Metatranscriptomic Analysis of the Mouse Gut Microbiome Response to the Persistent Organic Pollutant 2,3,7,8-Tetrachlorodibenzofuran

 , ,
, ,
Abstract
:1. Introduction
2. Results and Discussion
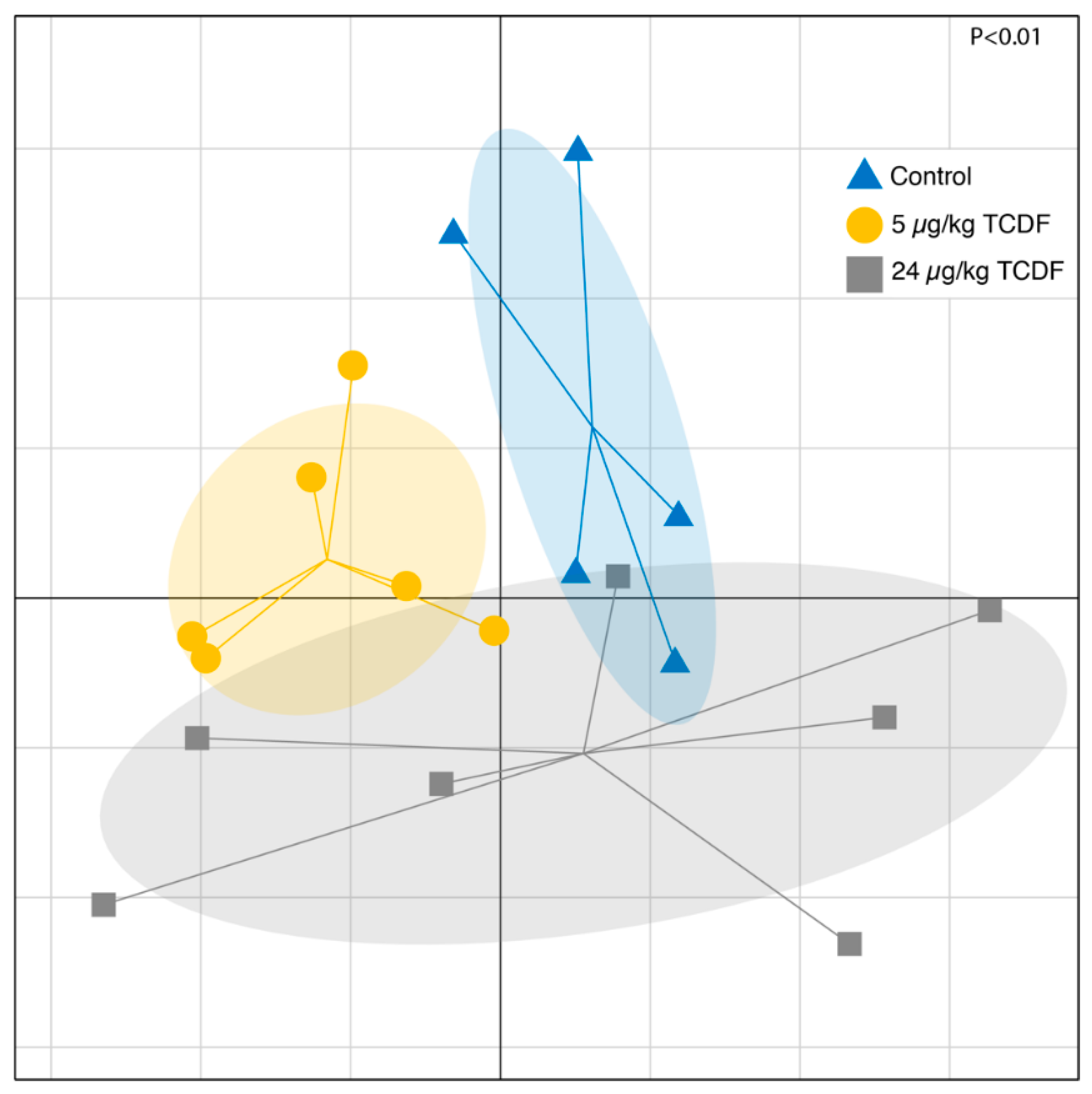
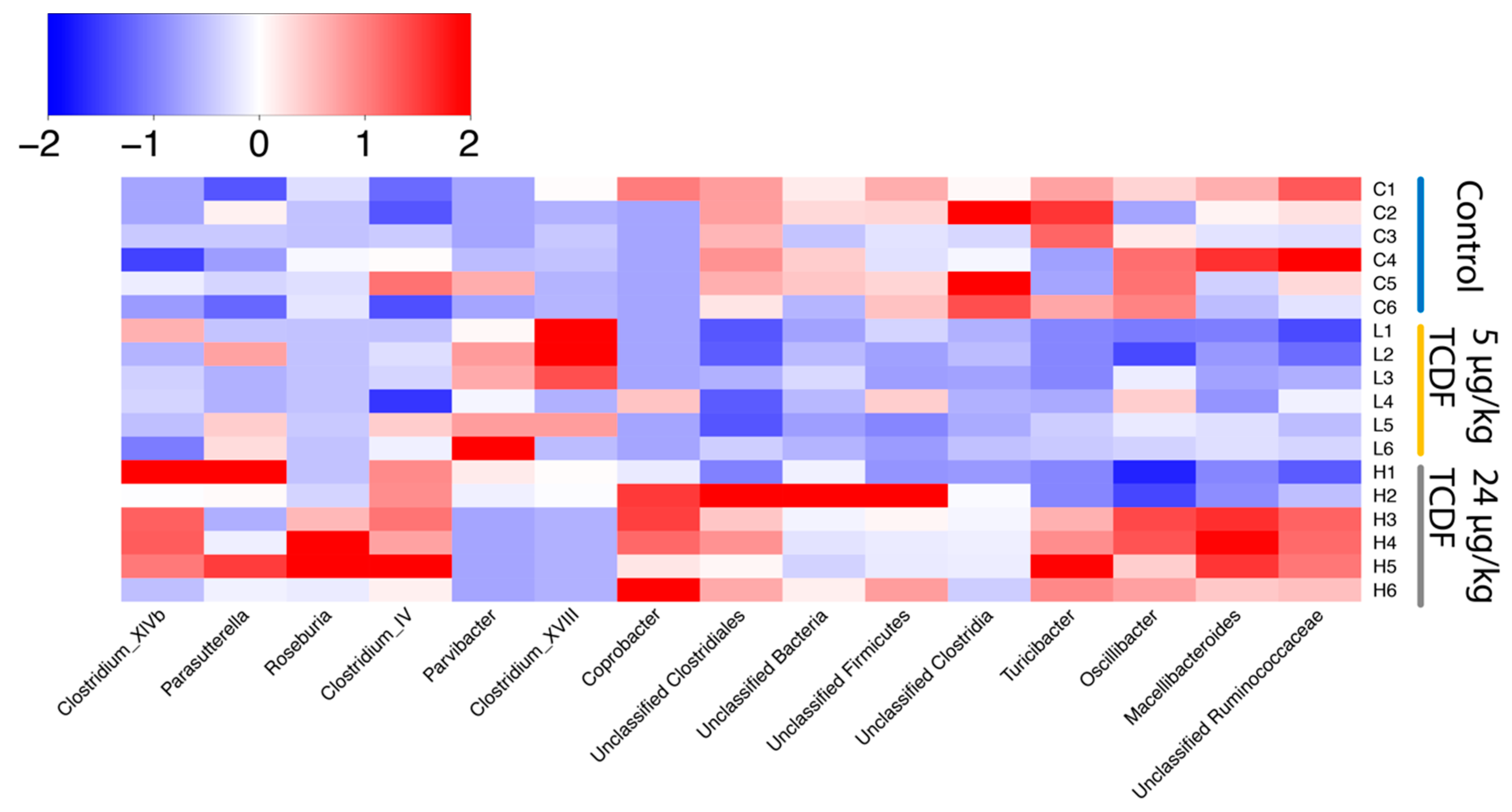
2.1. Bacterial Taxonomic Shifts After TCDF Treatment
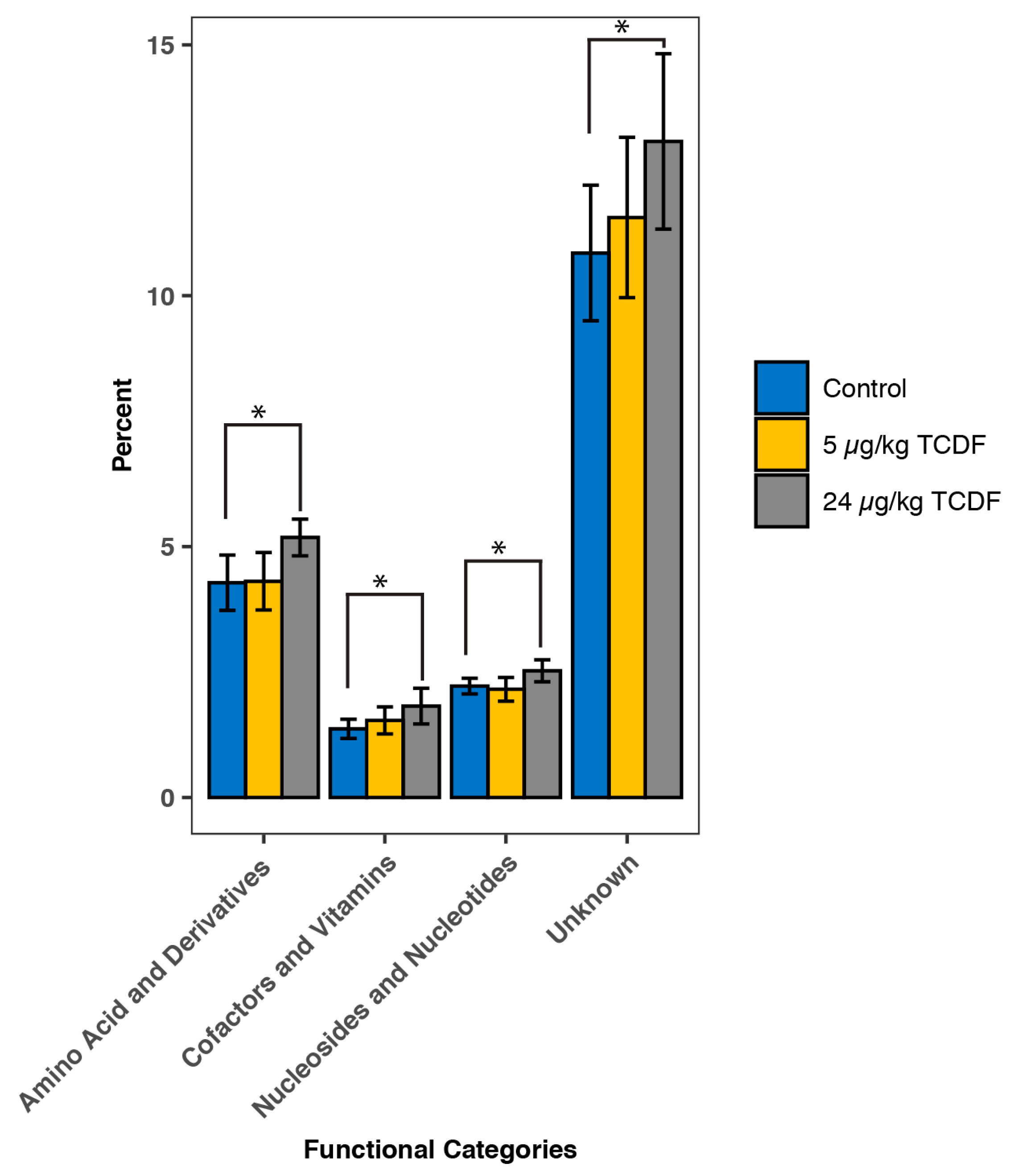
2.2. PICRUSt Functional Predictions
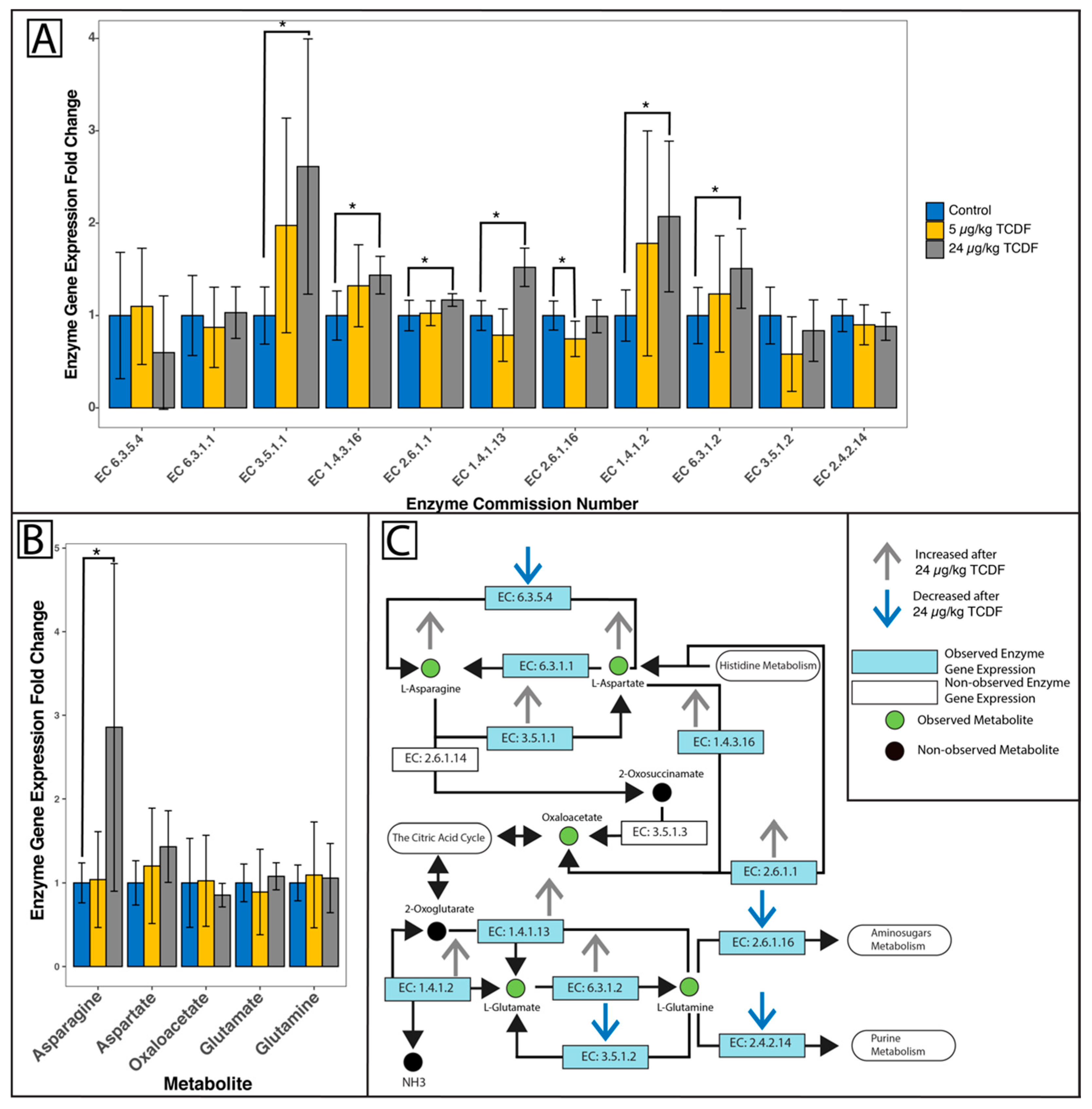
2.3. Shifts in the Metabolome after TCDF Treatment
2.4. Changes in Overall Microbiome Function
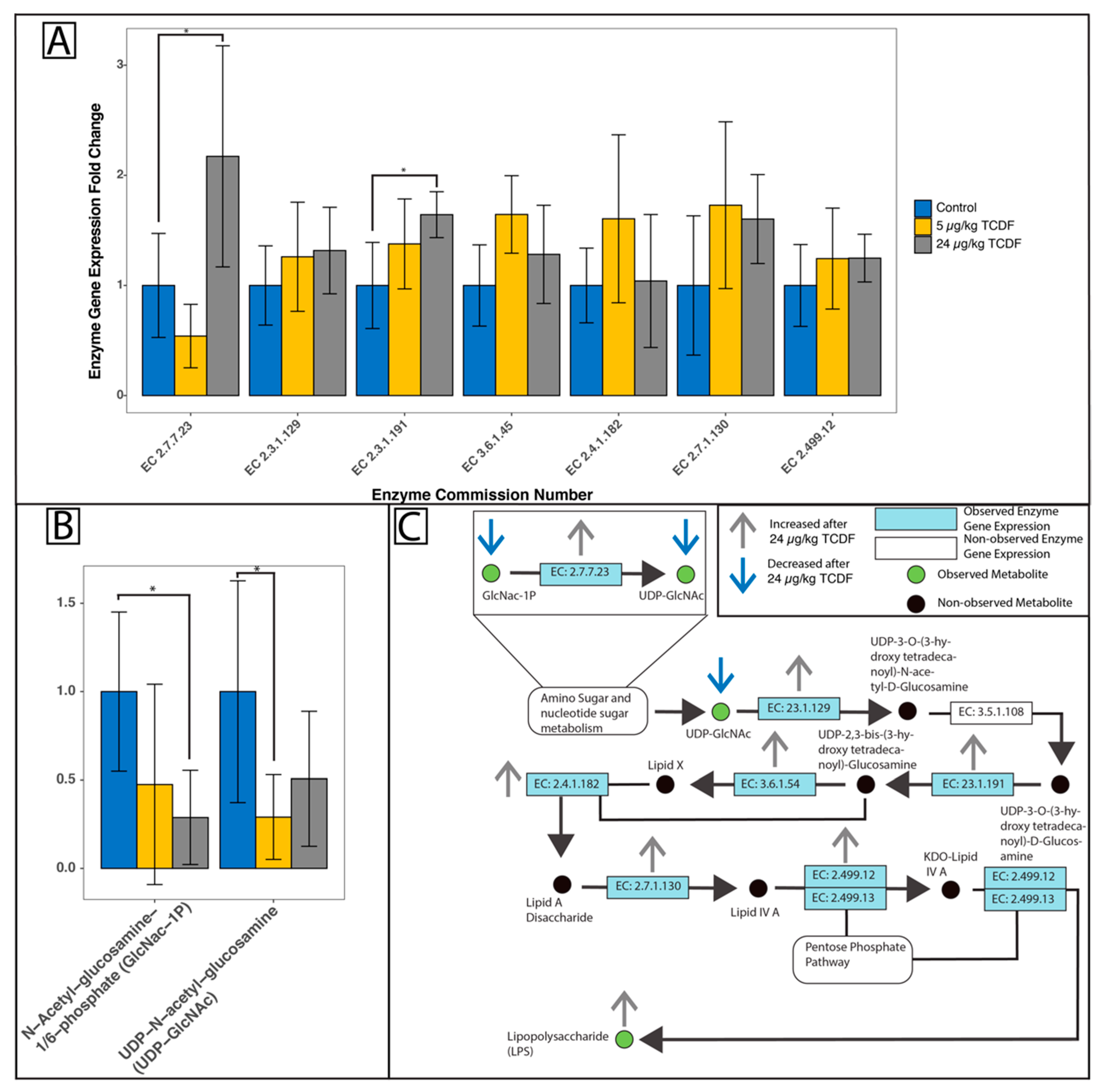
2.5. Modulation of Bacterial Pathways After TCDF Exposure
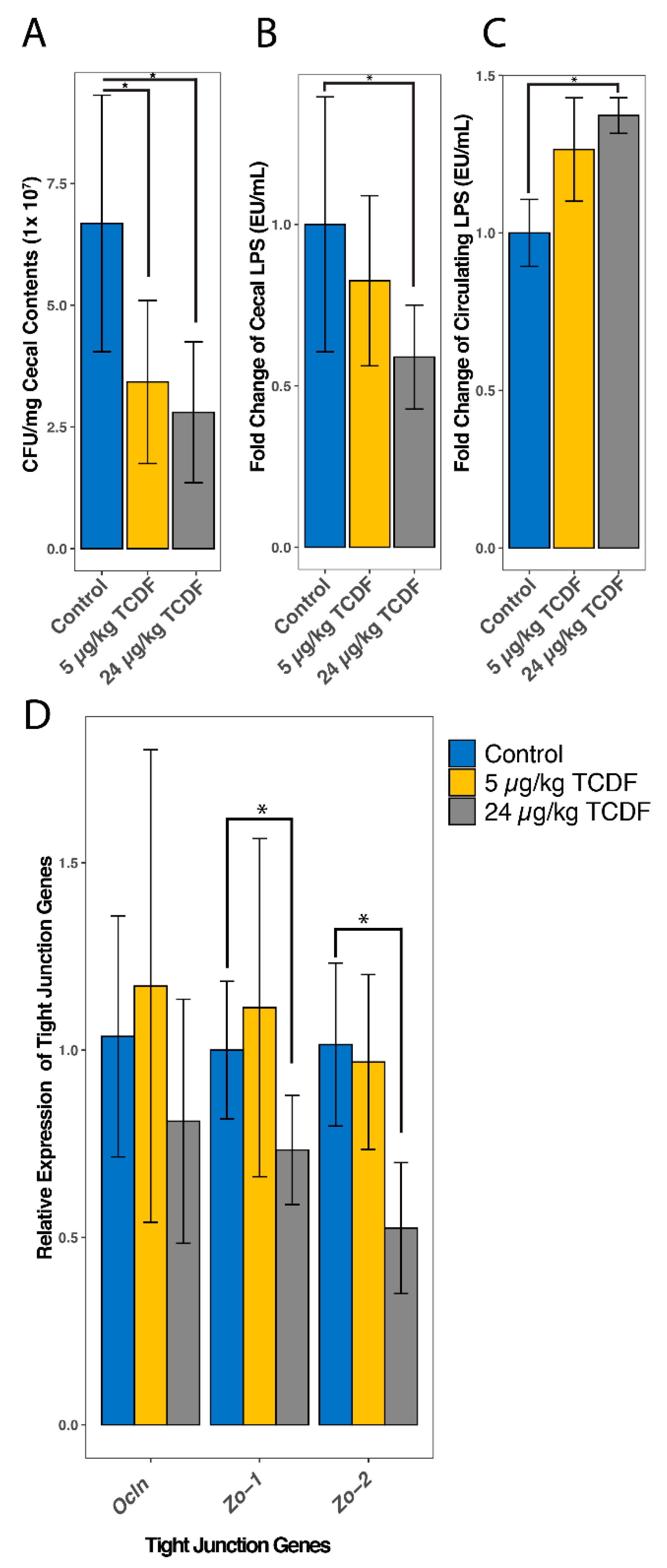
2.6. TCDF Exposure Causes an Increase in Circulating LPS
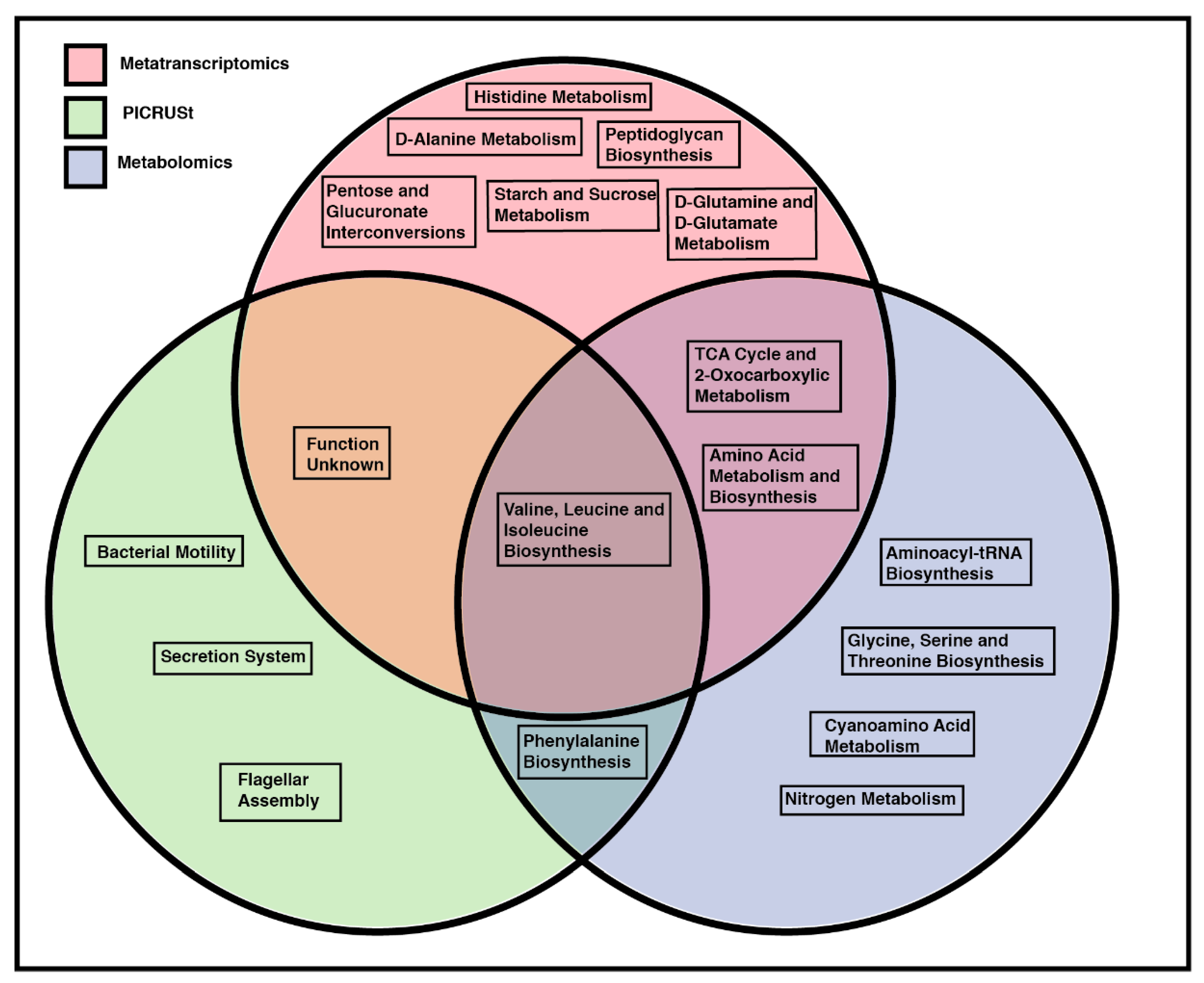
2.7. Overlapping Pathway Enrichment between Analytical Methods
3. Conclusions
4. Materials and Methods
4.1. Animals and Diets
4.2. 16S rRNA Gene Amplicon Sequencing and Analysis
4.3. LC-MS-Based Metabolomics
4.4. Metatranscriptomic Analysis
4.5. Lipopolysaccharide Detection
4.6. Total Bacteria Quantification
4.7. Quantitative Real-Time Polymerase Chain Reaction (qPCR) Analysis of Tight Junction Proteins
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Spanogiannopoulos, P.; Bess, E.N.; Carmody, R.N.; Turnbaugh, P.J. The microbial pharmacists within us: A metagenomic view of xenobiotic metabolism. Nat. Rev. Microbiol. 2016, 14, 273–287. [Google Scholar] [CrossRef] [PubMed]
- Swanson, H.I. Special section on drug metabolism and the microbiome—Commentary drug metabolism by the host and gut microbiota: A partnership or rivalry? Drug Metab. Dispos. 2015, 43, 1499–1504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pollet, R.M.; Agostino, E.H.D.; Walton, W.G.; Xu, Y.; Michael, S.; Biernat, K.A.; Pellock, S.J.; Patterson, L.M.; Benjamin, C.; Isenberg, H.N.; et al. An Atlas of β-Glucuronidases in the Human Intestinal Microbiome. Structure 2017, 25, 967–977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, Z.D.; Selwyn, F.P.; Cui, J.Y.; Klaassen, C.D. RNA-seq profiling of intestinal expression of xenobiotic processing genes in germ-free mice. Drug Metab. Dispos. 2017, 45, 1225–1238. [Google Scholar] [CrossRef]
- Selwyn, F.P.; Cheng, S.L.; Klaassen, C.D.; Cui, J.Y. Regulation of hepatic drug-metabolizing enzymes in germ-free mice by conventionalization and probiotics. Drug Metab. Dispos. 2016, 44, 262–274. [Google Scholar] [CrossRef]
- Nichols, R.G.; Hume, N.E.; Smith, P.B.; Peters, J.M.; Patterson, A.D. Omics Approaches To Probe Microbiota and Drug Metabolism Interactions. Chem. Res. Toxicol. 2016, 29, 1987–1997. [Google Scholar] [CrossRef]
- Xu, Z.; Knight, R. Dietary effects on human gut microbiome diversity. Br. J. Nutr. 2014, 113, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Takiishi, T.; Fenero, C.I.M.; Câmara, N.O.S. Intestinal barrier and gut microbiota: Shaping our immune responses throughout life. Tissue Barriers 2017, 5, e1373208. [Google Scholar] [CrossRef]
- Zhang, L.; Nichols, R.G.; Correll, J.; Murray, I.A.; Tanaka, N.; Smith, P.B.; Hubbard, T.D.; Sebastian, A.; Albert, I.; Hatzakis, E.; et al. Persistent Organic Pollutants Modify Gut Microbiota-Host Metabolic Homeostasis in Mice Through Aryl Hydrocarbon Receptor Activation. Environ. Health Perspect. 2015, 123, 679–688. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Nichols, R.G.; Patterson, A.D. The aryl hydrocarbon receptor as a moderator of host-microbiota communication. Curr. Opin. Toxicol. 2017, 2, 30–35. [Google Scholar] [CrossRef] [Green Version]
- Lefever, D.E.; Xu, J.; Chen, Y.; Huang, G.; Tamas, N.; Guo, T.L. TCDD modulation of gut microbiome correlated with liver and immune toxicity in streptozotocin (STZ)-induced hyperglycemic mice. Toxicol. Appl. Pharmacol. 2016, 304, 48–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stedtfeld, R.D.; Stedtfeld, T.M.; Fader, K.A.; Williams, M.R.; Bhaduri, P.; Quensen, J.; Zacharewski, T.R.; Tiedje, J.M.; Hashsham, S.A. TCDD influences reservoir of antibiotic resistance genes in murine gut microbiome. FEMS Microbiol. Ecol. 2017, 93, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Stockinger, B.; Meglio, P.D.; Gialitakis, M.; Duarte, J.H. The Aryl Hydrocarbon Receptor: Multitasking in the Immune System. Annu. Rev. Immunol. 2014, 32, 403–432. [Google Scholar] [CrossRef]
- Metidji, A.; Omenetti, S.; Crotta, S.; Li, Y.; Nye, E.; Ross, E.; Li, V.; Maradana, M.R.; Schiering, C.; Stockinger, B. The Environmental Sensor AHR Protects from Inflammatory Damage by Maintaining Intestinal Stem Cell Homeostasis and Barrier Integrity. Immunity 2018, 49, 353–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hubbard, T.D.; Murray, I.A.; Perdew, G.H. Indole and tryptophan metabolism: Endogenous and dietary routes to ah receptor activation. Drug Metab. Dispos. 2015, 43, 1522–1535. [Google Scholar] [CrossRef] [Green Version]
- Greenblum, S.; Turnbaugh, P.J.; Borenstein, E. Metagenomic systems biology of the human gut microbiome reveals topological shifts associated with obesity and inflammatory bowel disease. Proc. Natl. Acad. Sci. USA 2012, 109, 594–599. [Google Scholar] [CrossRef] [Green Version]
- Gutierrez-Orozco, F.; Thomas-Ahner, J.; Galley, J.; Bailey, M.; Clinton, S.; Lesinski, G.; Failla, M. Intestinal Microbial Dysbiosis and Colonic Epithelial Cell Hyperproliferation by Dietary α-Mangostin is Independent of Mouse Strain. Nutrients 2015, 7, 764–784. [Google Scholar] [CrossRef]
- Holmes, A.J.; Chew, Y.V.; Colakoglu, F.; Cliff, J.B.; Klaassens, E.; Read, M.N.; Solon-Biet, S.M.; McMahon, A.C.; Cogger, V.C.; Ruohonen, K.; et al. Diet-Microbiome Interactions in Health Are Controlled by Intestinal Nitrogen Source Constraints. Cell Metab. 2017, 25, 140–151. [Google Scholar] [CrossRef] [Green Version]
- Saad, M.J.A.; Santos, A.; Prada, P.O. Linking Gut Microbiota and Inflammation to Obesity and Insulin Resistance. Physiology 2016, 31, 283–293. [Google Scholar] [CrossRef]
- N’Jai, A.; Boverhof, D.R.; Dere, E.; Burgoon, L.D.; Tan, Y.S.; Rowlands, J.C.; Budinsky, R.A.; Stebbins, K.E.; Zacharewski, T.R. Comparative temporal toxicogenomic analysis of TCDD- and TCDF-mediated hepatic effects in immature female C57BL/6 mice. Toxicol. Sci. 2008, 103, 285–297. [Google Scholar] [CrossRef] [Green Version]
- Berg, M.V.D.; Birnbaum, L.S.; Denison, M.; Vito, M.D.; Feeley, M.; Fiedler, H.; Hakansson, H.; Hanberg, A.; Rose, M.; Safe, S.; et al. The 2005 World Health Organization Re-evaluation of Human and Mammalian Toxic Equivalency Factors for Dioxins and Dioxin-like Compounds. Toxicol. Sci. 2006, 93, 223–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eskenazi, B.; Warner, M.; Brambilla, P.; Signorini, S.; Ames, J.; Mocarelli, P. The Seveso accident: A look at 40 years of health research and beyond. Environ. Int. 2018, 121, 71–84. [Google Scholar] [CrossRef] [PubMed]
- Petriello, M.C.; Hoffman, J.B.; Vsevolozhskaya, O.; Morris, A.J.; Hennig, B. Dioxin-like PCB 126 increases intestinal inflammation and disrupts gut microbiota and metabolic homeostasis. Environ. Pollut. 2018, 242, 1022–1032. [Google Scholar] [CrossRef] [PubMed]
- Fox, E.; Reid-Bayliss, K.; Emond, M.; Loeb, L. Accuracy of Next Generation Sequencing Platforms. Next Gener. Seq. Appl. 2014, 1, 1000106. [Google Scholar] [CrossRef] [Green Version]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [Green Version]
- Xia, J.; Wishart, D.S. MSEA: A web-based tool to identify biologically meaningful patterns in quantitative metabolomic data. Nucleic Acids Res. 2010, 38, 71–77. [Google Scholar] [CrossRef] [Green Version]
- Chong, J.; Xia, J. MetaboAnalystR: An R package for flexible and reproducible analysis of metabolomics data. Bioinformatics (Oxf. Engl.) 2018, 34, 4313–4314. [Google Scholar] [CrossRef] [Green Version]
- Chong, J.; Yamamoto, M.; Xia, J. MetaboAnalystR 2.0: From raw spectra to biological insights. Metabolites 2019, 9, 57. [Google Scholar] [CrossRef] [Green Version]
- Dooley, R.K.; Holsapple, M.P. Elucidation of cellular targets responsible for 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD)-induced suppression of antibody responses: I. The role of the B lymphocyte. Immunopharmacology 1988, 16, 167–175. [Google Scholar] [CrossRef]
- Sulentic, C.E.W.; Holsapple, M.P.; Kaminski, N.E. Aryl Hydrocarbon Receptor-Dependent Suppression by 2,3,7,8-Tetrachlorodibenzo- p -dioxin of IgM Secretion in Activated B Cells. Mol. Pharmacol. 2018, 53, 623–629. [Google Scholar] [CrossRef] [Green Version]
- Sulentic, C.E.W.; Kaminski, N.E. The long winding road toward understanding the molecular mechanisms for B-cell suppression by 2,3,7,8-tetrachlorodibenzo-p-dioxin. Toxicol. Sci. 2011, 120 (Suppl. 1), S171–S191. [Google Scholar] [CrossRef] [PubMed]
- Kovalova, N.; Manzan, M.; Crawford, R.; Kaminski, N.E. Role of Aryl Hydrocarbon Receptor Polymorphisms on TCDD- mediated CYP1B1 Induction and IgM Suppression by Human B cells. Toxicol. Appl. Pharmacol. 2016, 309, 15–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feehily, C.; Karatzas, K.A.G. Role of glutamate metabolism in bacterial responses towards acid and other stresses. J. Appl. Microbiol. 2013, 114, 11–24. [Google Scholar] [CrossRef] [PubMed]
- Mailloux, R.J.; Singh, R.; Brewer, G.; Auger, C.; Lemire, J.; Appanna, V.D. α-ketoglutarate dehydrogenase and glutamate dehydrogenase work in tandem to modulate the antioxidant α-ketoglutarate during oxidative stress in Pseudomonas fluorescens. J. Bacteriol. 2009, 191, 3804–3810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peltier, M.R.; Arita, Y.; Klimova, N.G.; Gurzenda, E.M.; Koo, H.C.; Murthy, A.; Lerner, V.; Hanna, N. 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD) enhances placental inflammation. J. Reprod. Immunol. 2013, 98, 10–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, F.; Yan, B.; Wood, G.; Viluksela, M.; Rozman, K.K. Cytokines (IL-1β and TNFα) in relation to biochemical and immunological effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in rats. Toxicology 1997, 116, 9–16. [Google Scholar] [CrossRef]
- Han, M.; Liu, X.; Liu, S.; Su, G.; Fan, X.; Chen, J.; Yuan, Q.; Xu, G. 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD) induces hepatic stellate cell (HSC) activation and liver fibrosis in C57BL6 mouse via activating Akt and NF-κB signaling pathways. Toxicol. Lett. 2017, 273, 10–19. [Google Scholar] [CrossRef]
- Cani, P.D.; Bibiloni, R.; Knauf, C.; Neyrinck, A.M.; Delzenne, N.M. Changes in Gut Microbiota Control Metabolic Endotoxemia-Induced Inflammation in High-Fat Diet–Induced Obesity and Diabetes in Mice. Diabetes 2008, 57, 1470–1481. [Google Scholar] [CrossRef] [Green Version]
- Cao, S.; Zhang, Q.; Wang, C.C.; Wu, H.; Jiao, L.; Hong, Q.; Hu, C. LPS challenge increased intestinal permeability, disrupted mitochondrial function and triggered mitophagy of piglets. Innate Immun. 2018, 24, 221–230. [Google Scholar] [CrossRef] [Green Version]
- Fader, K.A.; Nault, R.; Zhang, C.; Kumagai, K.; Harkema, J.R.; Zacharewski, T.R. 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD)-elicited effects on bile acid homeostasis: Alterations in biosynthesis, enterohepatic circulation, and microbial metabolism. Sci. Rep. 2017, 7, 5921. [Google Scholar] [CrossRef]
- Kozich, J.J.; Westcott, S.L.; Baxter, N.T.; Highlander, S.K.; Schloss, P.D. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the miseq illumina sequencing platform. Appl. Environ. Microbiol. 2013, 79, 5112–5120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nichols, R.G.; Cai, J.; Murray, I.A.; Koo, I.; Smith, P.B.; Perdew, G.H.; Patterson, A.D. Structural and Functional Analysis of the Gut Microbiome for Toxicologists. Curr. Protoc. Toxicol. 2018, 78, e54. [Google Scholar] [CrossRef] [PubMed]
- Chang, Q.; Luan, Y.; Sun, F. Variance adjusted weighted UniFrac: A powerful beta diversity measure for comparing communities based on phylogeny. BMC Bioinform. 2011, 12, 118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langille, M.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.; Clemente, J.; Burkepile, D.; Vega Thurber, R.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef]
- Cai, J.; Nichols, R.G.; Koo, I.; Kalikow, Z.A.; Zhang, L.; Tian, Y.; Zhang, J.; Smith, P.B.; Patterson, A.D. Multiplatform Physiologic and Metabolic Phenotyping Reveals Microbial Toxicity. mSystems 2018, 3, e00123-18. [Google Scholar] [CrossRef] [Green Version]
- Tsugawa, H.; Cajka, T.; Kind, T.; Ma, Y.; Higgins, B.; Ikeda, K.; Kanazawa, M.; VanderGheynst, J.; Feihn, O.; Arita, M. MS-DIAL: Data Independent MS/MS Deconvolution for Comprehensive Metabolome Analysis. Nat. Methods 2015, 347, 882–886. [Google Scholar] [CrossRef]
- Westreich, S.T.; Treiber, M.L.; Mills, D.A.; Korf, I.; Lemay, D.G. SAMSA2: A standalone metatranscriptome analysis pipeline. BMC Bioinform. 2018, 19, 175. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Kobert, K.; Flouri, T.; Stamatakis, A. PEAR: A fast and accurate Illumina Paired-End reAd mergeR. Bioinformatics 2014, 30, 614–620. [Google Scholar] [CrossRef] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Kopylova, E.; Noé, L.; Touzet, H. SortMeRNA: Fast and accurate filtering of ribosomal RNAs in metatranscriptomic data. Bioinformatics 2012, 28, 3211–3217. [Google Scholar] [CrossRef]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Tatusova, T.; Ciufo, S.; Fedorov, B.; O’Neill, K.; Tolstoy, I. RefSeq microbial genomes database: New representation and annotation strategy. Nucleic Acids Res. 2014, 42, D553–D559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pathway | p Value | Upregulated (↑) or Downregulated (↓) |
|---|---|---|
| Arginine and proline metabolism | 0.01 | ↓ |
| Amino sugar and nucleotide sugar metabolism | 0.02 | ↓ |
| Otherion coupled transporters | 0.02 | ↓ |
| Lysine biosynthesis | 0.02 | ↓ |
| Glyoxylate and dicarboxylate metabolism | 0.02 | ↓ |
| Starch and sucrose metabolism | 0.02 | ↓ |
| Cysteine and methionine metabolism | 0.02 | ↓ |
| Valine leucine and isoleucine degradation | 0.02 | ↑ |
| Secretion system | 0.02 | ↑ |
| Bacterial motility proteins | 0.02 | ↑ |
| Flagellar assembly | 0.02 | ↑ |
| Function unknown | 0.04 | ↑ |
| Phenylalanine metabolism | 0.004 | ↑ |
| Metabolite Name | Control | 5 µg/kg TCDF | 24 µg/kg TCDF |
|---|---|---|---|
| 3-Phosphoglycerate | 7.92 ± 6.00 | 3.31 ± 3.07 | 1.86 ± 0.77 * |
| 6-Phospho-D-gluconate | 0.29 ± 0.27 | 0.11 ± 0.14 | 0.03 ± 0.02 * |
| Aconitate | 2.83 ± 1.23 | 1.82 ± 1.03 | 1.57 ± 0.47 * |
| Anthranilate | 0.38 ± 0.09 | 0.37 ± 0.12 | 0.30 ± 0.04 * |
| Asparagine | 2.46 ± 0.59 | 2.56 ± 1.41 | 7.03 ± 4.81 * |
| CDP | 0.16 ± 0.06 | 0.09 ± 0.09 | 0.07 ± 0.04 * |
| Citrulline | 133.18 ± 31.27 | 67.80 ± 60.84 * | 165.57 ± 53.93 |
| dGDP | 1.42 ± 0.52 | 1.22 ± 1.22 | 0.70 ± 0.37 * |
| Dihydroxy-acetone-phosphate | 3.81 ± 2.69 | 2.52 ± 2.83 | 0.95 ± 0.60 * |
| FMN | 3.38 ± 0.94 | 1.74 ± 1.26 * | 1.41 ± 0.68 * |
| Fumarate | 7.47 ± 2.42 | 13.08 ± 7.26 | 15.21 ± 4.50 * |
| GDP | 0.36 ± 0.10 | 0.28 ± 0.23 | 0.18 ± 0.08 * |
| Gluconolactone | 10.20 ± 7.46 | 3.20 ± 1.68 * | 5.64 ± 5.66 |
| Glycine | 15.34 ± 3.61 | 17.72 ± 10.54 | 21.71 ± 4.50 * |
| Hydroxyproline/Aminolevulinate | 3.42 ± 0.84 | 2.31 ± 1.39 | 1.30 ± 0.35 * |
| IMP | 0.77 ± 0.37 | 0.38 ± 0.29 | 0.30 ± 0.26 * |
| Leucine/Isoleucine | 331.04 ± 99.91 | 447.25 ± 261.53 | 599.00 ± 134.45 * |
| N-Acetyl-glucosamine-1/6-phosphate | 26.03 ± 11.66 | 12.33 ± 14.70 | 4.36 ± 4.34 * |
| Phosphoenolpyruvate | 0.89 ± 0.71 | 0.36 ± 0.31 | 0.17 ± 0.09 * |
| Prephenate | 1.33 ± 0.62 | 1.22 ± 0.52 | 0.73 ± 0.17 * |
| Proline | 56.59 ± 21.32 | 63.79 ± 44.10 | 83.57 ± 20.20 * |
| Pyridoxamine | 0.26 ± 0.06 | 0.24 ± 0.10 | 0.18 ± 0.06 * |
| Pyruvate | 8.32 ± 4.57 | 3.84 ± 1.97 * | 6.02 ± 3.53 |
| Serine | 33.77 ± 7.94 | 42.06 ± 25.03 | 53.34 ± 13.06 * |
| Glycerol-3-phosphate | 64.65 ± 30.11 | 55.85 ± 72.70 | 25.35 ± 16.89 * |
| Threonine/Homoserine | 60.19 ± 12.41 | 59.96 ± 32.22 | 85.89 ± 11.08 * |
| Tryptophan | 105.14 ± 26.96 | 128.27 ± 73.36 | 167.80 ± 39.14 * |
| UDP | 0.24 ± 0.08 | 0.17 ± 0.16 | 0.12 ± 0.08 * |
| UDP-D-glucose | 0.15 ± 0.08 | 0.07 ± 0.05 * | 0.06 ± 0.06 * |
| UDP-N-acetyl-glucosamine | 0.12 ± 0.07 | 0.04 ± 0.03 * | 0.05 ± 0.05 |
| Xanthine | 30.00 ± 8.01 | 14.65 ± 9.71 * | 21.14 ± 9.89 |
| Xanthosine | 55.04 ± 12.35 | 30.42 ± 24.52 * | 26.54 ± 15.38 * |
| Xanthosine-5-phosphate | 0.73 ± 0.40 | 0.36 ± 0.31 | 0.27 ± 0.22 * |
| Pathway | P Value | Q Value | Enriched (↑) or Depleted (↓) |
|---|---|---|---|
| Aminoacyl-tRNA biosynthesis * | 2.06 × 10−6 | 1.64 × 10−4 | ↑ |
| Glycine, serine and threonine metabolism * | 1.89 × 10−5 | 7.55 × 10−4 | ↑ |
| Phenylalanine, tyrosine, and tryptophan biosynthesis * | 2.82 × 10−4 | 0.007 | ↑ |
| Cyanoamino acid metabolism * | 8.77 × 10−4 | 0.02 | ↑ |
| Nitrogen metabolism * | 0.001 | 0.02 | ↑ |
| Citrate cycle (TCA cycle) * | 0.002 | 0.02 | ↑ |
| Valine, leucine and isoleucine biosynthesis * | 0.004 | 0.05 | ↑ |
| Glycerolipid metabolism | 0.007 | 0.07 | ↓ |
| Sulfur metabolism | 0.02 | 0.18 | ↑ |
| Purine metabolism | 0.02 | 0.20 | ↓ |
| Alanine, aspartate, and glutamate metabolism | 0.03 | 0.25 | ↑ |
| Pyrimidine metabolism | 0.04 | 0.25 | ↓ |
| Pathway | P Value | Q Value | Enriched (↑) or Depleted (↓) |
|---|---|---|---|
| Biosynthesis of amino acids | 1.24 × 10−17 | 1.84 × 10−15 | ↑ |
| Amino sugar and nucleotide sugar metabolism | 1.01 × 10−5 | 7.47 × 10−4 | ↑ |
| One carbon pool by folate | 7.05 × 10−5 | 3.48 × 10−4 | ↑ |
| Histidine metabolism | 3.5 × 10−4 | 0.01 | ↑ |
| Polyketide sugar unit biosynthesis | 5.58 × 10−8 | 0.01 | ↓ |
| Alanine, aspartate and glutamate metabolism | 6.05 × 10−4 | 0.0124 | ↑ |
| Pantothenate and CoA biosynthesis | 6.49 × 10−4 | 0.01 | ↓ |
| Pentose and glucuronate interconversions | 6.72 × 10−4 | 0.01 | ↑ |
| Pyruvate metabolism | 0.001 | 0.02 | ↓ |
| Terpenoid backbone biosynthesis | 0.001 | 0.02 | ↑ |
| Carbon fixation in photosynthetic organisms | 0.001 | 0.02 | ↓ |
| Peptidoglycan biosynthesis | 0.001 | 0.02 | ↑ |
| Starch and sucrose metabolism | 0.003 | 0.03 | ↑ |
| D-Alanine metabolism | 0.004 | 0.04 | ↑ |
| 2-Oxocarboxylic acid metabolism | 0.004 | 0.04 | ↑ |
| Valine, leucine and isoleucine biosynthesis | 0.004 | 0.04 | ↑ |
| Vitamin B6 metabolism | 0.005 | 0.04 | ↑ |
| Streptomycin biosynthesis | 0.005 | 0.04 | ↑ |
| D-Glutamine and D-glutamate metabolism | 0.006 | 0.05 | ↑ |
| Drug metabolism—other enzymes | 0.006 | 0.05 | ↑ |
| Carbon fixation pathways in prokaryotes | 0.007 | 0.05 | ↑ |
| Propanoate metabolism | 0.02 | 0.103 | ↓ |
| Carbapenem biosynthesis | 0.02 | 0.15 | ↓ |
| Novobiocin biosynthesis | 0.02 | 0.15 | ↑ |
| Glycolysis/Gluconeogenesis | 0.03 | 0.18 | ↓ |
| Lipopolysaccharide biosynthesis | 0.03 | 0.19 | ↑ |
| Cysteine and methionine metabolism | 0.04 | 0.20 | ↑ |
| Nicotinate and nicotinamide metabolism | 0.04 | 0.20 | ↑ |
| Arginine and proline metabolism | 0.04 | 0.20 | ↑ |
| Lysine biosynthesis | 0.04 | 0.20 | ↑ |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nichols, R.G.; Zhang, J.; Cai, J.; Murray, I.A.; Koo, I.; Smith, P.B.; Perdew, G.H.; Patterson, A.D. Metatranscriptomic Analysis of the Mouse Gut Microbiome Response to the Persistent Organic Pollutant 2,3,7,8-Tetrachlorodibenzofuran. Metabolites 2020, 10, 1. https://doi.org/10.3390/metabo10010001
Nichols RG, Zhang J, Cai J, Murray IA, Koo I, Smith PB, Perdew GH, Patterson AD. Metatranscriptomic Analysis of the Mouse Gut Microbiome Response to the Persistent Organic Pollutant 2,3,7,8-Tetrachlorodibenzofuran. Metabolites. 2020; 10(1):1. https://doi.org/10.3390/metabo10010001
Chicago/Turabian StyleNichols, Robert G., Jingtao Zhang, Jingwei Cai, Iain A. Murray, Imhoi Koo, Philip B. Smith, Gary H. Perdew, and Andrew D. Patterson. 2020. "Metatranscriptomic Analysis of the Mouse Gut Microbiome Response to the Persistent Organic Pollutant 2,3,7,8-Tetrachlorodibenzofuran" Metabolites 10, no. 1: 1. https://doi.org/10.3390/metabo10010001