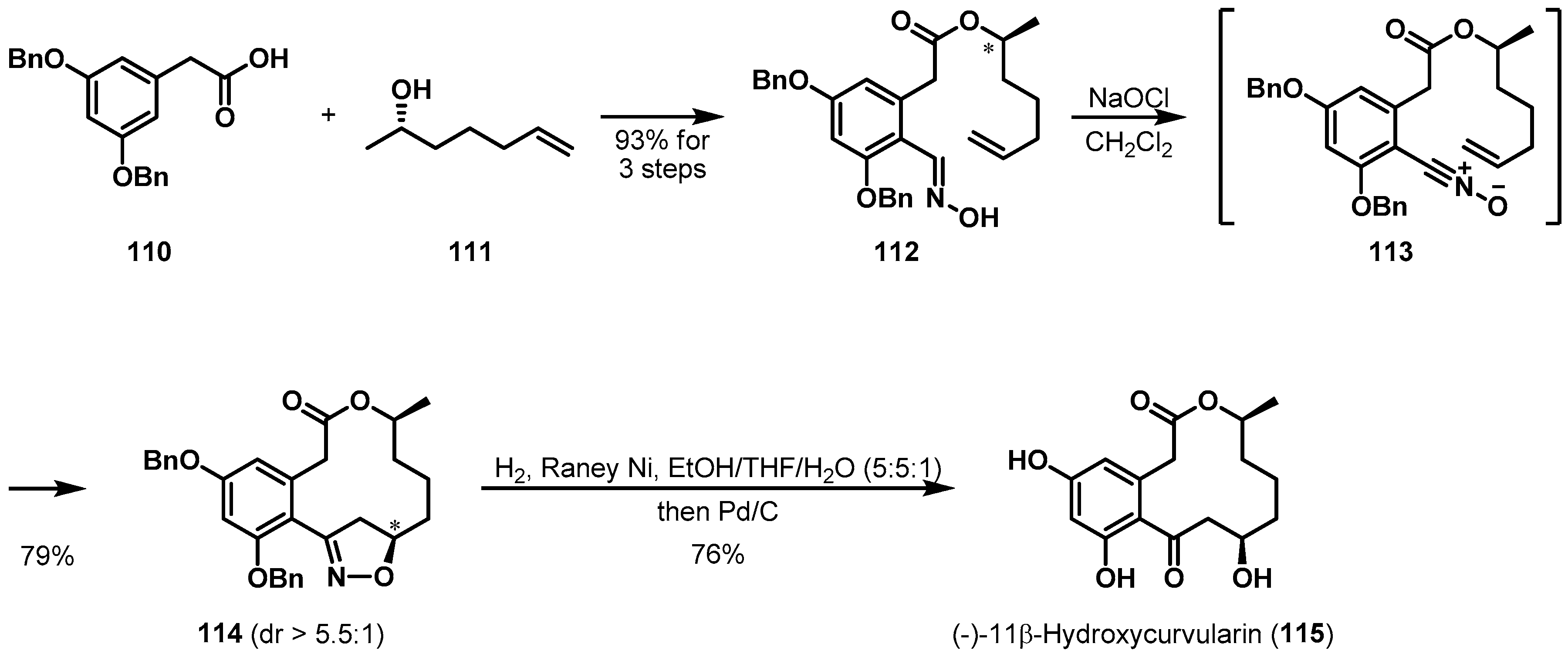
Intramolecular nitrile oxide-olefin cycloaddition (INOC) was utilized to form the macrocyclic moiety of (−)-11β-hydroxycurvularin (115) (Scheme 17). This polyketide was isolated from Alternaria tomato and contains a 3,5-dihydroxyphenylacetic acid skeleton [69]. Lee et al. described a remote stereoinductive fashion of the key cyclization to synthesize the natural product [70]. Oxime 112, the substrate of nitrile oxide 113, was efficiently prepared from commercially available 110 by esterification, formylation, and oxime formation. Then oxime 112 was subjected to an intramolecular nitrile oxide cycloaddition reaction. It is well known that isolated alkene group has decreased reactivity compared to enone group and that regio- and stereoselectivity can be problematic in large ring formation with remote stereocenter [71]. Unexpectedly, the desired isomer 114 was produced in 79% yield with good diastereoselectivity. This reaction was an example of unprecedented remote stereoinduction and concomitant macrocyclization.
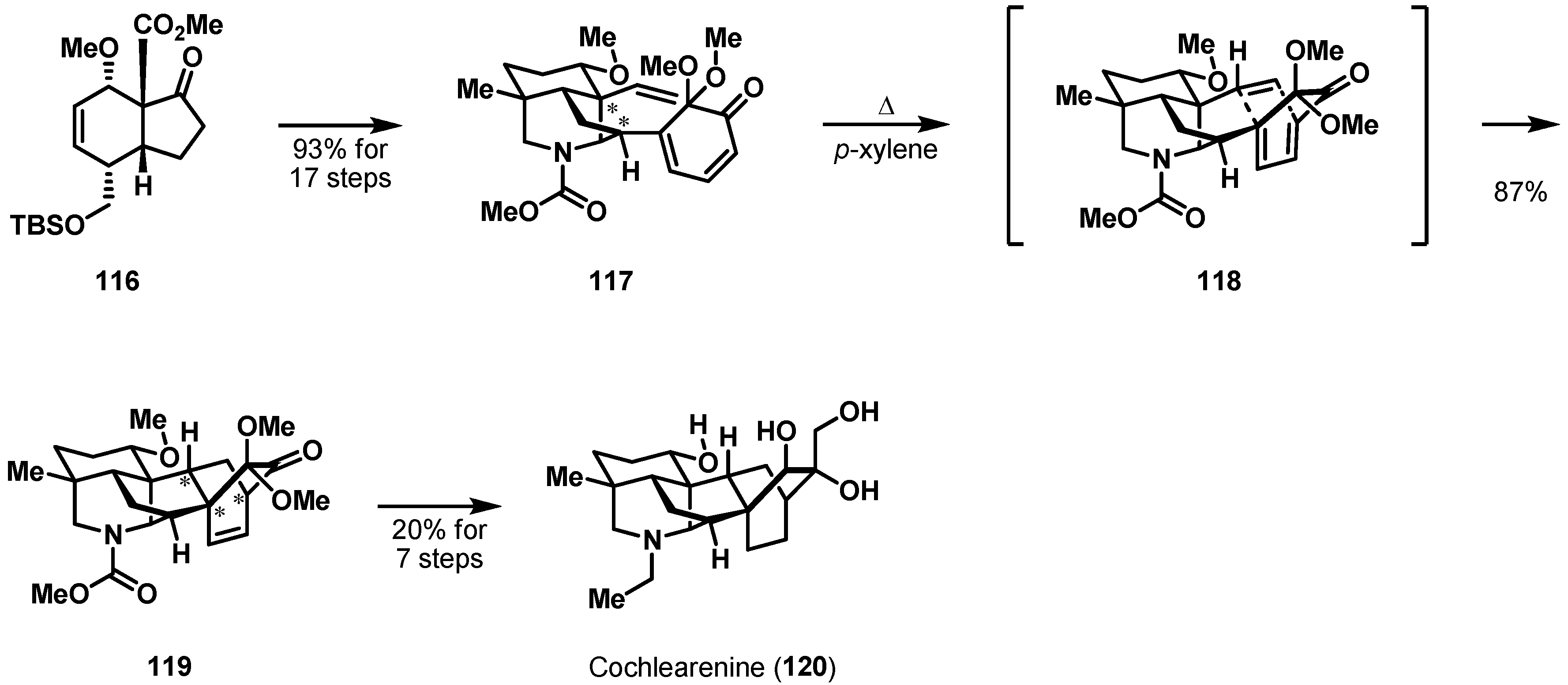
More recently, unified syntheses of denudatine-type diterpenoid alkaloids were disclosed by Sarpong et al. [72]. Among these alkaloids, cochlearenine (120) consists of a highly complex bicyclo[2.2.2] structural architecture and exhibits a bradycardic activity in guinea pig atria [73,74]. Synthesis of cyclization precursor 117 began with the known bicycle 116, which was effectively prepared using Diels-Alder cycloaddition [75] (Scheme 18). Subjecting a solution of dienone 117 in p-xylene to heat resulted in the exclusive formation of hexacycle 119, which contains the whole ring backbone of 120. The excellent diastereoselectivity was determined by a substrate-controlled transition state. Total synthesis of cochlearenine (120) was completed in seven steps from hexacycle 119.
6. Radical Cyclizations
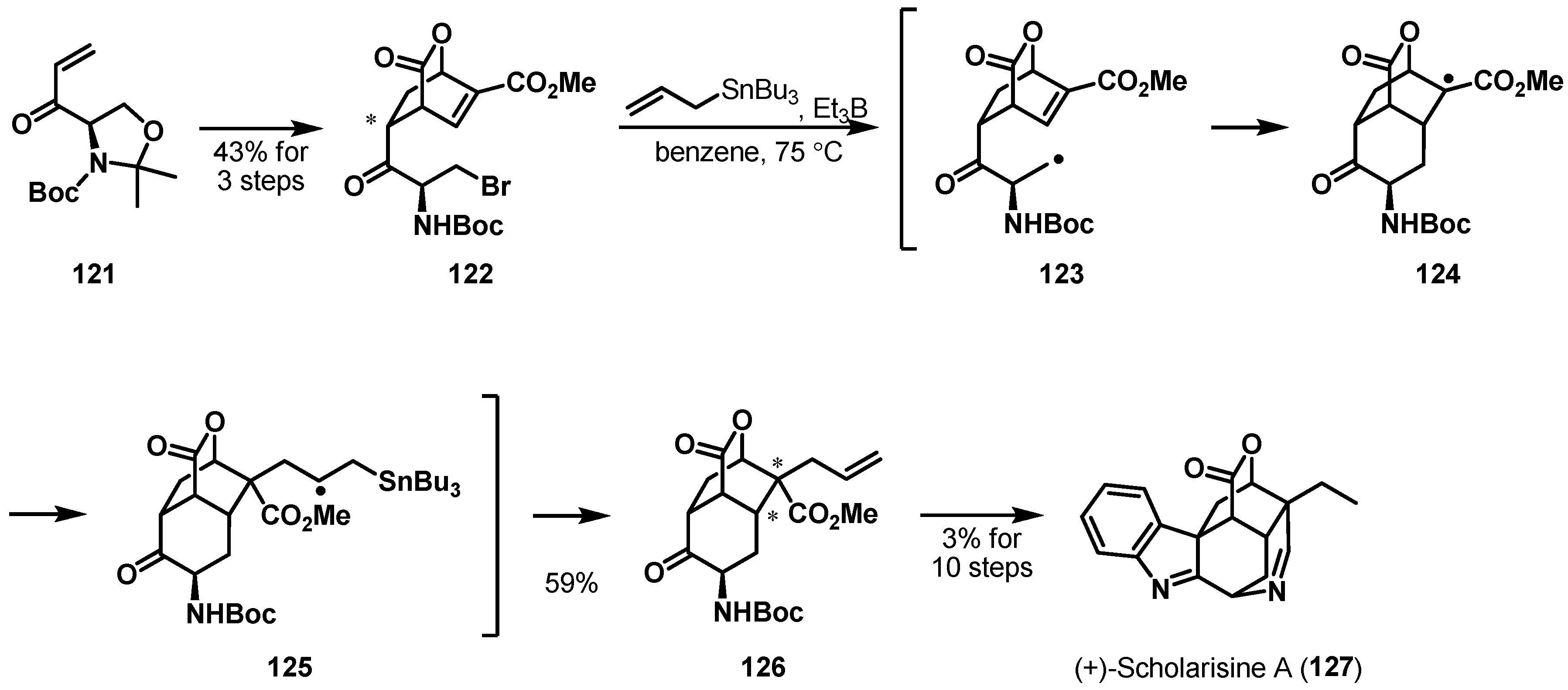
Radical cyclizations have served as a useful strategy to create various ring systems. As shown in Scheme 19, a total synthesis of the akuammiline alkaloid (+)-scholarisine A (127) was completed by Snyder et al. [76]. The structurally unique alkaloid, which was isolated from Alstonia scholaris, contains an indolenine fused to a strained carbocyclic cage and several tertiary and quaternary stereogenic centers [77]. To access the cage architecture of 126, a tandem 6-exo-trig radical cyclization/Keck allylation was explored. The key precursor 122 of the tandem reaction was efficiently prepared in three steps from acrylate 121 [78]. Substrate 122 was smoothly cyclized using allyltributylstannane and the radical initiator Et3B [79] in benzene at 75 °C, providing the desired 126 in 59% yield as a single diastereomer. The transformation likely progressed in the mechanistic fashion shown in Scheme 19 through the initial radical formation of 123, 6-exo-trig cyclization onto the Michael accepter of 124, and allylation of the resulting 125. The newly incorporated stereogenic centers were influenced by the asymmetric geometry of bicyclic lactone 122.
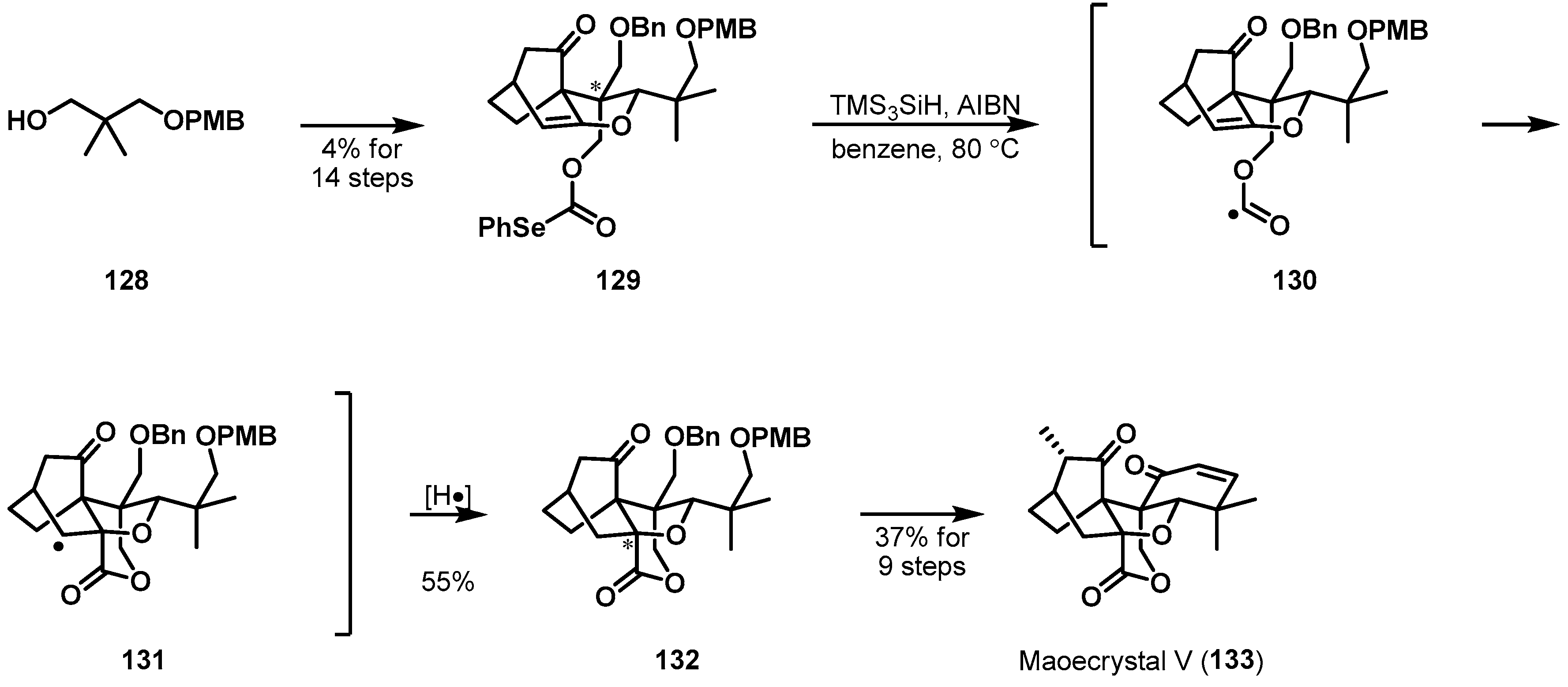
Another remarkable radical cyclization was reported by Zakarian et al. (Scheme 20) [80]. Maoecrystal V (133), a cytotoxic diterpenoid, was discovered in 2004 from Isodon eriocalyx, a Chinese medicinal herb [81]. Structurally, three contiguous quaternary stereocenters are compactly arranged on a pentacyclic framework; the complex molecular architecture provides an intriguing synthetic challenge. Key precursor 129, having two quaternary stereocenters, was synthesized from alcohol 128 through a C–H-insertion and a [4 + 2] cycloaddition. With phenylselenocarbonate 129 in hand, a substrate-controlled radical cyclization was explored. To a solution of substrate 129 in benzene at 80 °C was slowly added a mixture of TMS3SiH, as a less efficient hydrogen atom donor reagent, and AIBN, resulting in the formation of lactone 132 (55% yield). The asymmetric cyclization successfully generated the third quaternary stereocenter of 132 through the initially generated formyl radical 130.
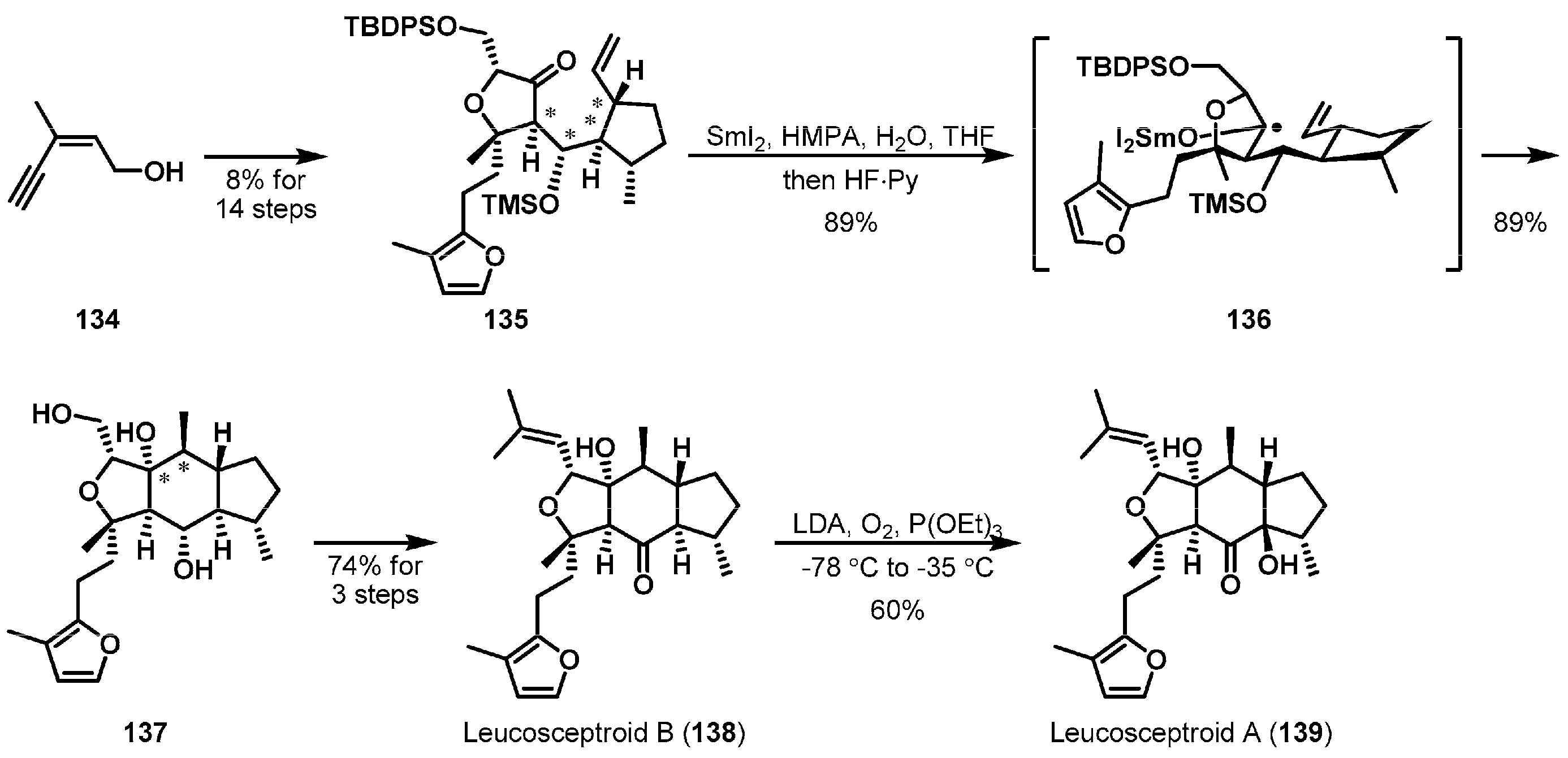
Ma et al. described the total syntheses of leucosceptroids A (139) and B (138) by applying a SmI2-mediated intramolecular ketyl-olefin radical cyclization (Scheme 21) [82]. These two sesterterpenoids were separated from glandilar trichomes of Leucosceptrum canum [83]. These intriguing compounds contain a common tricyclic hydrindane ring skeleton and eight contiguous stereogenic centers. Advanced intermediate 135 was smoothly accessed from commercially available enynol 134. Exposure of 135 to samarium(II) iodide gave rise to fused tricycle 137 through ketyl radical species 136 in a substrate-controlled manner. The α-OTMS unit of 136 played a critical role to promote selective 6-exo-cyclization, which competed with 7-endo-cyclization, by blocking the chelation of the free hydroxy group to SmI2, and high diastereoselectivity by decreasing the steric repulsion with the olefin. Further transformations efficiently led to completion of the syntheses of leucosceptroids A (139) and B (138).
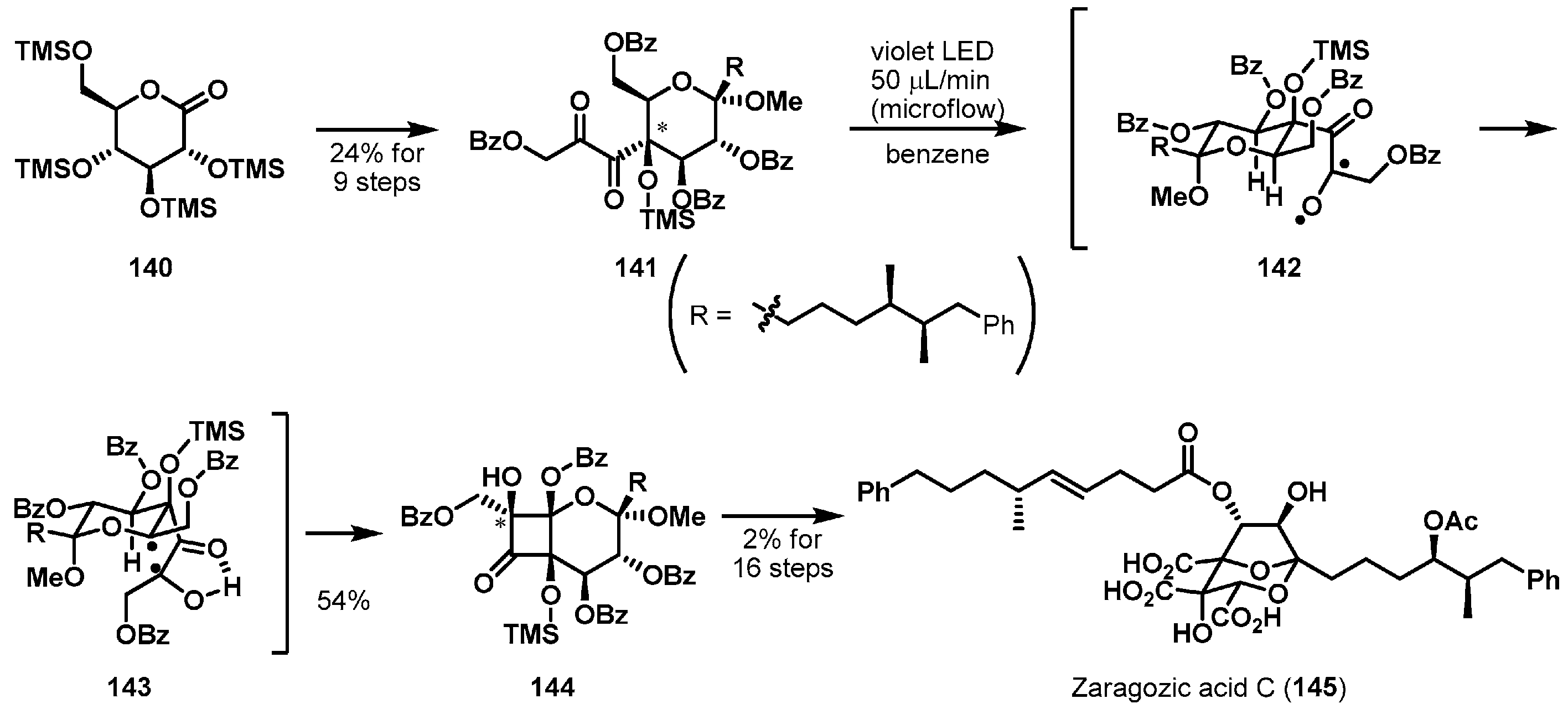
More recently, a remarkable photochemical C–H acylation was utilized to cyclize a complicated ring system (Scheme 22). Inoue et al. presented a total synthesis of zaragozic acid C (145) [84], a potent inhibitor of mammalian squalene synthase [85]. Natural acid 145 is characterized by a dioxabicyclo[3.2.1]octane architecture with an array of six stereocenters. Pivotal radical cyclization precursor 141 was produced from commercially available gluconolactone derivative 140. Irradiation of highly oxygenated substrate 141 with violet LED light excited the 1,2-diketone moiety and the resulting 1,2-biradical 142 spontaneously generated 1,4-biradical 143 via a hydrogen atom abstraction of the proximal ethereal C–H bond. The facile C–C bond formation of the 1,4-biradical 143 stereoselectively afforded the desired bicycle 144 (54% yield) by avoiding steric repulsions between the bulky substituents. Careful functional group transformations provide final product 145.
7. Conclusions
Substrate-controlled asymmetric cyclizations provide organic chemists with a powerful tool for the construction of optically active ring frameworks of natural products. In this review, a remarkable variety of cyclizations were classified by reaction type, such as anionic, cationic, transition metal-mediated, pericyclic, and radical reaction and discussed in detail. Various ring systems of natural products and their newly generated stereocenters were successfully established and defined. To conclude, asymmetric cyclization induced by the chiral nature of the substrate is in continuous development for the synthesis of natural products and related compounds.
Acknowledgments
This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT & Future Planning (2015R1C1A1A02036681) and the Bio & Medical Technology Development Program of the National Research Foundation (NRF) funded by the Ministry of Science, ICT & Future Planning, Republic of Korea (NRF-2016M3A9A5916225).
Author Contributions
All authors contributed to and approved the final manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| Ac | Acetyl |
| AIBN | Azobisisobutyronitrile |
| Bn | Benzyl |
| Boc | t-Butoxycarbonyl |
| Bu | Butyl |
| Bz | Benzoyl |
| cod | 1,5-Cyclooctadiene |
| CSA | Camphorsulfonic acid |
| dba | Dibenzylideneacetone |
| DBU | 1,8-Diazabicyclo[5.4.0]undec-7-ene |
| DCE | 1,2-Dichloroethane |
| DDQ | 2,3-Dichloro-5,6-dicyano-1,4-benzoquinone |
| DMDO | Dimethyldioxirane |
| DMF | N,N-Dimethylformamide |
| dppb | 1,4-Bis(diphenylphosphino)butane |
| Et | Ethyl |
| HMPA | Hexamethylphosphoramide |
| IBX | o-Iodoxybenzoic acid |
| LDA | Lithium diisopropylamide |
| LED | Light emitting diode |
| LHMDS | Lithium bis(trimethylsilyl)amide |
| Me | Methyl |
| MS | Molecular sieves |
| Ms | Mesyl |
| Ph | Phenyl |
| PMB | 4-Methoxybenzyl |
| PPTS | Pyridinium p-toluenesulfonate |
| Pr | Propyl |
| Py | Pyridine |
| TBDPS | t-Butyldiphenylsilyl |
| TBS | t-Butyldimethylsilyl |
| TES | Triethylsilyl |
| THF | Tetrahydrofuran |
| TMS | Trimethylsilyl |
| Ts | p-Toluenesulfonyl |
References
- Kobayashi, J.; Kubota, T. The Daphniphyllum alkaloids. Nat. Prod. Rep. 2009, 26, 936–962. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z. Muscarine, imidazole, oxazole, and thiazole alkaloids. Nat. Prod. Rep. 2011, 28, 1143–1191. [Google Scholar] [CrossRef] [PubMed]
- Kulcitki, V.; Harghel, P.; Ungur, N. Unusual cyclic terpenoids with terminal pendant prenyl moieties: From occurrence to synthesis. Nat. Prod. Rep. 2014, 31, 1686–1720. [Google Scholar] [CrossRef] [PubMed]
- Hanson, J.R. Diterpenoids of terrestrial origin. Nat. Prod. Rep. 2015, 32, 1654–1663. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Morris-Natschke, S.L.; Lee, K.H. Clerodane diterpenes: Sources, structures, and biological activities. Nat. Prod. Rep. 2016, 33, 1166–1226. [Google Scholar] [CrossRef] [PubMed]
- Moyano, A.; Rios, R. Asymmetric organocatalytic cyclization and cycloaddition reactions. Chem. Rev. 2011, 111, 4703–4832. [Google Scholar] [CrossRef] [PubMed]
- Gulder, T.; Baran, P.S. Strained cyclophane natural products: Macrocyclization at its limits. Nat. Prod. Rep. 2012, 29, 899–934. [Google Scholar] [CrossRef] [PubMed]
- Nicolaou, K.C.; Hale, C.R.H.; Nilewski, C.; Ioannidou, H.A. Constructing molecular complexity and diversity: Total synthesis of natural products of biological and medicinal importance. Chem. Soc. Rev. 2012, 41, 5185–5238. [Google Scholar] [CrossRef] [PubMed]
- Newhouse, T.; Baran, P.S.; Hoffmann, R.W. The economies of synthesis. Chem. Soc. Rev. 2009, 38, 3010–3021. [Google Scholar] [CrossRef] [PubMed]
- Paek, S.-M.; Jeong, M.; Jo, J.; Heo, Y.M.; Han, Y.T.; Yun, H. Recent advances in substrate-controlled asymmetric induction derived from chiral pool α-amino acids for natural product synthesis. Molecules 2016, 21, 951. [Google Scholar] [CrossRef] [PubMed]
- Kreis, L.M.; Carreira, E.M. Total synthesis of (−)-dendrobine. Angew. Chem. Int. Ed. 2012, 51, 3436–3439. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Keimatsu, I.; Ito, K. Über die alkaloide der chinesischen droge “Chin-Shih-Hu”. (II. Mitteil.). Über dendrobin. (I). J. Pharm. Soc. Jpn. 1932, 52, 1049–3439. (In German) [Google Scholar] [CrossRef]
- Suzuki, H.; Keimatsu, I.; Ito, K. Über die Alkaloide der chinesischen droge “Chin-Shin-Hu” (III. Mitteil.). Über das dendrobin. (II). J. Pharm. Soc. Jpn. 1934, 54, 801–819. (In German) [Google Scholar] [CrossRef]
- Costa, J.S.; Dias, A.G.; Anholeto, A.L.; Monteiro, M.D.; Patrocinio, V.L.; Costa, P.R.R. Syn-selective Michael addition of nitromethane derivatives to enoates derived from (R)-(+)-glyceraldehyde acetonide. J. Org. Chem. 1997, 62, 4002–4006. [Google Scholar] [CrossRef]
- Harada, S.; Sakai, T.; Takasu, K.; Yamada, K.; Yamamoto, Y.; Tomioka, K. Total synthesis of (−)-kopsinine by an asymmetric one-pot [N+2+3] cyclization. Chem. Asian J. 2012, 7, 2196–2198. [Google Scholar] [CrossRef] [PubMed]
- Crow, W.D.; Michael, M. The alkaloids of Kopsia longflora Merrill I. isolation of the alkaloids. Aust. J. Chem. 1955, 8, 129–135. [Google Scholar] [CrossRef]
- Crow, W.D.; Michael, M. Alkaloids of the Australian Apocynaceae: Kopsia longiflora Merr. III. preliminary Degradation of the Alkaloids. Aust. J. Chem. 1962, 15, 130–138. [Google Scholar] [CrossRef]
- Ravindar, K.; Caron, P.-Y.; Deslongchamps, P. Total synthesis of (+)-cassaine utilizing an anionic polycyclization strategy. Org. Lett. 2013, 15, 6270–6273. [Google Scholar] [CrossRef] [PubMed]
- Dalma, G. New alkaloids in Erythrophleum guineense. Ann. Chim. Appl. 1935, 25, 569–571. [Google Scholar]
- Caron, P.-Y.; Deslongchamps, P. Versatile strategy to access tricycles related to quassinoids and triterpenes. Org. Lett. 2010, 12, 508–511. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.-H.; Martinez, M.D.; Shenvi, R.A. An eight-step gram-scale synthesis of (−)-jiadifenolide. Nat. Chem. 2015, 7, 604–607. [Google Scholar] [CrossRef] [PubMed]
- Kubo, M.; Okada, C.; Huang, J.-M.; Harada, K.; Hioki, H.; Fukuyama, Y. Novel pentacyclic seco-prezizaane-type sesquiterpenoids with neurotrophic properties from Illicium jiadifengpi. Org. Lett. 2009, 11, 5190–5193. [Google Scholar] [CrossRef] [PubMed]
- Hirasawa, Y.; Morita, H.; Shiro, M.; Kobayashi, J. Sieboldine A, a novel tetracyclic alkaloid from Lycopodium sieboldii, inhibiting acetylcholinesterase. Org. Lett. 2003, 5, 3991–3993. [Google Scholar] [CrossRef] [PubMed]
- Canham, S.M.; France, D.J.; Overman, L.E. Total synthesis of (+)-sieboldine A. J. Am. Chem. Soc. 2010, 132, 7876–7877. [Google Scholar] [CrossRef] [PubMed]
- Canham, S.M.; France, D.J.; Overman, L.E. Total synthesis of (+)-sieboldine A: Evolution of a pinacol-terminated cyclization strategy. J. Org. Chem. 2013, 78, 9–34. [Google Scholar] [CrossRef] [PubMed]
- Sennhenn, P.; Gabler, B.; Helmchen, G. Enantiomerically pure cycloalkenylacetic acid derivatives via Pd-catalyzed asymmetric allylic alkylation and subsequent enantiomeric enrichment via iodolactones. Tetrahedron Lett. 1994, 35, 8595–8598. [Google Scholar] [CrossRef]
- Ernst, M.; Helmchen, G. A new synthesis route to enantiomerically pure jasmonoids. Angew. Chem. Int. Ed. 2002, 41, 4054–4056. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Cheng, X. Enantioselective total synthesis of (−)-zampanolide, a potent microtubule-stabilizing agent. Org. Lett. 2011, 13, 4108–4111. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.K.; Cheng, X.; Bai, R.; Hamel, E. Total synthesis of potent antitumor macrolide (−)-zampanolide: An oxidative intramolecular cyclization-based strategy. Eur. J. Org. Chem. 2012, 2012, 4130–4139. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, J.; Higa, T. Zampanolide, a new cytotoxic macrolide from a marine sponge. Tetrahedron Lett. 1996, 37, 5535–5538. [Google Scholar] [CrossRef]
- Field, J.J.; Singh, A.J.; Kanakkanthara, A.; Halafihi, T.; Northcote, P.T.; Miller, J.H. Microtubule-stabilizing activity of zampanolide, a potent macrolide isolated from the Tongan marine sponge Cacospongia mycofijiensis. J. Med. Chem. 2009, 52, 7328–7332. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, M.; Ohkuma, T.; Inoue, S.; Sayo, N.; Kumobayashi, H.; Akutagawa, S.; Ohta, T.; Takaya, H.; Noyori, R. Homogeneous asymmetric hydrogenation of functionalized ketones. J. Am. Chem. Soc. 1988, 110, 629–631. [Google Scholar] [CrossRef]
- McCandlish, L.E.; Hanson, J.C.; Stout, G.H. The structures of two derivatives of bicyclo[3,3,1]nonane-2,4,9-trione. A natural product: Clusianone, C33H42O4, and trimethylated catechinic acid, C18H20O6. Acta Crystallogr. Sect. B 1976, 32, 1793–1801. [Google Scholar] [CrossRef]
- Boyce, J.H.; Porco, J.A., Jr. Asymmetric, stereodivergent synthesis of (−)-clusianone utilizing a biomimetic cationic cyclization. Angew. Chem. Int. Ed. 2014, 53, 7832–7837. [Google Scholar] [CrossRef] [PubMed]
- Azuma, M.; Yoshikawa, T.; Kogure, N.; Kitajima, M.; Takayama, H. Biogenetically inspired total syntheses of Lycopodium alkaloids, (+)-flabellidine and (−)-lycodine. J. Am. Chem. Soc. 2014, 136, 11618–11621. [Google Scholar] [CrossRef] [PubMed]
- Manske, R.H.F.; Marion, L. The alkaloids of Lycopodium species: I. Lycopodium complanatum L. Can. J. Res. 1942, 20b, 87–92. [Google Scholar] [CrossRef]
- Anet, F.A.L.; Eves, C.R. Lycodine, a new alkaloid of Lycopodium annotinum. Can. J. Chem. 1958, 36, 902–909. [Google Scholar] [CrossRef]
- Nakayama, A.; Kogure, N.; Kitajima, M.; Takayama, H. First asymmetric total syntheses of fawcettimine-type Lycopodium alkaloids, lycoposerramine-C and phlegmariurine-A. Org. Lett. 2009, 11, 5554–5557. [Google Scholar] [CrossRef] [PubMed]
- Bödeker, K. Lycopodin, das erste alkaloïd der Gefässkryptogamen. Liebigs Ann. Chem. 1881, 208, 363–367. [Google Scholar] [CrossRef]
- Ma, D.; Zhong, Z.; Liu, Z.; Zhang, M.; Xu, S.; Xu, D.; Song, D.; Xie, X.; She, X. Protecting-group-free total synthesis of (−)-lycopodine via phosphoric acid promoted alkyne aza-Prins cyclization. Org. Lett. 2016, 18, 4328–4331. [Google Scholar] [CrossRef] [PubMed]
- Kozak, J.A.; Dake, G.R. Total synthesis of (+)-fawcettidine. Angew. Chem. Int. Ed. 2008, 47, 4221–4223. [Google Scholar] [CrossRef] [PubMed]
- Inuki, S.; Iwata, A.; Oishi, S.; Fujii, N.; Ohno, H. Enantioselective total synthesis of (+)-lysergic acid, (+)-lysergol, and (+)-isolysergol by palladium-catalyzed domino cyclization of allenes bearing amino and bromoindolyl groups. J. Org. Chem. 2011, 76, 2072–2083. [Google Scholar] [CrossRef] [PubMed]
- Agurell, S. Isolysergol from saprophytic cultures of ergot. Acta Pharm. Suec. 1966, 3, 7–10. [Google Scholar] [PubMed]
- Mizuno, T.; Oonishi, Y.; Takimoto, M.; Sato, Y. Total synthesis of (−)-corynantheidine by nickel-catalyzed carboxylative cyclization of enynes. Eur. J. Org. Chem. 2011, 2011, 2606–2609. [Google Scholar] [CrossRef]
- Janot, M.-M.; Goutarel, R. La corynanthéidine, nouvel alcaloїde cristallisé isolé des écorces de Pseudocinchona africana. Aug. Chev. C. R. Acad. Sci. 1944, 218, 852–854. (In French) [Google Scholar]
- Bailey, P.D.; Hollinshead, S.P.; McLay, N.R. Exceptional stereochemical control in the Pictet-Spengler reaction. Tetrahedron Lett. 1987, 28, 5177–5180. [Google Scholar] [CrossRef]
- Seo, H.; Yun, H.; Lee, S.; Jang, J.; Han, Y.T.; Kim, D.-D.; Lee, J.; Suh, Y.G. Stereoselective synthesis of 7-epi-incarvilline. Org. Lett. 2013, 15, 531–533. [Google Scholar] [CrossRef] [PubMed]
- Chi, Y.-M.; Yan, W.-M.; Li, J.-S. An alkaloid from Incarvillea sinensis. Phytochemistry 1990, 29, 2376–2378. [Google Scholar] [CrossRef]
- Chi, Y.-M.; Yan, W.-M.; Chen, D.-C.; Hiroshi, N.; Yoichi, I.; Ushio, S. A monoterpene alkaloid from Incarvillea sinensis. Phytochemistry 1992, 31, 2930–2932. [Google Scholar] [CrossRef]
- Chi, Y.-M.; Hashimoto, F.; Yan, W.-M.; Nohara, T. Two alkaloids from Incarvillea sinensis. Phytochemistry 1995, 39, 1485–1487. [Google Scholar] [CrossRef]
- Lee, S.; Paek, S.-M.; Yun, H.; Kim, N.-J.; Suh, Y.-G. Enantioselective total synthesis of a natural iridoid. Org. Lett. 2011, 13, 3344–3347. [Google Scholar] [CrossRef] [PubMed]
- Acebey, L.; Sauvain, M.; Beck, S.; Moulis, C.; Gimenez, A.; Jullian, V. Bolivianine, a new sesterpene with an unusual skeleton from Hedyosmum angustifolium, and its isomer, isobolivianine. Org. Lett. 2007, 9, 4693–4696. [Google Scholar] [CrossRef] [PubMed]
- Yuan, C.; Du, B.; Yang, L.; Liu, B. Bioinspired total synthesis of bolivianine: A Diels-Alder/intramolecular hetero-Diels-Alder cascade approach. J. Am. Chem. Soc. 2013, 135, 9291–9294. [Google Scholar] [CrossRef] [PubMed]
- Du, B.; Yuan, C.; Yu, T.; Yang, L.; Yang, Y.; Liu, B.; Qin, S. Asymmetric total synthesis of onoseriolide, bolivianine, and isobolivianine. Chem. Eur. J. 2014, 20, 2613–2622. [Google Scholar] [CrossRef] [PubMed]
- Naruse, N.; Tenmyo, O.; Kawano, K.; Tomita, K.; Ohgusa, N.; Miyaki, T.; Konishi, M.; Oki, T. Fluvirucins A1, A2, B1, B2, B3, B4 and B5, new antibiotics active against influenza A virus I. Production, isolation, chemical properties and biological activities. J. Antibiot. 1991, 44, 733–740. [Google Scholar] [CrossRef] [PubMed]
- Naruse, N.; Tsuno, T.; Sawada, Y.; Konishi, M.; Oki, T. Fluvirucins A1, A2, B1, B2, B3, B4 and B5, new antibiotics active against influenza A virus II. Structural determination. J. Antibiot. 1991, 44, 741–755. [Google Scholar] [CrossRef] [PubMed]
- Naruse, N.; Konishi, M.; Oki, T. Fluvirucins A1, A2, B1, B2, B3, B4 and B5, new antibiotics active against influenza A virus III. The stereochemistry and absolute configuration of fluvirucin A1. J. Antibiot. 1991, 44, 756–761. [Google Scholar] [CrossRef] [PubMed]
- Tomita, K.; Oda, N.; Hoshino, Y.; Ohkusa, N.; Chikazawa, H. Fluvirucins A1, A2, B1, B2, B3, B4 and B5, new antibiotics active against influenza A virus IV. Taxonomy on the producing organisms. J. Antibiot. 1991, 44, 940–948. [Google Scholar] [CrossRef] [PubMed]
- Hegde, V.R.; Patel, M.G.; Gullo, V.P.; Ganguly, A.K.; Sarre, O.; Puar, M.S. Macrolactams: A new class of antifungal agents. J. Am. Chem. Soc. 1990, 112, 6403–6405. [Google Scholar] [CrossRef]
- Hegde, V.; Patel, M.; Horan, A.; Gullo, V.; Marquez, J.; Gunnarsson, I.; Gentile, F.; Loebenberg, D.; King, A. Macrolactams: A novel class of antifungal antibiotics produced by Actinomadura spp. SCC 1776 and SCC 1777. J. Antibiot. 1992, 45, 624–632. [Google Scholar] [CrossRef] [PubMed]
- Suh, Y.-G.; Lee, Y.-S.; Kim, S.-H.; Jung, J.-K.; Yun, H.; Jang, J.; Kim, N.-J.; Jung, J.-W. A stereo-controlled access to functionalized macrolactams via an aza-Claisen rearrangement. Org. Biomol. Chem. 2012, 10, 561–568. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.S.; Jung, J.W.; Kim, S.H.; Jung, J.K.; Paek, S.M.; Kim, N.J.; Chang, D.J.; Lee, J.; Suh, Y.G. First total synthesis and structural confirmation of fluvirucinine A2 via an iterative ring expansion strategy. Org. Lett. 2010, 12, 2040–2043. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.M.; Ai, J.; Zhou, L.L.; Chung, A.C.K.; Li, R.; Nie, J.; Fang, P.; Wang, X.L.; Luo, J.; Hu, Q.; et al. Lingzhiols, Unprecedented Rotary Door-Shaped Meroterpenoids as Potent and Selective Inhibitors of p-Smad3 from Ganoderma lucidum. Org. Lett. 2013, 15, 5488–5491. [Google Scholar] [CrossRef] [PubMed]
- Long, R.; Huang, J.; Shao, W.; Liu, S.; Lan, Y.; Gong, J.; Yang, Z. Asymmetric total synthesis of (−)-lingzhiol via a Rh-catalysed [3 + 2] cycloaddition. Nat. Commun. 2014, 5, 5707–5716. [Google Scholar] [CrossRef] [PubMed]
- Justik, M.W.; Koser, G.F. Oxidative rearrangements of arylalkenes with [hydroxy(tosyloxy)iodo]benzene in 95% methanol: A general, regiospecific synthesis of α-aryl ketones. Tetrahedron Lett. 2004, 45, 6159–6163. [Google Scholar] [CrossRef]
- González, D.F.; Brand, J.P.; Waser, J. Ethynyl-1,2-benziodoxol-3(1H)-one (EBX): An exceptional reagent for the ethynylation of keto, cyano, and nitro esters. Chem. Eur. J. 2010, 16, 9457–9461. [Google Scholar] [CrossRef] [PubMed]
- Hong, A.Y.; Vanderwal, C.D. A synthesis of alsmaphorazine B demonstrates the chemical feasibility of a new biogenetic hypothesis. J. Am. Chem. Soc. 2015, 137, 7306–7309. [Google Scholar] [CrossRef] [PubMed]
- Koyama, K.; Hirasawa, Y.; Nugroho, A.E.; Hosoya, T.; Hoe, T.C.; Chan, K.L.; Morita, H. Alsmaphorazines A and B, novel indole alkaloids from Alstonia pneumatophore. Org. Lett. 2010, 12, 4188–4191. [Google Scholar] [CrossRef] [PubMed]
- Hyeon, S.-B.; Ozaki, A.; Suzuki, A.; Tamura, S. Isolation of αβ-dehydrocurvularin and β-hydroxycurvularin from Alternaria tomato as sporulation-suppressing factors. Agric. Biol. Chem. 1976, 40, 1663–1664. [Google Scholar]
- Choe, H.; Pham, T.T.; Lee, J.Y.; Latif, M.; Park, H.; Kang, Y.K.; Lee, J. Remote stereoinductive intramolecular nitrile oxide cycloaddition: Asymmetric total synthesis and structure revision of (−)-11 β-hydroxycurvularin. J. Org. Chem. 2016, 81, 2612–2617. [Google Scholar] [CrossRef] [PubMed]
- Shing, T.K.M.; Zhong, Y.-L. Synthesis of medium-sized cyclic ethers from carbohydrates via an intramolecular nitrile oxide-alkene cycloaddtion strategy. Synlett 2006, 1205–1208. [Google Scholar] [CrossRef]
- Kou, K.G.M.; Li, B.X.; Lee, J.C.; Gallego, G.M.; Lebold, T.P.; DiPasquale, A.G.; Sarpong, R. Syntheses of denudatine diterpenoid alkaloids: Cochlearenine, N-ethyl-1α-hydroxy-17-veratroyldictyzine, and paniculamine. J. Am. Chem. Soc. 2016, 138, 10830–10833. [Google Scholar] [CrossRef] [PubMed]
- Kolak, U.; Öztürk, M.; Özgökçe, F.; Ulubelen, A. Norditerpene alkaloids from Delphinium linearilobum and antioxidant activity. Phytochemistry 2006, 67, 2170–2175. [Google Scholar] [CrossRef] [PubMed]
- Wada, K.; Kawahara, N. Diterpenoid and norditerpenoid alkaloids from the roots of Aconitum yesoense var. macroyesoense. Helv. Chim. Acta 2009, 92, 629–637. [Google Scholar] [CrossRef]
- Marth, C.J.; Gallego, G.M.; Lee, J.C.; Lebold, T.P.; Kulyk, S.; Kou, K.G.M.; Qin, J.; Lilien, R.; Sarpong, R. Network-analysis-guided synthesis of weisaconitine D and liljestrandinine. Nature 2015, 528, 493–498. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.W.; Snyder, S.A. A concise total synthesis of (+)-scholarisine A empowered by a unique C-H arylation. J. Am. Chem. Soc. 2013, 135, 12964–12967. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.H.; Tan, Q.G.; Liu, Y.P.; Feng, T.; Du, Z.Z.; Li, W.Q.; Luo, X.D. A cage-monoterpene indole alkaloid from Alstonia scholaris. Org. Lett. 2008, 10, 577–580. [Google Scholar] [CrossRef] [PubMed]
- Ageno, G.; Banfi, L.; Cascio, G.; Guanti, G.; Manghisi, E.; Riva, R.; Rocca, V. Enantiospecific and diastereoselective synthesis of 4,4-disubstituted-3-amino-2-azetidinones, starting from d-serine. Tetrahedron 1995, 51, 8121–8134. [Google Scholar] [CrossRef]
- Reddy, L.R.; Fournier, J.-F.; Reddy, B.V.S.; Corey, E.J. An efficient, stereocontrolled synthesis of a potent omuralide-salinosporin hybrid for selective proteasome inhibition. J. Am. Chem. Soc. 2005, 127, 8974–8976. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.; Gu, Z.; Zakarian, A. Total synthesis of maoecrystal V: Early-stage C-H functionalization and lactone assembly by radical cyclization. J. Am. Chem. Soc. 2013, 135, 14552–14555. [Google Scholar] [CrossRef] [PubMed]
- Li, S.-H.; Wang, J.; Niu, X.-M.; Shen, Y.-H.; Zhang, H.-J.; Sun, H.-D.; Li, M.-L.; Tian, Q.-E.; Lu, Y.; Cao, P.; et al. Maoecrystal V, cytotoxic diterpenoid with a novel C19 skeleton from Isodon eriocalyx (Dunn.) Hara. Org. Lett. 2004, 6, 4327–4330. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Liu, J.; Ma, D. Total synthesis of leucosceptroids A and B. Angew. Chem. Int. Ed. 2015, 54, 1298–1301. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.-H.; Luo, Q.; Niu, X.-M.; Xie, M.-J.; Zhao, X.; Schneider, B.; Gershenzon, J.; Li, S.-H. Glandular trichomes of Leucosceptrum canum harbor defensive sesterterpenoids. Angew. Chem. Int. Ed. 2010, 49, 4471–4475. [Google Scholar] [CrossRef] [PubMed]
- Kawamata, T.; Nagatomo, M.; Inoue, M. Total synthesis of zaragozic acid C: Implementation of photochemical C(sp3)–H acylation. J. Am. Chem. Soc. 2017, 139, 1814–1817. [Google Scholar] [CrossRef] [PubMed]
- Dufresne, C.; Wilson, K.E.; Zink, D.; Smith, J.; Bergstrom, J.D.; Kurtz, M.; Rew, D.; Nallin, M.; Jenkins, R.; Bartizal, K.; et al. The isolation and structure elucidation of zaragozic acid C, a novel potent squalene synthase inhibitor. Tetrahedron 1992, 48, 10221–10226. [Google Scholar] [CrossRef]
Scheme 1.
Total syntheses of (−)-dendrobine (8).

Scheme 2.
Total synthesis of (−)-kopsinine (17).

Scheme 3.
Total syntheses of (+)-cassaine (23).

Scheme 4.
Total synthesis of (−)-jiadifenolide (31).

Scheme 5.
Total synthesis of (+)-sieboldine A (37).

Scheme 6.
Total synthesis of (−)-zampanolide (42).

Scheme 7.
Total synthesis of (−)-clusianone (48).

Scheme 8.
Total syntheses of (+)-flabellidine (55) and (−)-lycodine (56).

Scheme 9.
Total synthesis of (−)-lycopodine (63).

Scheme 10.
Total synthesis of (+)-isolysergol (69).

Scheme 11.
Total synthesis of (−)-corynantheidine (76).

Scheme 12.
Synthesis of 7-epi-incarvilline (77).

Scheme 13.
Total synthesis of bolivianine (90).

Scheme 14.
Total synthesis of fluvirucinine A1 (95).

Scheme 15.
Total synthesis of (−)-lingzhiol (99).

Scheme 16.
Total synthesis of alsmaphorazine B (108).

Scheme 17.
Total synthesis of (−)-11β-hydroxycurvularin (115).

Scheme 18.
Total synthesis of cochlearenine (120).

Scheme 19.
Total synthesis of (+)-scholarisine (127).

Scheme 20.
Total synthesis of maoecrystal V (133).

Scheme 21.
Total syntheses of leucosceptroids A (139) and B (138).

Scheme 22.
Total synthesis of zaragozic acid C (145).

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Jo, J.; Kim, S.-H.; Han, Y.T.; Kwak, J.-H.; Yun, H. Recent Advances in Substrate-Controlled Asymmetric Cyclization for Natural Product Synthesis. Molecules 2017, 22, 1069. https://doi.org/10.3390/molecules22071069
AMA Style
Jo J, Kim S-H, Han YT, Kwak J-H, Yun H. Recent Advances in Substrate-Controlled Asymmetric Cyclization for Natural Product Synthesis. Molecules. 2017; 22(7):1069. https://doi.org/10.3390/molecules22071069
Chicago/Turabian StyleJo, Jeyun, Seok-Ho Kim, Young Taek Han, Jae-Hwan Kwak, and Hwayoung Yun. 2017. "Recent Advances in Substrate-Controlled Asymmetric Cyclization for Natural Product Synthesis" Molecules 22, no. 7: 1069. https://doi.org/10.3390/molecules22071069