
Coevolution Theory of the Genetic Code at Age Forty: Pathway to Translation and Synthetic Life



Abstract
:1. Introduction
2. Origin of the Gene
Under conditions of active synthesis of RNA-like replicators, accelerated template-directed synthesis of RNA-like replicators, and the presence of a huge population of random RNA-like duplexes in the environment, functional RNA-like aptamers/ribozymes will be selectively amplified by their cognate metabolites in the environment through the replicator induction by metabolite (REIM) mechanism based on the metabolic expansion equation, leading to the appearance of novel RNA-like ribozymes catalytically acting on the metabolites to form novel metabolites and thereby expand metabolism.
3. Origin of Messenger RNA
3.1. Self-rARS Template (SART)
3.2. Direct RNA Template (DRT)
3.3. Intermediate Acceptor Template (IMAT)
- (I)
- finding a cognate RNA acceptor for each amino acid to be employed in the peptide prosthetic groups on fRNAs;
- (II)
- finding a cognate rARS to join each amino acid to its cognate RNA acceptor; and
- (III)
- switching the original binding sites on the template designed for amino acids to binding sites for RNA acceptors of amino acids.
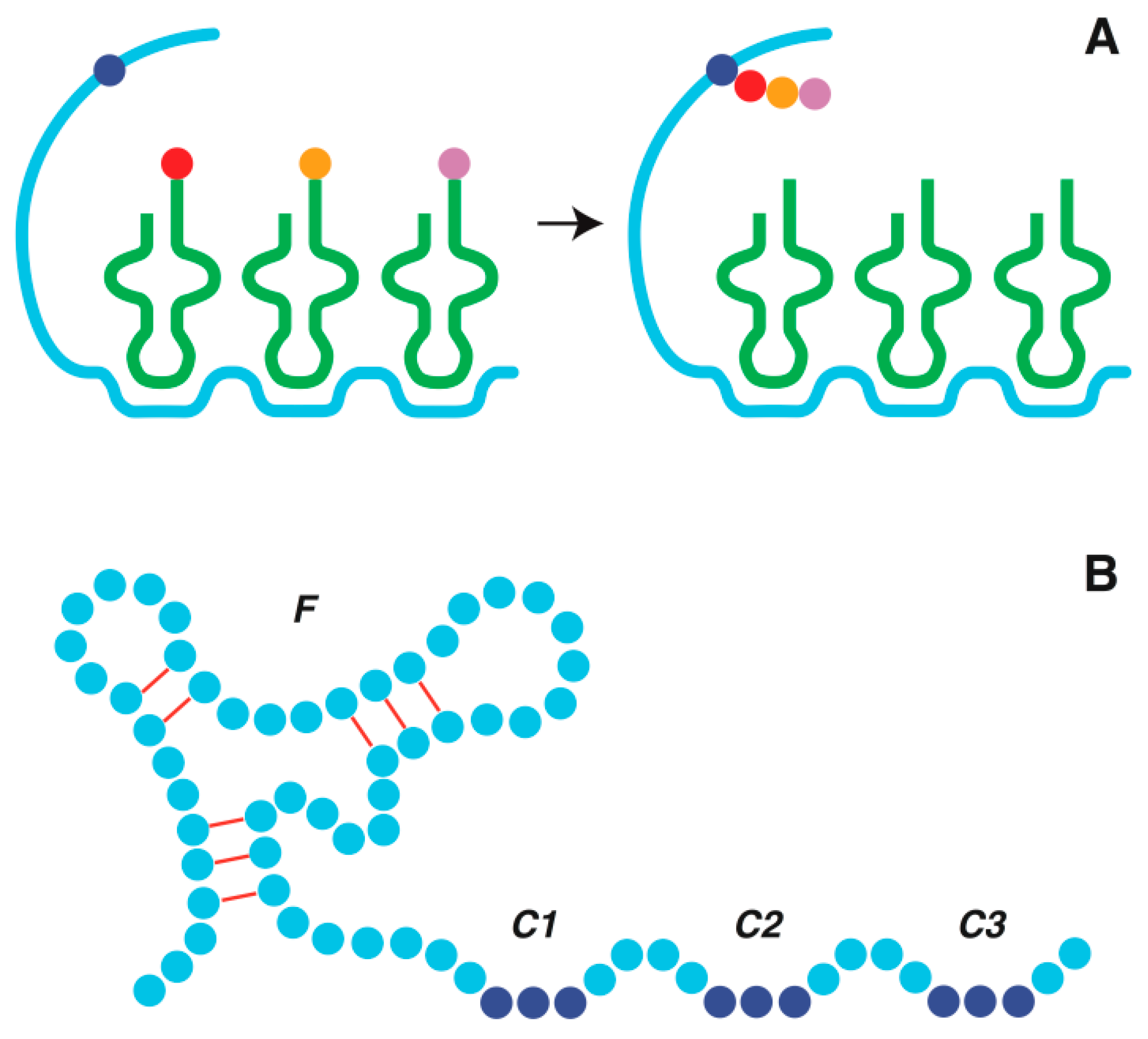
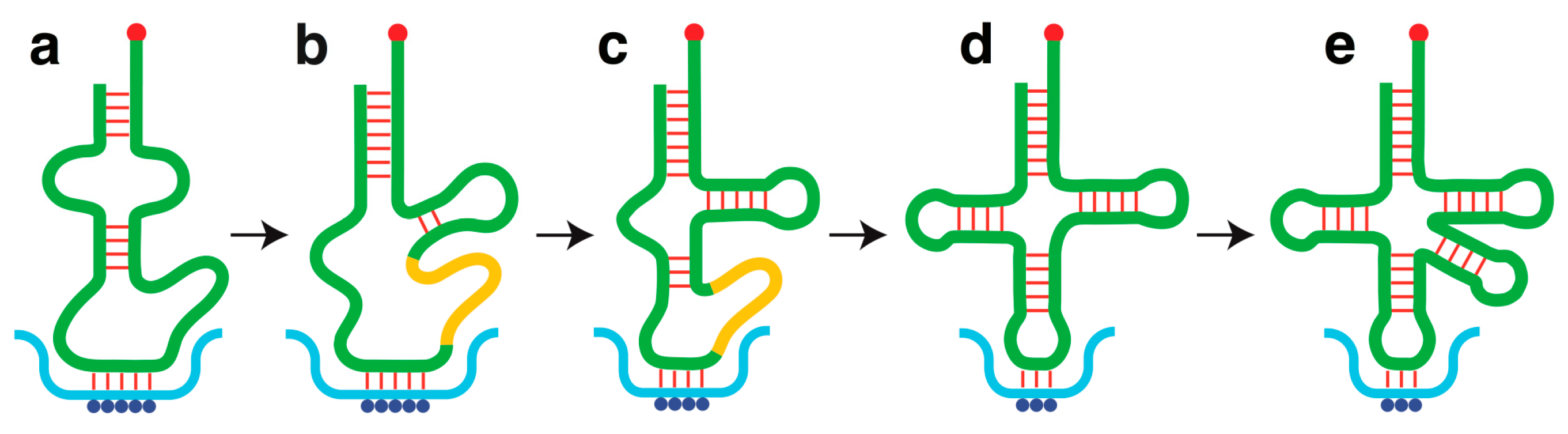
4. Origin of Transfer RNA
5. Origin of Genetic Code
- (i)
- (ii)
- The single codon assignments to Met and Trp strongly indicate that they are late arrivals supplied by biosynthesis.
- (iii)
- The 20 encoded amino acids give rise to 190 pairs. The Cys-Trp pair ranks as the chemically most unlike pair, with the largest chemical distance of 215 compared to the minimum distance of 5 for the Leu-Ile pair, yet they are assigned codons in the same UGN box, which provides an unambiguous biosynthetic signal that the UGN codons are former Ser codons that have been apportioned to the Ser biosynthetic products Cys and Trp [88]. This biosynthetic signal is validated by the remarkable discoveries of allocation of part use of the UGA codon to selenoCys (Sec) via pretran synthesis of Sec-tRNA from Ser-tRNA [89], and the allocation of UGY codons to Cys via pretran synthesis of Cys-tRNA from Sep-tRNA [90].
- (iv)
- Phe and Tyr as in the case of Trp and His are easily degraded by UV radiation: They were > 50% destroyed by irradiation for 48 h at pH 7 under an energy flux of 1.8 mW/cm2 [91]. However, whereas Gln and Asn could not hide from thermal degradation, prebiotically synthesized Phe and Tyr might find some shielding from UV radiation behind rocks or in ocean depths.
6. Origin of Extant Life
7. Origins of Intron and Triplet Codon
7.1. Exon Shuffling
7.2. Exon Regulation
7.3. Exon Diversification
- (a)
- Expansion of the anticodon repertoire of the genetic code at different stages of tRNA evolution so that ample anticodons were made available to the evolving tRNAs as the tRNAome underwent expansion with recruitment of new anticodons.
- (b)
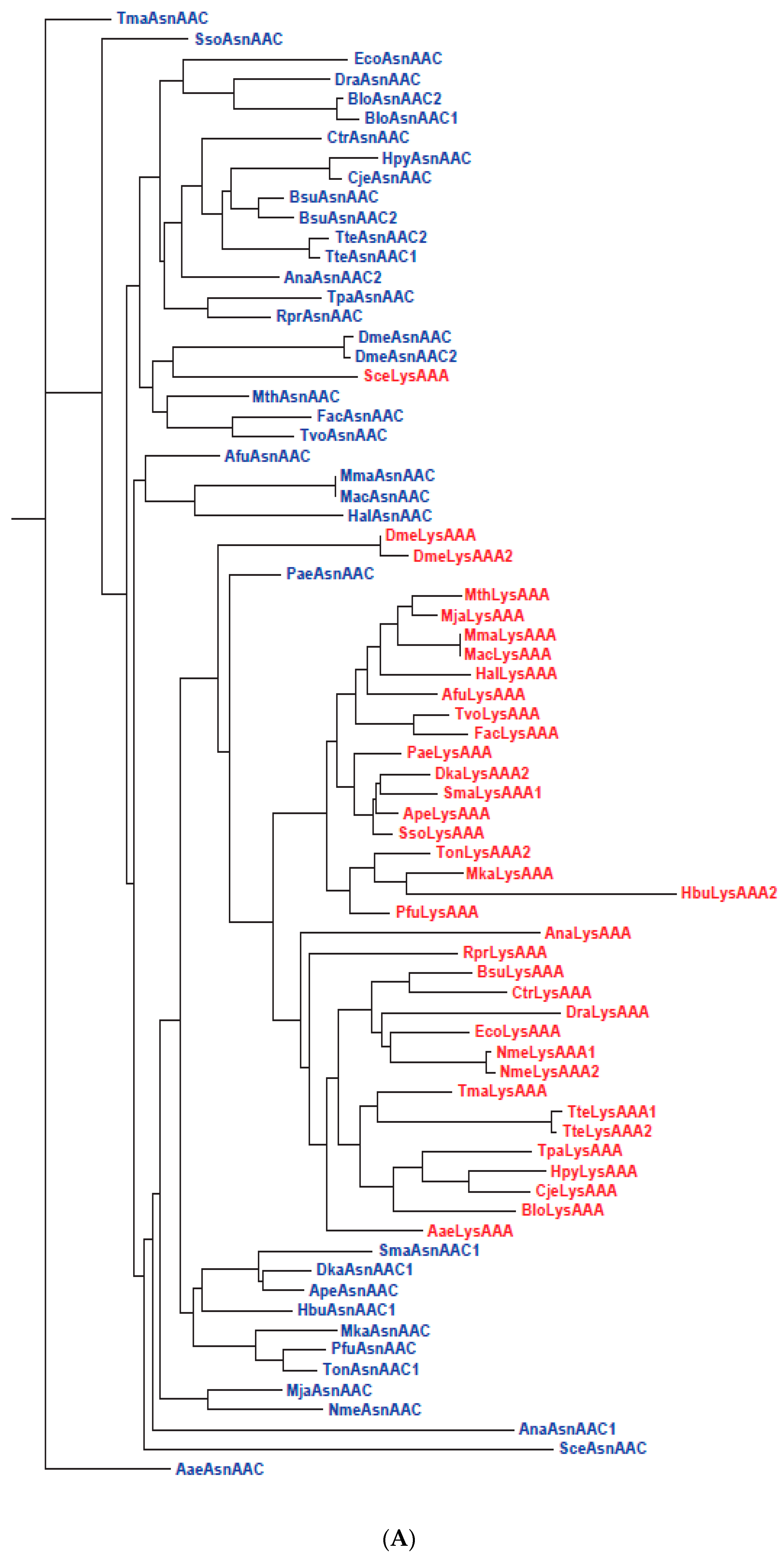
- Continual variation of the loop sequence with concomitant variation in loop nucleoside modifications facilitated the fulfillment of wobble base pairing requirements. Notably, the estimated LUCA genome contained a number of nucleoside modifying enzymes [160], and the tRNAs of LUCA-proximal Methanopyrus is enriched with modified nucleosides including ac6A, which represents a “minimalist” nucleoside modification where the amino acid moiety in t6A is replaced by an acetyl function. The discovery of ac6A and two minimalist wyeosine-family nucleosides from Archaea suggests that tRNA nucleoside modifications are simpler in Archaea than in Bacteria or Eukarya [137,138], thus contributing evidence Line 29 to Table 2 in support of the primitivity of Archaea.
- (c)
- Progressive enhancement of the codon-anticodon association constant on account of optimizations in anticodon loop sequence and nucleoside modifications, thereby enabling a reduction in anticodon size and complexity down to three bases to establish the triplet codons and anticodons of the modern genetic code (Figure 2).
8. Origin of Wobble
9. Origins of Biological Domains
9.1. Anticodon Strategy
9.2. Membrane Lipids
9.3. Nuclear Membrane
10. Origin of Synthetic Life
11. Discussion
- Stage 1. Prebiotic synthesis
- Stage 2. Functional RNA selection by metabolite
- Stage 3. RNA World
- Stage 4. Peptidated RNA World
- Stage 5. Coevolution of genetic code and amino acid biosynthesis
- Stage 6. Last universal common ancestor
- Stage 7. Darwinian evolution
- Stage 8. Synthetic life
11.1. Amino Acid-RNA Cooperation
11.2. Side Chain Imperative
11.3. Paralogs from Code Expansion
11.4. Feedback for Near Perfection
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Orgel, L.E. Evolution of the genetic apparatus. J. Mol. Biol. 1968, 38, 381–393. [Google Scholar] [CrossRef]
- Kruger, K.; Grabowski, P.J.; Zaug, A.J.; Sands, J.; Gottschling, D.E.; Cech, T.R. Self-splicing RNA: autoexcision and autocyclization of the ribosomal RNA intervening sequence of Tetrahymena. Cell 1982, 31, 147–157. [Google Scholar] [CrossRef]
- Guerrier-Takada, C.; Gardiner, K.; Marsh, T.; Pace, N.; Altman, S. The RNA moiety of ribonuclease P is the catalytic subunit of the enzyme. Cell 1983, 35, 849–857. [Google Scholar] [CrossRef]
- Gilbert, W. The RNA World. Nature 1986, 319, 618. [Google Scholar] [CrossRef]
- Joyce, G.F.; Orgel, L.E. Prospects for understanding the origin of the RNA World. In The RNA World, 2nd ed.; Gesteland, R.F., Cech, T.R., Atkins, J.F., Eds.; Cold Spring Harbor Laboratory Press: New York, NY, USA, 1999; pp. 49–77. [Google Scholar]
- Wong, J.T. Introduction. In Prebiotic Evolution and Astrobiology; Wong, J.T., Lazcano, A., Eds.; Landes Bioscience: Austin, TX, USA, 2009; pp. 1–9. [Google Scholar]
- Szostak, J.W. The eightfold path to non-enzymatic RNA replication. J. Syst. Chem. 2012, 3. [Google Scholar] [CrossRef]
- Wong, J.T. Emergence of life: From functional RNA selection to natural selection and beyond. Front. Biosci. (Landmark Ed.) 2014, 19, 1117–1150. [Google Scholar] [CrossRef] [PubMed]
- Abel, D.L. The capabilities of chaos and complexity. Int. J. Mol. Sci. 2009, 10, 247–291. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.T. Biomolecules. In Prebiotic Evolution and Astrobiology; Wong, J.T., Lazcano, A., Eds.; Landes Bioscience: Austin, TX, USA, 2009; pp. 65–75. [Google Scholar]
- Shock, E.L. Constraints on the origins of organic compounds in hydrothermal systems. Orig. Life Evol. Biosph. 1990, 20, 331–367. [Google Scholar] [CrossRef]
- Pizzarello, S. Meteorites and the chemistry that preceded life’s origin. In Prebiotic Evolution and Astrobiology; Wong, J.T., Lazcano, A., Eds.; Landes Bioscience: Austin, TX, USA, 2009; pp. 46–51. [Google Scholar]
- Monnard, P.A.; Kanavarioti, A.; Deamer, D.W. Eutectic phase polymerization of activated ribonucleotide mixtures yields quasi-equimolar incorporation of purine and pyrimidine nucleobases. J. Am. Chem. Soc. 2003, 125, 13734–13740. [Google Scholar] [CrossRef] [PubMed]
- Vlassov, A.V.; Kazakov, S.A.; Johnston, B.H.; Landweber, L.F. The RNA World on ice: A new scenario for the emergence of RNA information. J. Mol. Evol. 2005, 61, 264–273. [Google Scholar] [CrossRef] [PubMed]
- Attwater, J.; Wochner, A.; Holliger, P. In-ice evolution of RNA polymerase ribozyme activity. Nat. Chem. 2013, 5, 1011–1018. [Google Scholar] [CrossRef] [PubMed]
- Rajamani, S.; Vlassov, A.; Benner, S.; Coombs, A.; Plasagasti, F.; Deamer, D. Lipid-assisted synthesis of RNA-like polymers from mononucleotides. Orig. Life Evol. Biosph. 2008, 38, 57–74. [Google Scholar] [CrossRef] [PubMed]
- Ferris, J.P. Montmorillonite-catalyzed formation of RNA oligomers: The possible role of catalysis in the origin of life. Phil. Tran. R. Soc. B 2006, 361, 1777–1786. [Google Scholar] [CrossRef] [PubMed]
- Ellington, A.; Szostak, J.W. In vitro selection of RNA molecules that bind specific ligands. Nature 1990, 346, 818–822. [Google Scholar] [CrossRef] [PubMed]
- Wilson, D.S.; Szostak, J.W. In vitro selection of functional nucleic acids. Annu. Rev. Biochem. 1999, 68, 611–647. [Google Scholar] [CrossRef] [PubMed]
- Mojzsis, S.J.; Krishnamurthy, R.; Arrhenius, G. Before RNA and after: Geophysical and geochemical constraints on molecular evolution. In The RNA World, 2nd ed.; Gesteland, R.F., Cech, T.R., Atkins, J.F., Eds.; Cold Spring Harbor Laboratory Press: New York, NY, USA, 1999; pp. 1–47. [Google Scholar]
- Hughes, R.A.; Ellington, A.D. Ribozymes and the evolution of metabolism. In Prebiotic Evolution and Astrobiology; Wong, J.T., Lazcano, A., Eds.; Landes Bioscience: Austin, TX, USA, 2009; pp. 87–93. [Google Scholar]
- Cech, T.R.; Golden, B.L. Building a catalytic active site using only RNA. In The RNA World, 2nd ed.; Gesteland, R.F., Cech, T.R., Atkins, J.F., Eds.; Cold Spring Harbor Laboratory Press: New York, NY, USA, 1999; pp. 321–349. [Google Scholar]
- Wong, J.T.; Xue, H. Self-perfecting evolution of heteropolymer building blocks and sequences as the basis of life. In Fundamentals of Life; Palyi, G., Zucchi, C., Caglioti, L., Eds.; Elsevier: Paris, France, 2002; pp. 473–494. [Google Scholar]
- Lane, B.G. Historical perspectives on RNA nucleoside modifications. In Modification and Editing of RNA; Grosjean, H., Benne, R., Eds.; ASM Press: Washington, DC, USA, 1998; pp. 1–20. [Google Scholar]
- Wong, J.T. Origin of genetically encoded protein synthesis: A model based on selection for RNA peptidation. Orig. Life Evol. Biosph. 1991, 21, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Harada, K.; Martin, S.S.; Tan, R.; Frankel, A.D. Molding a peptide into an RNA site by in vivo evolution. Proc. Natl. Acad. Sci. USA 1997, 94, 11887–11892. [Google Scholar] [CrossRef] [PubMed]
- Atsumi, S.; Ikawa, Y.; Shiraishi, H.; Inoue, T. Design and development of a catalytic ribonucleoprotein. EMBO J. 2001, 20, 5453–5460. [Google Scholar] [CrossRef] [PubMed]
- Noller, H.F. The driving force for molecular evolution of translation. RNA 2004, 10, 1833–1837. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, T.W. Properties of human mitochondrial ribosomes. IUBMB Life 2003, 55, 505–513. [Google Scholar] [CrossRef] [PubMed]
- Schuenemann, D.; Gupta, S.; Persello-Cartieaux, F.; Klimyuk, V.I.; Jones, J.D.G.; Nusssaume, L.; Hoffman, N.E. A novel signal recognition particle targets light-harvesting proteins to the thylakoid membranes. Proc. Natl. Acad. Sci. USA 1998, 95, 10312–10316. [Google Scholar] [CrossRef] [PubMed]
- Kurland, C.G. The RNA dreamtime: Modern cells feature proteins that might have supported a prebiotic polypeptide world but nothing indicates that RNA World ever was. BioEssays 2010, 32, 866–871. [Google Scholar] [CrossRef] [PubMed]
- Di Giulio, M. On the RNA World: Evidence in favor of an early ribonucleopeptide world. J. Mol. Evol. 1997, 45, 571–578. [Google Scholar] [CrossRef] [PubMed]
- Cech, T. Crawling out of the RNA World. Cell 2009, 136, 599–602. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Franklyn, C.; Carter, C.W., Jr. Aminoacylating urzymes challenege the RNA World hypothesis. J. Biol. Chem. 2013, 288, 26856–26863. [Google Scholar] [CrossRef] [PubMed]
- Caetano-Anolles, G.; Seufferheld, M.J. The coevolutionary roots of biochemistry and cellular organization challenge the RNA World paradigm. J. Microbiol. Biotechnol. 2013, 23, 152–177. [Google Scholar] [CrossRef] [PubMed]
- Smith, T.F.; Lee, J.C.; Gutell, R.R.; Hartman, H. The origin and evolution of the ribosome. Biol. Direct 2008, 3. [Google Scholar] [CrossRef] [PubMed]
- Harish, A.; Caetano-Anolles, G. Ribosomal history reveals origins of protein synthesis. PLoS ONE 2012, 7, e32776. [Google Scholar] [CrossRef] [PubMed]
- Benner, S.A. Paradoxes in the origin of life. Orig. Life Evol. Biosph. 2014, 44, 339–343. [Google Scholar] [CrossRef] [PubMed]
- Bjork, G.R. Biosynthesis and function of modified nucelosides. In tRNA: Structure, Biosynthesis and Function; Söll, D., Rajbandary, U.L., Eds.; ASM Press: Washington, DC, USA, 1995; pp. 165–205. [Google Scholar]
- Wong, J.T. Kinetics of Enzyme Mechanisms; Academic Press: London, UK, 1975; pp. 73–78. [Google Scholar]
- Yarus, M. Amino acids as RNA ligands: A direct-RNA-template theory for the code’s origin. J. Mol. Evol. 1998, 47, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Illangasekare, M.; Sanchez, G.; Nickles, T.; Yarus, M. Aminoacyl-RNA synthesis catalyzed by an RNA. Science 1995, 267, 643–647. [Google Scholar] [CrossRef] [PubMed]
- Lohse, P.A.; Szostak, J.W. Ribozyme-catalysed amino-acid transfer reactions. Nature 1996, 381, 442–444. [Google Scholar] [CrossRef] [PubMed]
- Illangasekare, M.; Yarus, M. Small-molecule-substrate interactions with a self-aminoacylating ribozyme. J. Mol. Biol. 1997, 268, 631–639. [Google Scholar] [CrossRef] [PubMed]
- Jenne, A.; Famulok, M. A novel ribozyme with ester transferase activity. Chem. Biol. 1998, 5, 23–34. [Google Scholar] [CrossRef]
- Illangasekare, M.; Yarus, M. A tiny RNA that catalyzes both aminoacyl-RNA and peptidyl-RNA synthesis. RNA 1999, 5, 1482–1489. [Google Scholar] [CrossRef] [PubMed]
- Lee, N.; Bessho, Y.; Wei, K.; Szostak, J.W.; Suga, H. Ribozyme-catalyzed tRNA aminoacylation. Nat. Struct. Biol. 2000, 7, 28–33. [Google Scholar] [PubMed]
- Saito, H.; Kourouklis, D.; Suga, H. An in vitro evolved precursor tRNA with aminoacylation activity. EMBO J. 2001, 20, 1797–1806. [Google Scholar] [CrossRef] [PubMed]
- Lee, N.; Suga, H. A minihelix-loop RNA acts as a trans-aminoacylation catalyst. RNA 2001, 7, 1043–1051. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Huang, F. Ribozyme-catalyzed aminoacylation from CoA thioesters. Biochemistry 2005, 44, 4582–4590. [Google Scholar] [CrossRef] [PubMed]
- Turk, R.M.; Chumachenko, N.V.; Yarus, M. Multiple translational products from a five-nucleotide ribozyme. Proc. Natl. Acad. Sci. USA 2010, 107, 4585–4589. [Google Scholar] [CrossRef] [PubMed]
- Suga, H.; Hayashi, G.; Terasaka, N. The RNA origin of transfer RNA aminoacylation and beyond. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2011, 366, 2959–2964. [Google Scholar] [CrossRef] [PubMed]
- Yarus, M.; Widmann, J.J.; Knight, R. RNA-amino acid binding: A stereochemical era for the genetic code. J. Mol. Evol. 2009, 69, 406–429. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Li, N.; Ellington, A.D. Ribozyme catalysis of metabolism in the RNA World. Chem. Biodivers 2007, 4, 633–655. [Google Scholar] [CrossRef] [PubMed]
- Rodin, A.S.; Szathmary, E.; Rodin, S.N. On origin of genetic code and tRNA before translation. Biol. Direct 2011, 6. [Google Scholar] [CrossRef] [PubMed]
- Breaker, R.R. Riboswitches and the RNA World. Cold Spring Harb. Perspect. Biol. 2012, 4. [Google Scholar] [CrossRef] [PubMed]
- Serganov, A.; Patel, D.J. Metabolite recognition principles and molecular mechanisms underlying riboswitch function. Annu. Rev. Biophys. 2012, 41, 343–370. [Google Scholar] [CrossRef] [PubMed]
- Puglisi, J.D.; Tan, R.; Calnan, B.J.; Frankel, A.D.; Williamson, J.R. Conformation of the TAR RNA-arginine complex by NMR spectroscopy. Science 1992, 257, 76–80. [Google Scholar] [CrossRef] [PubMed]
- Thiebe, R.; Harbers, K.; Zachau, H.G. Aminoacylation of fragment combinations from yeast tRNAPhe. Eur. J. Biochem. 1972, 26, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Schimmel, P.; Giege, R.; Moras, D.; Yokoyama, S. An operational RNA code for amino acids and possible relationship to genetic code. Proc. Natl. Acad. Sci. USA 1993, 90, 8763–8768. [Google Scholar] [CrossRef] [PubMed]
- Schimmel, P.; Henderson, B. Possible role of aminoacyl-RNA complexes in noncoded peptide synthesis and origin of coded synthesis. Proc. Natl. Acad. Sci. USA 1994, 91, 11283–11286. [Google Scholar] [CrossRef] [PubMed]
- Di Giulio, M. Was it an ancient gene codifying for a hairpin RNA that, by means of direct duplication, gave rise to the primitive transfer-RNA molecule. J. Theor. Biol. 1995, 177, 95–101. [Google Scholar] [CrossRef]
- Tamura, K. Origins and early evolution of the tRNA molecule. Life 2015, 5, 1687–1699. [Google Scholar] [CrossRef] [PubMed]
- Szathmary, E. Coding coenzyme handles: A hypothesis for the origin of the genetic code. Proc. Natl. Acad. Sci. USA 1993, 90, 9916–9920. [Google Scholar] [CrossRef] [PubMed]
- Lambowitz, A.M.; Caprara, M.G.; Zimmerly, S.; Perlman, P.S. Group I and Group II ribozymes as RNPs: Clues to the past and guides to the future. In The RNA World, 2nd ed.; Gesteland, R.F., Cech, T.R., Atkins, J.F., Eds.; Cold Spring Harbor Laboratory Press: New York, NY, USA, 1999; pp. 451–485. [Google Scholar]
- Toor, N.; Hausner, G.; Zimmerly, S. Coevolution of group II intron RNA structures with their intron-encoded reverse transcriptase. RNA 2001, 7, 1142–1152. [Google Scholar] [CrossRef] [PubMed]
- Caetano-Anolles, G.; Sun, F.J. The natural history of transfer RNA and its interactions with the ribosome. Front. Genet. 2014, 5. [Google Scholar] [CrossRef]
- Caetano-Anolles, G.; Nasir, A.; Zhou, K.; Caetano-Anolles, D.; Mittenthal, J.E.; Sun, F.J.; Kim, K.M. Archaea: The first domain of diversified life. Archaea 2014, 2014. Article ID 590214. [Google Scholar] [CrossRef] [PubMed]
- Caetano-Anolles, G.; Kim, K.M.; Caetano-Anolles, D. The phylogenomic roots of modern biochemistry: Origins of proteins, cofactors and protein biosynthesis. J. Mol. Evol. 2012, 74, 1–34. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Cech, T.R. Peptidyl-transferase ribozymes: tRANs reactions, structural characterization and ribosomal RNA-like features. Chem. Biol. 1998, 5, 539–553. [Google Scholar] [CrossRef]
- Grosjean, H.; Soll, D.G.; Crothers, D.M. Studies of the complex between transfer RNAs with complementary anticodons. I. Origins of enhanced affinity between complementary triplets. J. Mol. Biol. 1976, 103, 499–519. [Google Scholar] [CrossRef]
- Xue, H.; Shen, W.; Giege, R.; Wong, J.T. Identity elements of tRNA(Trp). Identification and evolutionary conservation. J. Biol. Chem. 1993, 268, 9316–9322. [Google Scholar] [PubMed]
- Guo, Q.; Gong, Q.; Grosjean, H.; Zhu, G.; Wong, J.T.; Xue, H. Recognition by tryptophanyl-tRNA synthetases of discriminator base on the tRNATrp from three biological domains. J. Biol. Chem. 2002, 277, 14343–14349. [Google Scholar] [CrossRef] [PubMed]
- Maizels, N.; Weiner, A.M. The genomic tag hypothesis: What molecular fossils tell us about the evolution of tRNA. In The RNA World, 2nd ed.; Gesteland, R.F., Cech, T.R., Atkins, J.F., Eds.; Cold Spring Harbor Laboratory Press: New York, NY, USA, 1999; pp. 79–111. [Google Scholar]
- Smit, A.F.; Riggs, A.D. MIRs are classic, tRNA-derived SINEs that amplified before the mammalian radiation. Nucleic Acids Res. 1995, 23, 98–102. [Google Scholar] [CrossRef] [PubMed]
- Murnane, J.P.; Morales, J.F. Use of a mammalian interspersed repetitive (MIR) element in the coding and processing sequences of mammalian genes. Nucleic Acids Res. 1995, 23, 2837–2839. [Google Scholar] [CrossRef] [PubMed]
- Krull, M.; Petrusma, M.; Makalowski, W.; Brosius, J.; Schmitz, J. Functional persistence of exonized mammalian-wide interspersed repeat elements (MIRs). Genome Res. 2007, 17, 1139–1145. [Google Scholar] [CrossRef] [PubMed]
- Jjingo, D.; Conley, A.B.; Wang, J.; Marino-Ramirez, L.; Lunyak, V.V.; Jordan, I.K. Mammalian-wide interspersed repeat (MIR)-derived enhancers and the regulation of human gene expression. Mob. DNA 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Akins, R.A.; Kelley, R.L.; Lambowitz, A.M. Characterization of mutant mitochondrial plasmids of Neurospora spp. that have incorporated tRNAs by reverse transcription. Mol. Cell Biol. 1989, 9, 678–691. [Google Scholar] [CrossRef] [PubMed]
- Brosius, J. Echoes from the past—Are we still in an RNP world? Cytogenet. Genome Res. 2005, 110, 8–24. [Google Scholar] [CrossRef] [PubMed]
- Touchon, M.; Rocha, E.P. Causes of insertion sequences abundance in prokaryotic genomes. Mol. Biol. Evol. 2007, 24, 969–981. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.T. A co-evolution theory of the genetic code. Proc. Natl. Acad. Sci. USA 1975, 72, 1909–1912. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.T. Coevolution theory of the genetic code at age thirty. Bioessays 2005, 27, 416–425. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.T. Genetic code. In Prebiotic Evolution and Astrobiology; Wong, J.T., Lazcano, A., Eds.; Landes Bioscience: Austin, TX, USA, 2009; pp. 110–119. [Google Scholar]
- Weber, A.L.; Miller, S.L. Reasons for the occurrence of the twenty coded protein amino acids. J. Mol. Evol. 1981, 17, 273–284. [Google Scholar] [CrossRef] [PubMed]
- Parker, E.T.; Cleaves, H.J.; Callahan, M.P.; Dworkin, J.P.; Glavin, D.P.; Lazcano, A.; Bada, J.L. Prebiotic dynthesis of methionine and other sulfur-containing compounds on the primitive Earth: A contemporary reassessment based on an unpublished 1958 Stanley Miller experiment. Orig. Life Evol. Biosph. 2011, 41, 201–212. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.T.; Bronskill, P.M. Inadequacy of prebiotic synthesis as origin of proteinous amino acids. J. Mol. Evol. 1979, 13, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.T. Role of minimization of chemical distances between amino acids in the evolution of the genetic code. Proc. Natl. Acad. Sci. USA 1980, 77, 1083–1086. [Google Scholar] [CrossRef] [PubMed]
- Commans, S.; Bock, A. Selenocysteine inserting tRNAs: An overview. FEMS Microbiol. Rev. 1999, 23, 335–351. [Google Scholar] [CrossRef] [PubMed]
- Sauerwald, A.; Zhu, W.; Major, T.A.; Roy, H.; Palioura, S.; Jahn, D.; Whitman, W.B.; Yates, J.R., 3rd; Ibba, M.; Soll, D. RNA-dependent cysteine biosynthesis in archaea. Science 2005, 307, 1969–1972. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.T. Evolution and mutation of the amino acid code. In Dynamics of Biochemical Systems; Ricard, J., Cornish-Bowden, A., Eds.; Plenum Press: New York, NY, USA, 1983; pp. 247–258. [Google Scholar]
- Wong, J.T. Coevolution of the genetic code and amino acid biosynthesis. Trends Biochem. Sci. 1981, 16, 33–35. [Google Scholar] [CrossRef]
- Kobayashi, K.; Tsuchiya, M.; Oshima, T.; Yanagawa, H. Abiotic synthesis of amino acids and imidazole by proton irradiation of simulated primitive earth atmosphere. Orig. Life Evol. Biosph. 1990, 20, 99–109. [Google Scholar] [CrossRef]
- Kobayashi, K.; Kaneko, T.; Saito, T.; Oshima, T. Amino acid formation in gas mixtures by high energy particle irradiation. Orig. Life Evol. Biosph. 1998, 28, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Pizzarello, S.; Holmes, W. Nitrogen-containing compounds in two CR2 meteorites: 15N composition, molecular distribution and precursor molecules. Geochim. Cosmochim. Acta 2009, 73, 2150–2162. [Google Scholar] [CrossRef]
- Wong, J.T. The evolution of a universal genetic code. Proc. Natl. Acad. Sci. USA 1976, 73, 2336–2340. [Google Scholar] [CrossRef] [PubMed]
- Freeland, S.J.; Hurst, L.D. The genetic code is one in a million. J. Mol. Evol. 1998, 47, 238–248. [Google Scholar] [CrossRef] [PubMed]
- Knight, R.; Landweber, L.; Yarus, M. Tests of a stereochemical genetic code. In Translation Mechanisms; Lapointe, J., Brakier-Gingras, L., Eds.; Landes Bioscience: Austin, TX, USA, 2003; pp. 115–128. [Google Scholar]
- Shepherd, J.C.W. Fossil remnants of a primeval genetic code in all forms of life? Trends Biochem. Sci. 1984, 9, 8–10. [Google Scholar] [CrossRef]
- Watson, J.D.; Hopkins, N.H.; Roberts, J.W.; Steitz, J.A.; Weiner, A.M. Molecular Biology of the Gene, 4th ed.; Benjamin Cummings: San Francisco, CA, USA, 1987; pp. 459–462. [Google Scholar]
- Hartman, H. Speculations on the origin of the genetic code. J. Mol. Evol. 1995, 40, 541–544. [Google Scholar] [CrossRef] [PubMed]
- Ikehara, K.; Omori, Y.; Arai, R.; Hirose, A. A novel theory on the origin of the genetic code: A GNC-SNS hypothesis. J. Mol. Evol. 2002, 54, 530–538. [Google Scholar] [CrossRef] [PubMed]
- Di Giulio, M. An extension of the coevolution theory of the genetic code. Biol. Direct 2008, 3. [Google Scholar] [CrossRef] [PubMed]
- Higgs, P.G. A four-column theory for the origin of the genetic code: Tracing the evolutionary pathways that gave rise to an optimized code. Biol. Direct 2009, 4. [Google Scholar] [CrossRef] [PubMed]
- Marck, C.; Grosjean, H. tRNomics: Analysis of tRNA genes from 50 genomes of Eukarya, Archaea, and Bacteria reveals anticodon-sparing strategies and domain-specific features. RNA 2002, 8, 1189–1232. [Google Scholar] [CrossRef]
- Schattner, P.; Brooks, A.N.; Lowe, T.M. The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 2005, 33, W686–W689. [Google Scholar] [CrossRef] [PubMed]
- Felsenstein, J. PHYLIP—Phylogeny Inference Package (Version 3.2). Cladistics 1989, 5, 164–166. [Google Scholar]
- Woese, C.R.; Kandler, O.; Wheelis, M.L. Towards a natural system of organisms: Proposal for the domains Archaea, Bacteria, and Eucarya. Proc. Natl. Acad. Sci. USA 1990, 87, 4576–4579. [Google Scholar] [CrossRef] [PubMed]
- Pennisi, E. Is it time to uproot the tree of life? Science 1999, 284, 1305–1307. [Google Scholar] [CrossRef] [PubMed]
- Xue, H.; Tong, K.L.; Marck, C.; Grosjean, H.; Wong, J.T. Transfer RNA paralogs: Evidence for genetic code-amino acid biosynthesis coevolution and an archaeal root of life. Gene 2003, 310, 59–66. [Google Scholar] [CrossRef]
- Xue, H.; Ng, S.K.; Tong, K.L.; Wong, J.T. Congruence of evidence for a Methanopyrus-proximal root of life based on transfer RNA and aminoacyl-tRNA synthetase genes. Gene 2005, 360, 120–130. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.T.; Chen, J.; Mat, W.K.; Ng, S.K.; Xue, H. Polyphasic evidence delineating the root of life and roots of biological domains. Gene 2007, 403, 39–52. [Google Scholar] [CrossRef] [PubMed]
- Tong, K.L.; Wong, J.T. Anticodon and wobble evolution. Gene 2004, 333, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Slesarev, A.I.; Mezhevaya, K.V.; Makarova, K.S.; Polushin, N.N.; Shcherbinina, O.V.; Shakhova, V.V.; Belova, G.I.; Aravind, L.; Natale, D.A.; Rogozin, I.B.; et al. The complete genome of hyperthermophile Methanopyrus kandleri AV19 and monophyly of archaeal methanogens. Proc. Natl. Acad. Sci. USA 2002, 99, 4644–4649. [Google Scholar] [CrossRef] [PubMed]
- Battistuzzi, F.U.; Feijao, A.; Hedges, S.B. A genomic timescale of prokaryote evolution: Insights into the origin of methanogenesis, phototrophy, and the colonization of land. BMC Evol. Biol. 2004, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Giulio, M. A methanogen hosted the origin of the genetic code. J. Theor. Biol. 2009, 260, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Raymond, J.; Segre, D. The effect of oxygen on biochemical networks and the evolution of complex life. Science 2006, 311, 1764–1767. [Google Scholar] [CrossRef] [PubMed]
- Archetti, M.; Di Giulio, M. The evolution of the genetic code took place in an anaerobic environment. J. Theor. Biol. 2007, 245, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Stetter, K.O. Hyperthermophiles in the history of life. Ciba Found. Symp. 1996, 202, 1–23. [Google Scholar] [PubMed]
- Di Giulio, M. The universal ancestor and the ancestor of bacteria were hyperthermophiles. J. Mol. Evol. 2003, 57, 721–730. [Google Scholar] [CrossRef] [PubMed]
- Gaucher, E.A.; Govindarajan, S.; Ganesh, O.K. Palaeotemperature trend for Precambrian life inferred from resurrected proteins. Nature 2008, 451, 704–707. [Google Scholar] [CrossRef] [PubMed]
- Groussin, M.; Gouy, M. Adaptation to environmental temperature is a major determinant of molecular evolutionary rates in archaea. Mol. Biol. Evol. 2011, 28, 2661–2674. [Google Scholar] [CrossRef] [PubMed]
- Schwartzman, D.W.; Lineweaver, C.H. The hyperthermophilic origin of life revisited. Biochem. Soc. Trans. 2004, 32, 168–171. [Google Scholar] [CrossRef] [PubMed]
- Di Giulio, M. A comparison of proteins from Pyrococcus furiosus and Pyrococcus abyssi: Barophily in the physicochemical properties of amino acids and in the genetic code. Gene 2005, 346, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Di Giulio, M. Structuring of the genetic code took place at acidic pH. J. Theor. Biol. 2005, 237, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Bernhardt, H.S.; Tate, W.P. Primordial soup or vinaigrette: Did the RNA World evolve at acidic pH? Biol. Direct 2012, 7. [Google Scholar] [CrossRef] [PubMed]
- Leigh, J.A. Evolution of energy metabolism. In Biodiversity of Microbial Life; Staley, J.T., Reysenbach, A.L., Eds.; Wiley-Liss: New York, NY, USA, 2001; pp. 103–120. [Google Scholar]
- Falkowski, P.G. Evolution. Tracing oxygen’s impsrint on earth’s metabolic evolution. Science 2006, 311, 1724–1725. [Google Scholar] [CrossRef] [PubMed]
- Shock, E.L. High-temperature life without photosynthesis as a model for Mars. J. Geophys. Res. 1997, 102, 23687–23694. [Google Scholar] [CrossRef] [PubMed]
- Takai, K.; Nakamura, K. Archaeal diversity and community development in deep-sea hydrothermal vents. Curr. Opin. Microbiol. 2011, 14, 282–291. [Google Scholar] [CrossRef] [PubMed]
- Sun, F.J.; Caetano-Anolles, G. Evolutionary patterns in the sequence and structure of transfer RNA: Early origins of archaea and viruses. PLoS Comput. Biol. 2008, 4, e1000018. [Google Scholar] [CrossRef] [PubMed]
- Sun, F.J.; Caetano-Anolles, G. The evolutionary history of the structure of 5S ribosomal RNA. J. Mol. Evol. 2009, 69, 430–443. [Google Scholar] [CrossRef] [PubMed]
- Sun, F.J.; Caetano-Anolles, G. The ancient history of the structure of ribonuclease P and the early origins of Archaea. BMC Bioinform. 2010, 11. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Jiang, Y.Y.; Kim, K.M.; Qu, G.; Ji, H.F.; Mittenthal, J.E.; Zhang, H.Y.; Caetano-Anolles, G. A universal molecular clock of protein folds and its power in tracing the early history of aerobic metabolism and planet oxygenation. Mol. Biol. Evol. 2011, 28, 567–582. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.M.; Caetano-Anolles, G. The evolutionary history of protein fold families and proteomes confirms that the archaeal ancestor is more ancient than the ancestors of other superkingdoms. BMC Evol. Biol. 2012, 12. [Google Scholar] [CrossRef] [PubMed]
- Nasir, A.; Kim, K.M.; Caetano-Anolles, G. A phylogenomic census of molecular functions identifies modern thermophilic archaea as the most ancient form of cellular life. Archaea 2014, 2014. [Google Scholar] [CrossRef] [PubMed]
- Sauerwald, A.; Sitaramaiah, D.; McCloskey, J.A.; Soll, D.; Crain, P.F. N6-Acetyladenosine: A new modified nucleoside from Methanopyrus kandleri tRNA. FEBS Lett. 2005, 579, 2807–2810. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Sitaramaiah, D.; Noon, K.R.; Guymon, R.; Hashizume, T.; McCloskey, J.A. Structures of two new “minimalist” modified nucleosides from archaeal tRNA. Bioorg. Chem. 2004, 32, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Rogozin, I.B.; Carmel, L.; Csuros, M.; Koonin, E.V. Origin and evolution of spliceosomal introns. Biol. Direct 2012, 7. [Google Scholar] [CrossRef] [PubMed]
- Edgell, D.R.; Belfort, M.; Shub, D.A. Barriers to intron promiscuity in bacteria. J. Bacteriol. 2000, 182, 5281–5289. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, Y.; Yokobori, S.; Inaba, T.; Yamagishi, A.; Oshima, T.; Kawarabayasi, Y.; Kikuchi, H.; Kita, K. Introns in protein-coding genes in Archaea. FEBS Lett. 2002, 510, 27–30. [Google Scholar] [CrossRef]
- Lykke-Andersen, J.; Aagaard, C.; Semionenkov, M.; Garrett, R.A. Archaeal introns: Splicing, intercellular mobility and evolution. Trends Biochem. Sci. 1997, 22, 326–331. [Google Scholar] [CrossRef]
- Trotta, C.R.; Abelson, J. tRNA spicing: An RNA add-on or an ancient reaction? In The RNA World, 2nd ed.; Gesteland, R.F., Cech, T.R., Atkins, J.F., Eds.; Cold Spring Harbor Laboratory Press: New York, NY, USA, 1999; pp. 561–584. [Google Scholar]
- Sugahara, J.; Kikuta, K.; Fujishima, K.; Yachie, N.; Tomita, M.; Kanai, A. Comprehensive analysis of archaeal tRNA genes reveals rapid increase of tRNA introns in the order thermoproteales. Mol. Biol. Evol. 2008, 25, 2709–2716. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, W. Why genes in pieces. Nature 1978, 271. [Google Scholar] [CrossRef]
- Logsdon, J.M., Jr. The recent origins of spliceosomal introns revisited. Curr. Opin. Genet. Dev. 1998, 8, 637–648. [Google Scholar] [CrossRef]
- De Souza, S.J. The emergence of a synthetic theory of intron evolution. Genetica 2003, 118, 117–121. [Google Scholar] [CrossRef] [PubMed]
- Cavalier-Smith, T. Intron phylogeny: A new hypothesis. Trends Genet. 1991, 7, 145–148. [Google Scholar] [CrossRef]
- Yoshinari, S.; Itoh, T.; Hallam, S.J.; DeLong, E.F.; Yokobori, S.; Yamagishi, A.; Oshima, T.; Kita, K.; Watanabe, Y. Archaeal pre-mRNA splicing: A connection to hetero-oligomeric splicing endonuclease. Biochim. Biophys. Res. Commun. 2006, 346, 1024–1032. [Google Scholar] [CrossRef] [PubMed]
- Tocchini-Valentini, G.D.; Fruscoloni, P.; Tocchini-Valentini, G.P. Evolution of introns in the archaeal world. Proc. Natl. Acad. Sci. USA 2011, 108, 4782–4787. [Google Scholar] [CrossRef] [PubMed]
- Marck, C.; Grosjean, H. Identification of BHB splicing motifs in intron-containing tRNAs from 18 archaea: Evolutionary implications. RNA 2003, 9, 1516–1531. [Google Scholar] [CrossRef] [PubMed]
- Heinemann, I.U.; Soll, D.; Randau, L. Transfer RNA processing in archaea: Unusual pathways and enzymes. FEBS Lett. 2010, 584, 303–309. [Google Scholar] [CrossRef] [PubMed]
- Su, A.A.; Tripp, V.; Randau, L. RNA-Seq analyses reveal the order of tRNA processing events and the maturation of C/D box and CRISPR RNAs in the hyperthermophile Methanopyrus kandleri. Nucleic Acids Res. 2013, 41, 6250–6258. [Google Scholar] [CrossRef] [PubMed]
- Rogers, J.H. The role of introns in evolution. FEBS Lett. 1990, 268, 339–343. [Google Scholar] [CrossRef]
- Hall, D.H.; Liu, Y.; Shub, D.A. Exon shuffling by recombination between self-splicing introns of bacteriophage T4. Nature 1989, 340, 575–576. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Wang, F.; Pun, F.W.; Mei, L.; Ren, L.; Yu, Z.; Ng, S.K.; Chen, J.; Tsang, S.Y.; Xue, H. Epigenetic regulation on GABRB2 isoforms expression: Developmental variations and disruptions in psychotic disorders. Schizophr. Res. 2012, 134, 260–266. [Google Scholar] [CrossRef] [PubMed]
- Xiong, H.Y.; Alipanahi, B.; Lee, L.J.; Bretschneider, H.; Merico, D.; Yuen, R.K.; Hua, Y.; Gueroussov, S.; Najafabadi, H.S.; Hughes, T.R.; et al. RNA splicing. The human splicing code reveals new insights into the genetic determinants of disease. Science 2015, 347. [Google Scholar] [CrossRef] [PubMed]
- Wessler, S.R.; Baran, G.; Varagona, M.; Dellaporta, S.L. Excision of Ds produces waxy proteins with a range of enzymatic activities. EMBO J. 1986, 5, 2427–2432. [Google Scholar] [PubMed]
- Alberts, B.; Bray, D.; Lewis, J.; Raff, M.; Roberts, K.; Watson, J.D. Molecular Biology of the Cell, 3rd ed.; Garland Publisher: New York, NY, USA, 1994; p. 1224. [Google Scholar]
- Mat, W.K.; Xue, H.; Wong, J.T. The genomics of LUCA. Front. Biosci. 2008, 13, 5605–5613. [Google Scholar] [CrossRef] [PubMed]
- Crick, F.H. Codon—Anticodon pairing: The wobble hypothesis. J. Mol. Biol. 1966, 19, 548–555. [Google Scholar] [CrossRef]
- Agris, P.F.; Vendeix, F.A.P.; Graham, W.D. tRNA’s wobble decoding of the genome: 40 years of modification. J. Mol. Biol. 2007, 366, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Rogalski, M.; Karcher, D.; Bock, R. Superwobbling facilitates translation with reduced tRNA sets. Nat. Struct. Mol. Biol. 2008, 15, 192–198. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, K.; Osawa, S. tRNA sequences and variations in the genetic code. In tRNA: Structure, Biosynthesis and Function; Söll, D., Rajbandary, U.L., Eds.; ASM Press: Washington, DC, USA, 1995; pp. 225–250. [Google Scholar]
- Gupta, R. Halobacterium volcanii tRNAs. Identification of 41 tRNAs covering all amino acids, and the sequences of 33 class I tRNAs. J. Biol. Chem. 1984, 259, 9461–9471. [Google Scholar] [PubMed]
- Agris, P.F. Wobble position modified nucleosides evolved to select transfer RNA codon recognition: A modified wobble hypothesis. Biochimie 1991, 73, 1345–1349. [Google Scholar] [CrossRef]
- Yarus, M. Translational efficiency of transfer RNA’s: Uses of an extended anticodon. Science 1995, 218, 645–652. [Google Scholar] [CrossRef]
- Curran, J.F. Modified nucleosides in translation. In Modification and Editing of RNA; Grosjean, H., Benne, R., Eds.; ASM Press: Washington, DC, USA, 1998; pp. 493–516. [Google Scholar]
- Rozenski, J.; Crain, P.F.; McCloskey, J.A. The RNA modification database: 1999 update. Nucl. Acid. Res. 1999, 27, 196–197. [Google Scholar] [CrossRef]
- Murphy, F.V.; Ramakrishnan, V. Structure of a purine-purine wobble base pair in the decoding center of the ribosome. Nat. Struct. Mol. Biol. 2004, 11, 1251–1252. [Google Scholar] [CrossRef] [PubMed]
- Rozov, A.; Demeshkina, N.; Khusainov, I.; Westhof, E.; Yusupov, M.; Yusupova, G. Novel base-pairing interactions at the tRNA wobble position crucial for accurate reading of the genetic code. Nat. Commun. 2015, 7, 10457. [Google Scholar] [CrossRef] [PubMed]
- Woese, C.R.; Fox, G.E. Phylogenetic structure of the prokaryotic domain: The primary kingdoms. Proc. Natl. Acad. Sci. USA 1977, 74, 5088–5090. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Polacek, N.; Vesper, O.; Staub, E.; Einfeldt, E.; Wilson, D.N.; Nierhaus, K.H. The highly conserved LepA is a ribosomal elongation factor that back-translocates the ribosome. Cell 2006, 127, 721–733. [Google Scholar] [CrossRef] [PubMed]
- Nierhaus, K.H.; (Max-Planck-Institut für Molekulare Genetik, Berlin, Germany). Personal communication, 2009.
- Wong, J.T. Root of Life. In Prebiotic Evolution and Astrobiology; Wong, J.T., Lazcano, A., Eds.; Landes Bioscience: Austin, TX, USA, 2009; pp. 120–144. [Google Scholar]
- Strickberger, M.W. Evolution, 2nd ed.; Jones & Bartlett: Burlington, VT, USA, 1996; p. 259. [Google Scholar]
- Crick, F.H. The origin of the genetic code. J. Mol. Biol. 1968, 38, 367–379. [Google Scholar] [CrossRef]
- Wong, J.T. Membership mutation of the genetic code: Loss of fitness by tryptophan. Proc. Natl. Acad. Sci. USA 1983, 80, 6303–6306. [Google Scholar] [CrossRef] [PubMed]
- Bronskill, P.M.; Wong, J.T. Suppression of fluorescence of tryptophan residues in proteins by replacement with 4-fluorotryptophan. Biochem. J. 1988, 249, 305–308. [Google Scholar] [CrossRef] [PubMed]
- Mat, W.K.; Xue, H.; Wong, J.T. Genetic code mutations: The breaking of a three billion year invariance. PLoS ONE 2010, 5, e12206. [Google Scholar] [CrossRef] [PubMed]
- Cowie, D.B.; Cohen, G.N. Biosynthesis by Escherichia coli of active altered proteins containing selenium instead of sulfur. Biochim. Biophys. Acta 1957, 26, 252–261. [Google Scholar] [CrossRef]
- Hendrickson, W.A.; Horton, J.R.; LeMaster, D.M. Selenomethionyl protein produced for analysis by multiwavelength anomalous diffraction (MAD): A vehicle for direct determination of three dimensional structure. EMBO J. 1990, 9, 1665–1672. [Google Scholar] [PubMed]
- Frank, P.; Licht, A.; Tullius, T.D.; Hodgson, K.O.; Pecht, I. A selenomethionine-containing azurin from an auxotroph of Pseudomonas aeruginosa. J. Biol. Chem. 1985, 260, 5518–5525. [Google Scholar] [PubMed]
- Hesman, T. Code breakers: Scientists are altering bacteria in a most fundamental way. Sci. News 2000, 157, 360–362. [Google Scholar] [CrossRef]
- Wong, J.T.; Xue, H. Synthetic genetic codes as the basis of synthetic life. In Chemical Synthetic Biology; Luisi, P.L., Chiarabelli, C., Eds.; Wiley: New York, NY, USA, 2010; pp. 178–199. [Google Scholar]
- Bacher, J.M.; Ellington, A.D. Selection and characterization of Escherichia coli variants capable of growth on an otherwise toxic tryptophan analogue. J. Bacteriol. 2001, 183, 5414–5425. [Google Scholar] [CrossRef] [PubMed]
- Bacher, J.M.; Bull, J.J.; Ellington, A.D. Evolution of phage with chemically ambiguous proteomes. BMC Evol. Biol. 2003, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bacher, J.M.; Hughes, R.A.; Tze-Fei Wong, J.; Ellington, A.D. Evolving new genetic codes. Trends Ecol. Evol. 2004, 19, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Hoesl, M.G.; Oehm, S.; Durkin, P.; Darmon, E.; Peil, L.; Aerni, H.R.; Rappsilber, J.; Rinehart, J.; Leach, D.; Soll, D.; et al. Chemical evolution of a bacterial proteome. Angew. Chem. Int. Ed. Engl. 2015, 54, 10030–10034. [Google Scholar] [CrossRef] [PubMed]
- Kwok, Y.; Wong, J.T. Evolutionary relationship between Halobacterium cutirubrum and eukaryotes determined by use of aminoacyl-tRNA synthetases as phylogenetic probes. Can. J. Biochem. 1980, 58, 213–218. [Google Scholar] [CrossRef] [PubMed]
- Santoro, S.W.; Anderson, J.C.; Lakshman, V.; Schultz, P.G. An archaebacteria-derived glutamyl-tRNA synthetase and tRNA pair for unnatural amino acid mutagenesis of proteins in Escherichia coli. Nucleic Acids Res. 2003, 31, 6700–6709. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.C.; Schultz, P.G. Adding new chemistries to the genetic code. Annu. Rev. Biochem. 2010, 79, 413–444. [Google Scholar] [CrossRef] [PubMed]
- Hoesl, M.G.; Budisa, N. Recent advances in genetic code engineering in Escherichia coli. Curr. Opin. Biotechnol. 2012, 23, 751–757. [Google Scholar] [CrossRef] [PubMed]
- Mehl, R.A.; Anderson, J.C.; Santoro, S.W.; Wang, L.; Martin, A.B.; King, D.S.; Horn, D.M.; Schultz, P.G. Generation of a bacterium with a 21 amino acid genetic code. J. Am. Chem. Soc. 2003, 125, 935–939. [Google Scholar] [CrossRef] [PubMed]
- Tian, H.; Deng, D.; Huang, J.; Yao, D.; Xu, X.; Gao, X. Screening system for orthogonal suppressor tRNAs based on the species-specific toxicity of suppressor tRNAs. Biochimie 2013, 95, 881–888. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.C.; Qi, L.; Yanofsky, C.; Arkin, A.P. Regulation of transcription by unnatural amino acids. Nat. Biotechnol. 2011, 29, 164–168. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhang, W.; Song, W.; Wang, Y.; Yu, Z.; Li, J.; Wu, M.; Wang, L.; Zang, J.; Lin, Q. A biosynthetic route to photoclick chemistry on proteins. J. Am. Chem. Soc. 2010, 132, 14812–14818. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Schmied, W.H.; Chin, J.W. Reprogramming the genetic code: From triplet to quadruplet codes. Angew. Chem. Int. Ed. Engl. 2012, 51, 2288–2297. [Google Scholar] [CrossRef] [PubMed]
- Lepthien, S.; Merkel, L.; Budisa, N. In vivo double and triple labeling of proteins using synthetic amino acids. Angew. Chem. Int. Ed. Engl. 2010, 49, 5446–5450. [Google Scholar] [CrossRef] [PubMed]
- Merkel, L.; Schauer, M.; Antranikian, G.; Budisa, N. Parallel incorporation of different fluorinated amino acids: On the way to “teflon” proteins. Chembiochem 2010, 11, 1505–1507. [Google Scholar] [CrossRef] [PubMed]
- Lajoie, M.J.; Rovner, A.J.; Goodman, D.B.; Aerni, H.R.; Haimovich, A.D.; Kuznetsov, G.; Mercer, J.A.; Wang, H.H.; Carr, P.A.; Mosberg, J.A.; et al. Genomically recoded organisms expand biological functions. Science 2013, 342, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Hancock, S.M.; Uprety, R.; Deiters, A.; Chin, J.W. Expanding the genetic code of yeast for incorporation of diverse unnatural amino acids via a pyrrolysyl-tRNA synthetase/tRNA pair. J. Am. Chem. Soc. 2010, 132, 14819–14824. [Google Scholar] [CrossRef] [PubMed]
- Nehring, S.; Budisa, N.; Wiltschi, B. Performance analysis of orthogonal pairs designed for an expanded eukaryotic genetic code. PLoS ONE 2012, 7, e31992. [Google Scholar] [CrossRef] [PubMed]
- Ye, S.; Riou, M.; Carvalho, S.; Paoletti, P. Expanding the genetic code in Xenopus laevis oocytes. Chembiochem 2013, 14, 230–235. [Google Scholar] [CrossRef] [PubMed]
- Shen, B.; Xiang, Z.; Miller, B.; Louie, G.; Wang, W.; Noel, J.P.; Gage, F.H.; Wang, L. Genetically encoding of unnatural amino acids in neural stem cells and optically reporting voltage-sensitive domain changes in differentiated neurons. Stem Cells 2011, 29, 1231–1240. [Google Scholar] [CrossRef] [PubMed]
- Thibodeaux, G.N.; Liang, X.; Moncivais, K.; Umeda, A.; Singer, O.; Alfonta, L.; Zhang, Z.J. Transforming a pair of orthogonal tRNA-aminoacyl-tRNA synthetase from Archaea to function in mammalian cells. PLoS ONE 2010, 5, e11263. [Google Scholar] [CrossRef] [PubMed]
- Greiss, S.; Chin, J.W. Expanding the genetic code of an animal. J. Am. Chem. Soc. 2011, 133, 14196–14199. [Google Scholar] [CrossRef] [PubMed]
- Parrish, A.R.; She, X.; Xiang, Z.; Coin, I.; Shen, Z.; Briggs, S.P.; Dillin, A.; Wang, L. Expanding the genetic code of Caenorhabditis elegans using bacterial aminoacyl-tRNA synthetase/tRNA pairs. ACS Chem. Biol. 2012, 7, 1292–1302. [Google Scholar] [CrossRef] [PubMed]
- Mandell, D.J.; Lajoie, M.J.; Mee, M.T.; Takeuchi, R.; Kuznetsov, G.; Norville, J.E.; Gregg, C.J.; Stoddard, B.L.; Church, G.M. Biocontainment of genetically modified organisms by synthetic protein design. Nature 2015, 518, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Rovner, A.J.; Haimovich, A.D.; Katz, S.R.; Li, Z.; Grome, M.W.; Gassaway, B.M.; Amiram, M.; Patel, J.R.; Gallagher, R.R.; Rinehart, J.; et al. Recoded organisms engineered to depend on synthetic amino acids. Nature 2015, 518, 89–93. [Google Scholar] [CrossRef] [PubMed]
- Lemeignan, P.; Sonigo, P.; Marliere, P. Phenotypic suppression by incorporation of an alien amino acid. J. Mol. Biol. 1993, 231, 161–166. [Google Scholar] [CrossRef] [PubMed]
- Marliere, P.; Patrouix, J.; Doring, V.; Herdewijn, P.; Tricot, S.; Cruveiller, S.; Bouzon, M.; Mutzel, R. Chemical evolution of a bacterium’s genome. Angew. Chem. Int. Ed. Engl. 2011, 50, 7109–7114. [Google Scholar] [CrossRef] [PubMed]
- Marliere, P. Charting the xenobiotic continent. In Proceedings of First Conference on Xenobiology, Genoa, Italy, 6–8 May 2014; p. 3.
- Acevedo-Rocha, C.G.; Budisa, N. On the road towards chemically modified organisms endowed with a genetic firewall. Angew. Chem. Int. Ed. Engl. 2011, 50, 6960–6962. [Google Scholar] [CrossRef] [PubMed]
- Yu, A.C.; Yim, A.K.; Mat, W.K.; Tong, A.H.; Lok, S.; Xue, H.; Tsui, S.K.; Wong, J.T.; Chan, T.F. Mutations enabling displacement of tryptophan by 4-fluorotryptophan as a canonical amino acid of the genetic code. Genome Biol. Evol. 2014, 6, 629–641. [Google Scholar] [CrossRef] [PubMed]
- Hammerling, M.J.; Ellefson, J.W.; Boutz, D.R.; Marcotte, E.M.; Ellington, A.D.; Barrick, J.E. Bacteriophages use an expanded genetic code on evolutionary paths to higher fitness. Nat. Chem. Biol. 2014, 10, 178–180. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.T. Question 6-Coevolution theory of the genetic code: A proven theory. Orig. Life Evol. Biosph. 2007, 37, 403–408. [Google Scholar] [CrossRef] [PubMed]
- Min, B.; Pelaschier, J.T.; Graham, D.E.; Tumbula-Hansen, D.; Soll, D. Transfer RNA-dependent amino acid biosynthesis: An essential route to asparagine formation. Proc. Natl. Acad. Sci. USA 2002, 99, 2678–2683. [Google Scholar] [CrossRef] [PubMed]
- Roy, H.; Becker, H.D.; Reinbolt, J.; Kern, D. When contemporary aminoacyl-tRNA synthetases invent their cognate amino acid metabolism. Proc. Natl. Acad. Sci. USA 2003, 100, 9837–9842. [Google Scholar] [CrossRef] [PubMed]
- Francklyn, C. tRNA synthetase paralogs: Evolutionary links in the transition from tRNA-dependent amino acid biosynthesis to de novo biosynthesis. Proc. Natl. Acad. Sci. USA 2003, 100, 9650–9652. [Google Scholar] [CrossRef] [PubMed]
- O’Donoghue, P.; Sethi, A.; Woese, C.R.; Luthy-Schulten, Z.A. The evolutionary history of Cys-tRNACys formation. Proc. Natl. Acad. Sci. USA 2005, 102, 19003–19008. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.M.; Qin, T.; Jiang, Y.Y.; Chen, L.L.; Xiong, M.; Caetano-Anolles, D.; Zhang, H.Y.; Caetano-Anolles, G. Protein domain structure uncovers the origin of aerobic metabolism and the rise of planetary oxygen. Structure 2012, 20, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Mojzsis, S.J.; Arrhenius, G.; McKeegan, K.D.; Harrison, T.M.; Nutman, A.P.; Friend, C.R. Evidence for life on Earth before 3800 million years ago. Nature 1996, 384, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Fournier, G.; Andam, C.P.; Gogarten, J.P. Ancient horizontal gene transfers and the last common ancestors. BMC Evol. Biol. 2015, 15. [Google Scholar] [CrossRef] [PubMed]
- Cohen, P. Life the Sequel. New Sci. 2000, 167, 33–36. [Google Scholar]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Evidence | Gly | Ala | Ser | Asp | Glu | Val | Leu | Ile | Pro | Thr | Phe | Tyr | Arg | His | Trp | Asn | Gln | Lys | Cys | Met | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Coevolution theory | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | [92] |
| Irradiated synthesis | + | + | + | + | + | + | + | + | + | + | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | NA | NA | [93,94] |
| Meteorite composition | + | + | + | + | + | + | + | + | + | + | + | + | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | [12,95] |
| Prebiotic synthesis | + | + | + | + | + | + | + | + | + | + | + | + | 0 | 0 | + | + | + | 0 | + | + | [85,86] |
| Electric discharge synthesis | + | + | + | + | + | + | + | + | + | + | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | + | [85,86] |
| Line No. | Type of Evidence * | Evidence for | Reference |
|---|---|---|---|
| 1 | Alloacceptor tRNA distances | A, M | [110] |
| 2 | Initiator-elongator tRNAMet distances | A, M | [111] |
| 3 | Anticodon usages | A, M | [113] |
| 4 | Aminoacyl-tRNA synthetase distances | A, M | [111] |
| 5 | Archaeal root of ValRS | A | [112] |
| 6 | Lack of GlnRS in Mka | M | [112,114] |
| 7 | Lack of AsnRS in Mka | M | [112,114] |
| 8 | Lack of CysRS in Mka | A, M | [112,114] |
| 9 | Lack of cytochromes in Mka | M | [112] |
| 10 | Early Euryarchaea-Crenarchaea separation | A, M | [115] |
| 11 | Mka as deep branching archaeon | M | [115] |
| 12 | Primitivity of methanogenesis | A, M | [115,116] |
| 13 | Primitivity of anaerobiosis | M | [117,118] |
| 14 | Primitivity of hyperthermophily | A, M | [119,120,121,122,123] |
| 15 | Primitivity of barophily | M | [124] |
| 16 | Primitivity of acidophily | M | [125,126] |
| 17 | Use of CO2 as electron acceptor | A, M | [127,128] |
| 18 | Chemolithotrophy | M | [112] |
| 19 | Hydrothermal vents as appropriate home for LUCA | M | [11,129,130] |
| 20 | Minimalist regulations | M | [114] |
| 21 | tRNA evolution pattern | A | [131] |
| 22 | 5S rRNA tree | A | [132] |
| 23 | Ribonuclease P tree | A | [133] |
| 24 | Protein fold tree | A | [134,135] |
| 25 | Proteome tree | A | [135] |
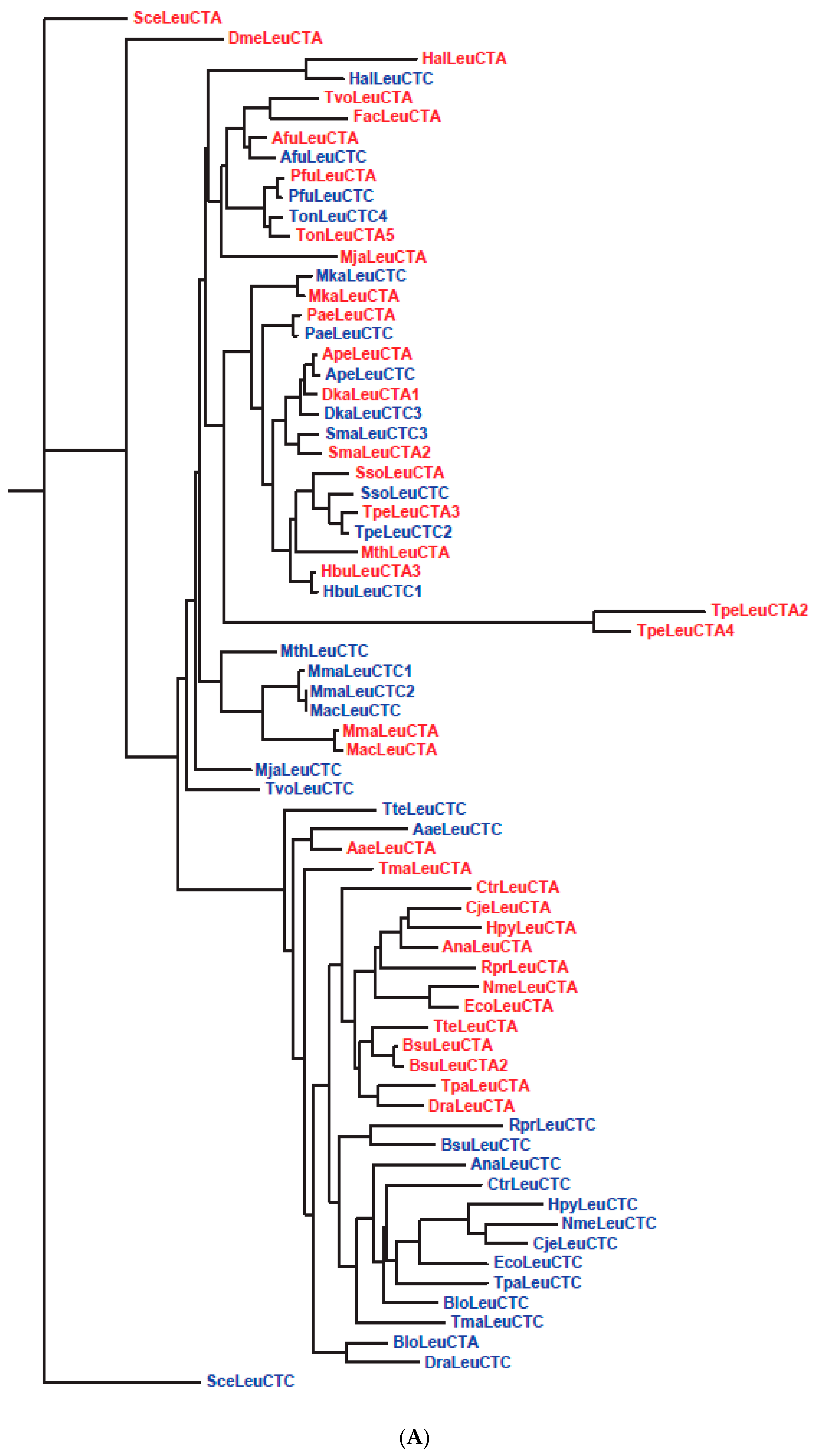
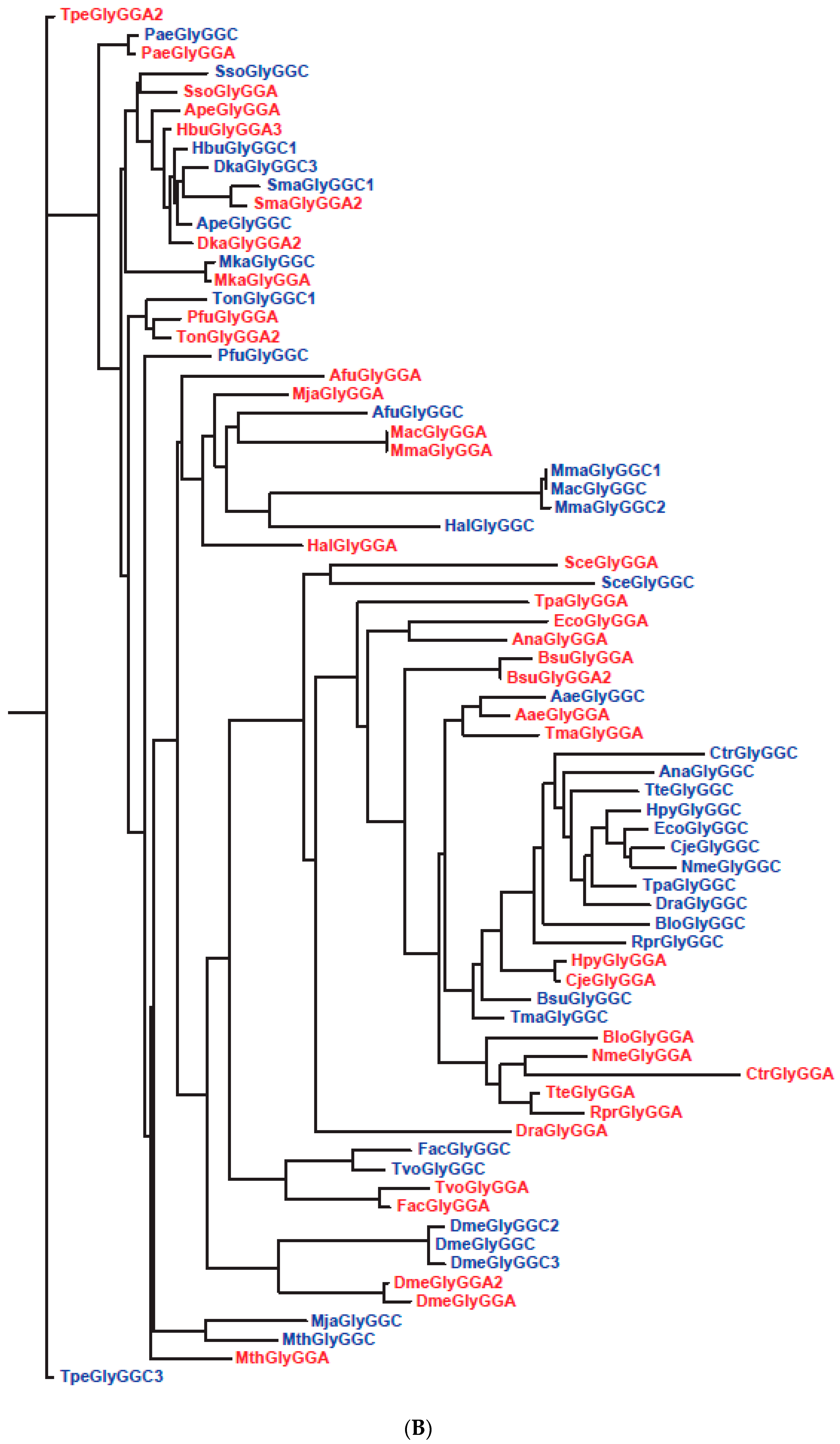
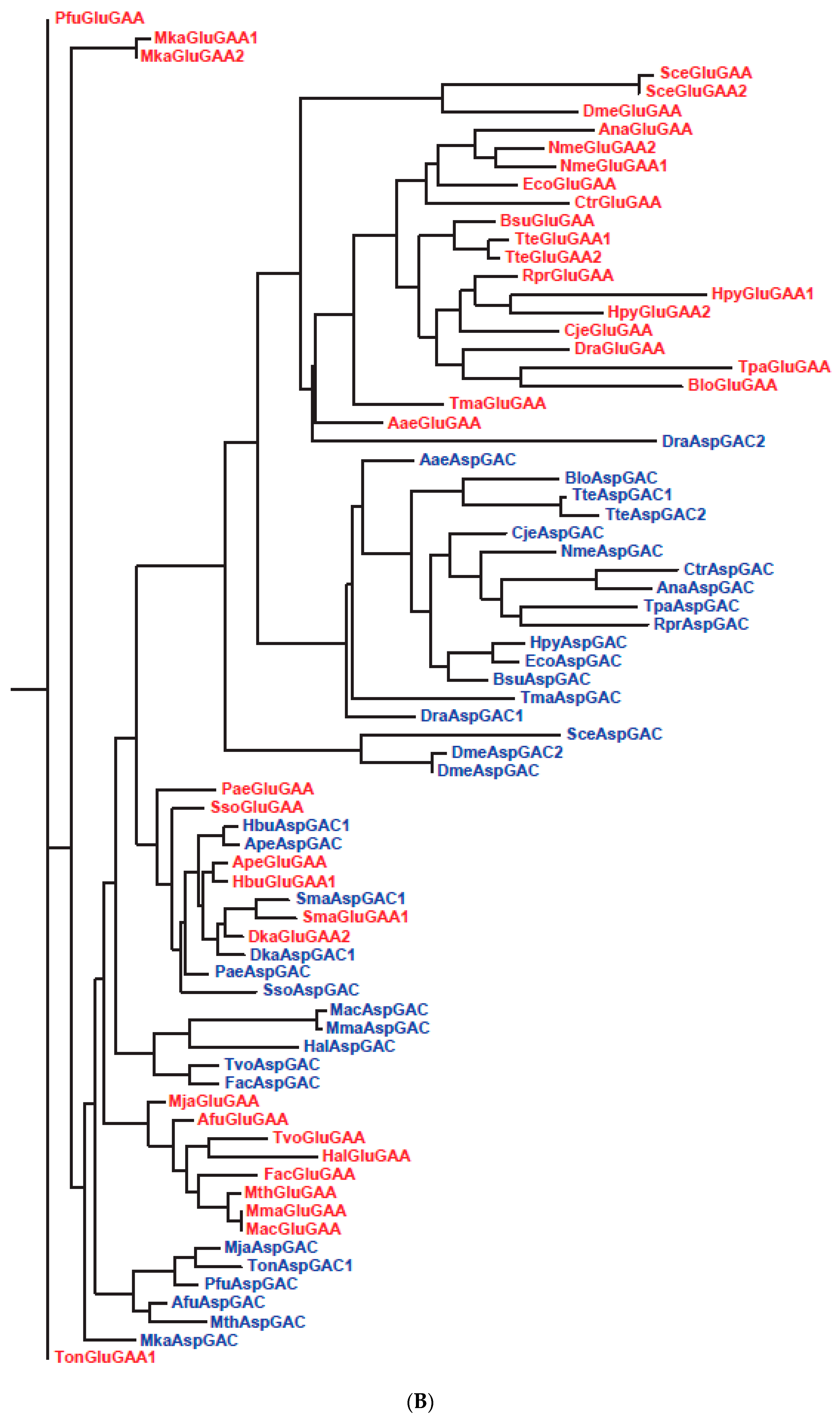
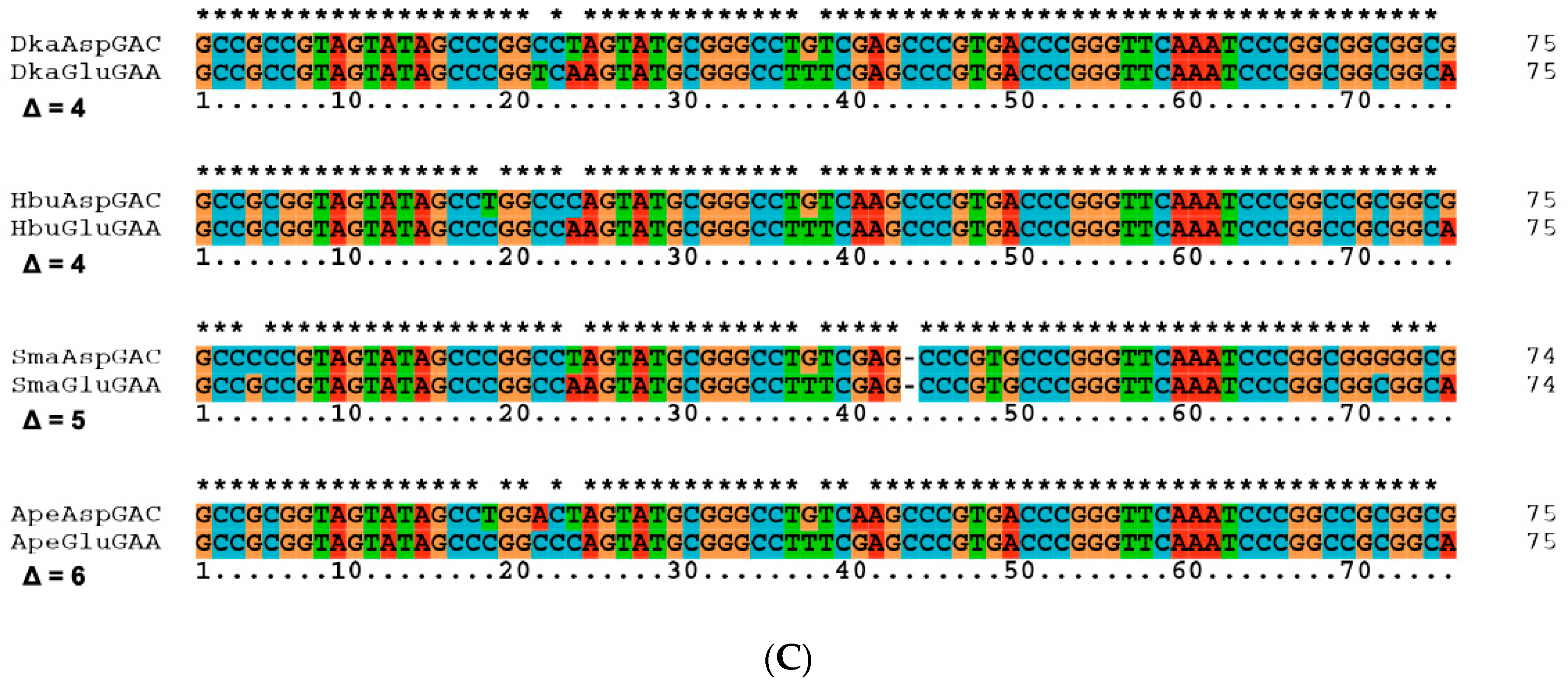
| 26 | Slow segregation of Asp and Glu tRNAs | A | Figure 4 and Figure S2 |
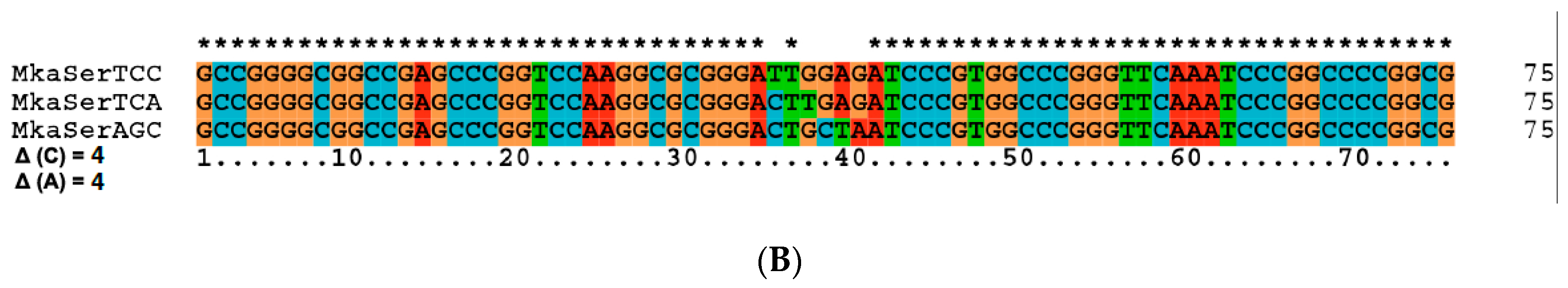
| 27 | Ser tRNA missing link | A, M | Figure 5 and Figure S3 |
| 28 | Gene ontology | A | [136] |
| 29 | Simplistic nucleoside modifications | A | [137,138] |
| Stage | Anticodon * | 3rd Codon Base Read | Main Users |
|---|---|---|---|
| I | UNN | U, C, A, G | Pre-LUCA organisms |
| II | GNN UNN | U, C A, G | Primitive Archaea |
| III | GNN UNN CNN | U, C A, G G | Majority Archaea |
| IV | GNN UNN CNN INN | U, C A, G G U, C, A | Bacteria, Eukarya |
| V | UNN | U, C, A, G in 1aa boxes | Mitochondria, chloroplasts, Mycoplasma, Stretoococcus, Borrelia, Lactococcus, etc. |
| Type * | Insertion | Altered Site | System | Ref. |
|---|---|---|---|---|
| o-Synthetic | NCAA | Proteome-wide | B. subtilis LC33, LC88, E.coli B7-3: 4FTrp, 5FTrp, 6FTrp; E. coli MT16-20: [3,2]Tpa | [178,180,186,187,188,189] |
| m-Synthetic | NCAA | Proteome-wide | B. subtilis HR15, HR23: 4FTrp | [178,180] |
| o-Synthetic | NCAA | Specific sites | E. coli, C. elegans etc.: p-aminoPhe, p-azidoPhe etc. | [191,192,193,194,195,196,197,198,199,200,201,202,203,204,205,206,207,208] |
| m-Synthetic | NCAA | Specific sites | E. coli C321ΔA: biphenylPhe etc.; E. coli thyA R126L: azaLeu | [209,210,211] |
| o-Synthetic | NCDN | Genome-wide | E.coli CLU5: 5-chloroU | [212,214] |
| m-Synthetic | NCDN | Genome-wide | E. coli CLU5 variant: 5-chloroU | [213] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wong, J.T.-F.; Ng, S.-K.; Mat, W.-K.; Hu, T.; Xue, H. Coevolution Theory of the Genetic Code at Age Forty: Pathway to Translation and Synthetic Life. Life 2016, 6, 12. https://doi.org/10.3390/life6010012
Wong JT-F, Ng S-K, Mat W-K, Hu T, Xue H. Coevolution Theory of the Genetic Code at Age Forty: Pathway to Translation and Synthetic Life. Life. 2016; 6(1):12. https://doi.org/10.3390/life6010012
Chicago/Turabian StyleWong, J. Tze-Fei, Siu-Kin Ng, Wai-Kin Mat, Taobo Hu, and Hong Xue. 2016. "Coevolution Theory of the Genetic Code at Age Forty: Pathway to Translation and Synthetic Life" Life 6, no. 1: 12. https://doi.org/10.3390/life6010012