Integrated Framework of the Immune-Defense Transcriptional Signatures in the Arabidopsis Shoot Apical Meristem

 , and
, and
Abstract
:1. Introduction
2. Results
2.1. Acquisition of the Arabidopsis Transcriptome Datasets Pertaining to SAM Cellular Populations and PAMPs-Treated Mesophyll Cells
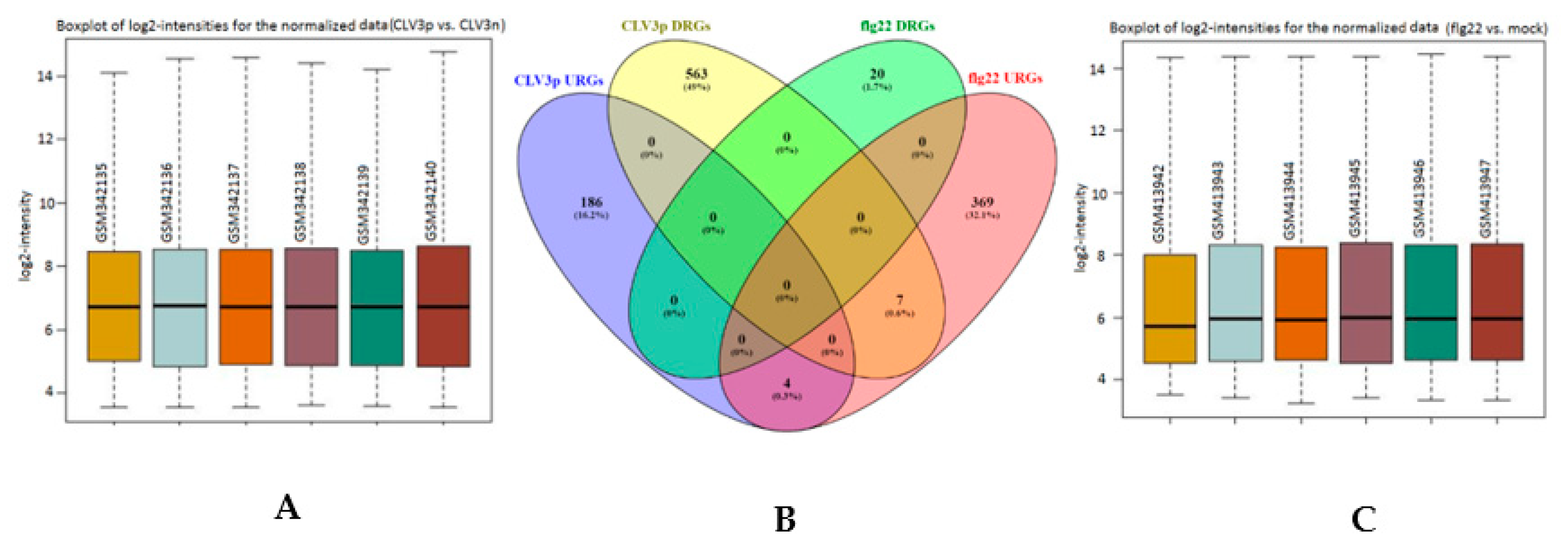
2.2. Normalization and Analysis of the SAM Cellular Transcriptomes and Functional Categorization of the DEGs (Differentially Expressed Genes)
2.3. GO-Based Enrichment Analysis of the SAM-Cellular Transcriptome for Immune-Related Functions
2.4. Transcriptome Guided SAM Interactome and Insights Into the Proteins of Immune-Related DEGs of the SAM-Cellular Populations
2.5. Transcriptional Basis of the Arabidopsis CLV3p-Triggered Genes in the SAM and its Comparison with flg22-Mediated Gene Expression in the Mesophyll Cells
3. Discussion
4. Material and Methods
4.1. Source of Transcriptomes (Gene Expression Data/Profiles): Gene Expression Omnibus (GEO)
4.2. R Packages
4.3. Voronoi Tree-Maps
4.4. The Gene Ontology Resource
4.5. CCSB Interactome Database
4.6. Cytoscape 3.7.1 and NetworkAnalyzer
4.7. Data Preprocessing, Analysis, and Filtering
4.8. Pathway Enrichment Analysis
4.9. Gene Ontology Enrichment Analysis for Immune-Related Processes
4.10. Network Construction, Visualization, and Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Szövényi, P.; Waller, M.; Kirbis, A. Evolution of the plant body plan. Curr. Top. Dev. Biol. 2019, 131, 1–34. [Google Scholar] [PubMed]
- Busch, W.; Miotk, A.; Ariel, F.D.; Zhao, Z.; Forner, J.; Daum, G.; Suzaki, T.; Schuster, C.; Schultheiss, S.J.; Leibfried, A.; et al. Transcriptional control of a plant stem cell niche. Dev. Cell 2010, 18, 841–853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aichinger, E.; Kornet, N.; Friedrich, T.; Laux, T. Plant stem cell niches. Annu. Rev. Plant Biol. 2012, 63, 615–636. [Google Scholar] [CrossRef] [PubMed]
- Hwang, I.; Sheen, J.; Muller, B. Cytokinin signaling networks. Annu. Rev. Plant Biol. 2012, 63, 353–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.; Chah, O.K.; Sheen, J. Stem-cell-triggered immunity through CLV3p-FLS2 signalling. Nature 2011, 473, 376–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.; Khatri, A.; Plotnikov, J.M.; Zhang, X.C.; Sheen, J. Complexity in differential peptide-receptor signaling: Response to Segonzac et Al. and Mueller et Al. commentaries. Plant Cell 2012, 24, 3177–3185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.; Chah, O.K.; Plotnikov, J.; Sheen, J. Stem cell signaling in immunity and development. Cold Spring Harb. Symp. Quant. Biol. 2012, 77, 75–81. [Google Scholar] [CrossRef]
- Naseem, M.; Wolfling, M.; Dandekar, T. Cytokinins for immunity beyond growth, galls and green islands. Trends Plant Sci. 2014, 19, 481–484. [Google Scholar] [CrossRef]
- Perales, M.; Reddy, G.V. Stem cell maintenance in shoot apical meristems. Curr. Opin. Plant Biol. 2012, 15, 10–16. [Google Scholar] [CrossRef]
- Song, X.F.; Yu, D.L.; Xu, T.T.; Ren, S.C.; Guo, P.; Liu, C.M. Contributions of individual amino acid residues to the endogenous CLV3 function in shoot apical meristem maintenance in Arabidopsis. Mol. Plant. 2012, 515–523. [Google Scholar] [CrossRef] [Green Version]
- Yadav, R.K.; Girke, T.; Pasala, S.; Xie, M.; Reddy, G.V. Gene expression map of the Arabidopsis shoot apical meristem stem cell niche. Proc. Natl. Acad. Sci. USA 2009, 106, 941–946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yadav, R.K.; Tavakkoli, M.; Xie, M.; Girke, T.; Reddy, G.V. A high-resolution gene expression map of the Arabidopsis shoot meristem stem cell niche. Development 2014, 141, 2735–2744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurakawa, T.; Ueda, N.; Maekawa, M.; Kobayashi, K.; Kojima, M.; Nagato, Y.; Sakakibara, H.; Kyozuka, J. Direct control of shoot meristem activity by a cytokinin-activating enzyme. Nature 2007, 445, 652–655. [Google Scholar] [CrossRef]
- Nishimura, C.; Ohashi, Y.; Sato, S.; Kato, T.; Tabata, S.; Ueguchi, C. Histidine kinase homologs that act as cytokinin receptors possess overlapping functions in the regulation of shoot and root growth in Arabidopsis. Plant Cell 2004, 16, 1365–1377. [Google Scholar] [CrossRef] [Green Version]
- Bartrina, I.; Otto, E.; Strnad, M.; Werner, T.; Schmulling, T. Cytokinin regulates the activity of reproductive meristems, flower organ size, ovule formation, and thus seed yield in Arabidopsis thaliana. Plant Cell 2011, 23, 69–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.; Li, L.; Macho, A.P.; Han, Z.; Hu, Z.; Zipfel, C.; Zhou, J.-M.; Chai, J. Structural basis for flg22-induced activation of the Arabidopsis FLS2-BAK1 immune complex. Science 2013, 342, 624–628. [Google Scholar] [CrossRef]
- Jones, J.D.; Dangl, J.L. The plant immune system. Nature 2006, 444, 323–329. [Google Scholar] [CrossRef] [Green Version]
- Mueller, K.; Chinchilla, D.; Albert, M.; Jehle, A.K.; Kalbacher, H.; Boller, T.; Felix, G. Contamination risks in work with synthetic peptides: Flg22 as an example of a pirate in commercial peptide preparations. Plant Cell 2012, 24, 3193–3197. [Google Scholar] [CrossRef] [Green Version]
- Segonzac, C.; Nimchuk, Z.L.; Beck, M.; Tarr, P.T.; Robatzek, S.; Meyerowitz, E.M.; Zipfel, C. The shoot apical meristem regulatory peptide CLV3 does not activate innate immunity. Plant Cell 2012, 24, 3186–3192. [Google Scholar] [CrossRef] [Green Version]
- Asai, T.; Tena, G.; Plotnikova, J.; Willmann, M.R.; Chiu, W.L.; Gomez-Gomez, L.; Boller, T.; Ausubel, F.M.; Sheen, J. MAP kinase signalling cascade in Arabidopsis innate immunity. Nature 2002, 415, 977–983. [Google Scholar] [CrossRef]
- Boller, T.; Felix, G. A renaissance of elicitors: Perception of microbe-associated molecular patterns and danger signals by pattern-recognition receptors. Annu. Rev. Plant Biol. 2009, 60, 379–406. [Google Scholar] [CrossRef] [PubMed]
- Boudsocq, M.; Willmann, M.R.; McCormack, M.; Lee, H.; Shan, L.; He, P.; Bush, J.; Cheng, S.H.; Sheen, J. Differential innate immune signaling via Ca sensor protein kinases. Nature 2010, 464, 418–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edgar, R.; Domrachev, M.; Lash, A.E. Gene expression omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for functional genomics data sets-update. Nucleic Acids Res. 2013, 41, D991–D995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, B.S.; Irizarry, R.A. A framework for oligonucleotide microarray preprocessing. Bioinformatics 2010, 26, 2363–2367. [Google Scholar] [CrossRef]
- Bolstad, B.M.; Irizarry, R.A.; Astrand, M.; Speed, T.P. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics 2003, 19, 185–193. [Google Scholar] [CrossRef] [Green Version]
- Irizarry, R.A.; Bolstad, B.M.; Collin, F.; Cope, L.M.; Hobbs, B.; Speed, T.P. Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res. 2003, 31, e15. [Google Scholar] [CrossRef]
- Irizarry, R.A.; Hobbs, B.; Collin, F.; Beazer-Barclay, Y.D.; Antonellis, K.J.; Scherf, U.; Speed, T.P. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 2003, 4, 249–264. [Google Scholar] [CrossRef] [Green Version]
- Liebermeister, W.; Noor, E.; Flamholz, A.; Davidi, D.; Bernhardt, J.; Milo, R. Visual account of protein investment in cellular functions. Proc. Natl. Acad. Sci. USA 2014, 111, 8488–8493. [Google Scholar] [CrossRef] [Green Version]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The gene ontology consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The gene ontology, C. The gene ontology resource: 20 years and still GOing strong. Nucleic Acids Res. 2019, 47, D330–D338. [Google Scholar]
- Mi, H.; Huang, X.; Muruganujan, A.; Tang, H.; Mills, C.; Kang, D.; Thomas, P.D. PANTHER version 11: Expanded annotation data from gene ontology and Reactome pathways, and data analysis tool enhancements. Nucleic Acids Res. 2017, 45, D183–D189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Assenov, Y.; Ramirez, F.; Schelhorn, S.E.; Lengauer, T.; Albrecht, M. Computing topological parameters of biological networks. Bioinformatics 2008, 24, 282–284. [Google Scholar] [CrossRef] [Green Version]
- Klaus, B.; Reisenauer, S. An end to end workflow for differential gene expression using Affymetrix microarrays. F1000Research 2016, 5, 1384. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Okuno, Y.; Hattori, M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004, 32, 277–280. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
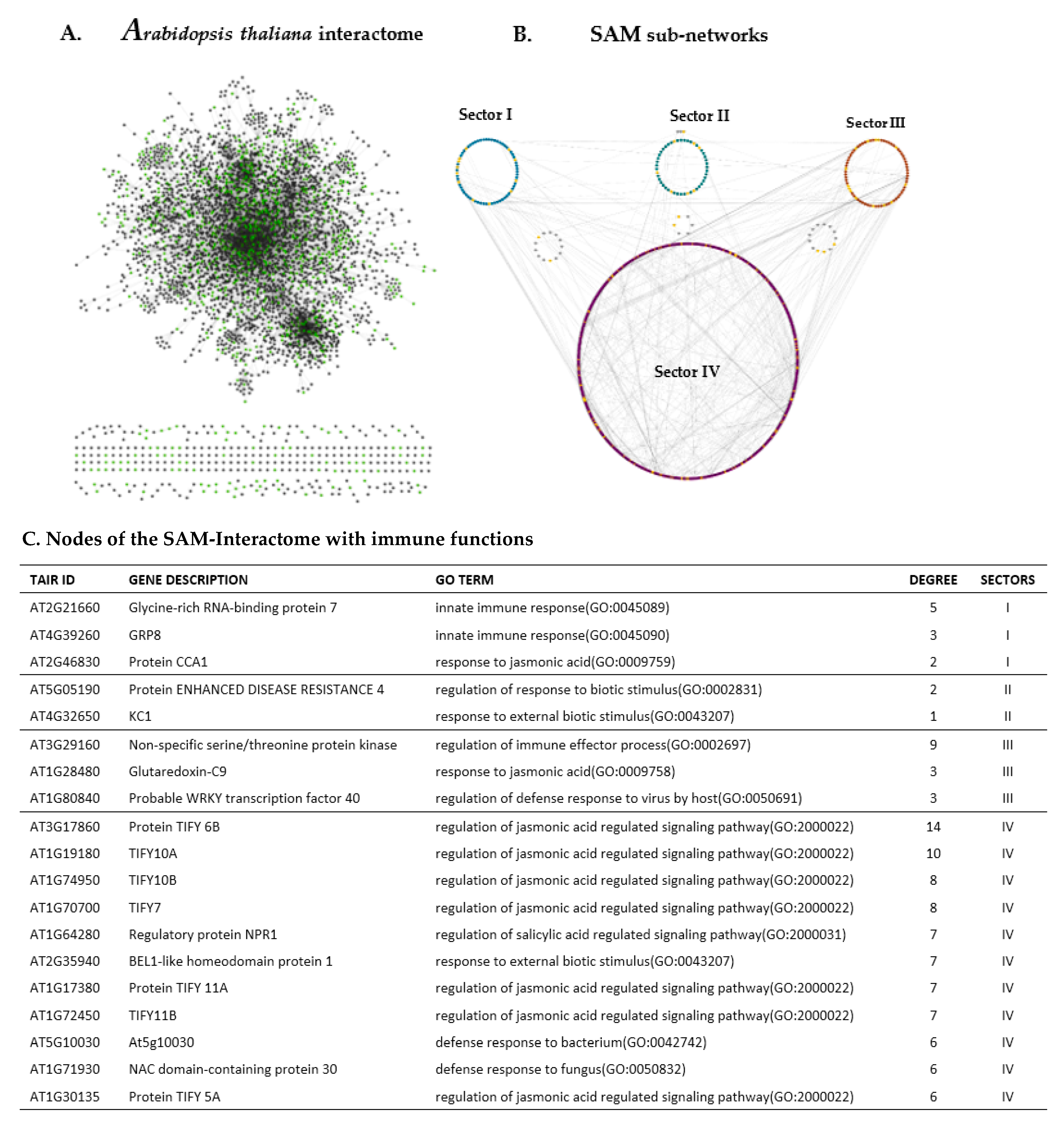{kind=link}
{kind=link}
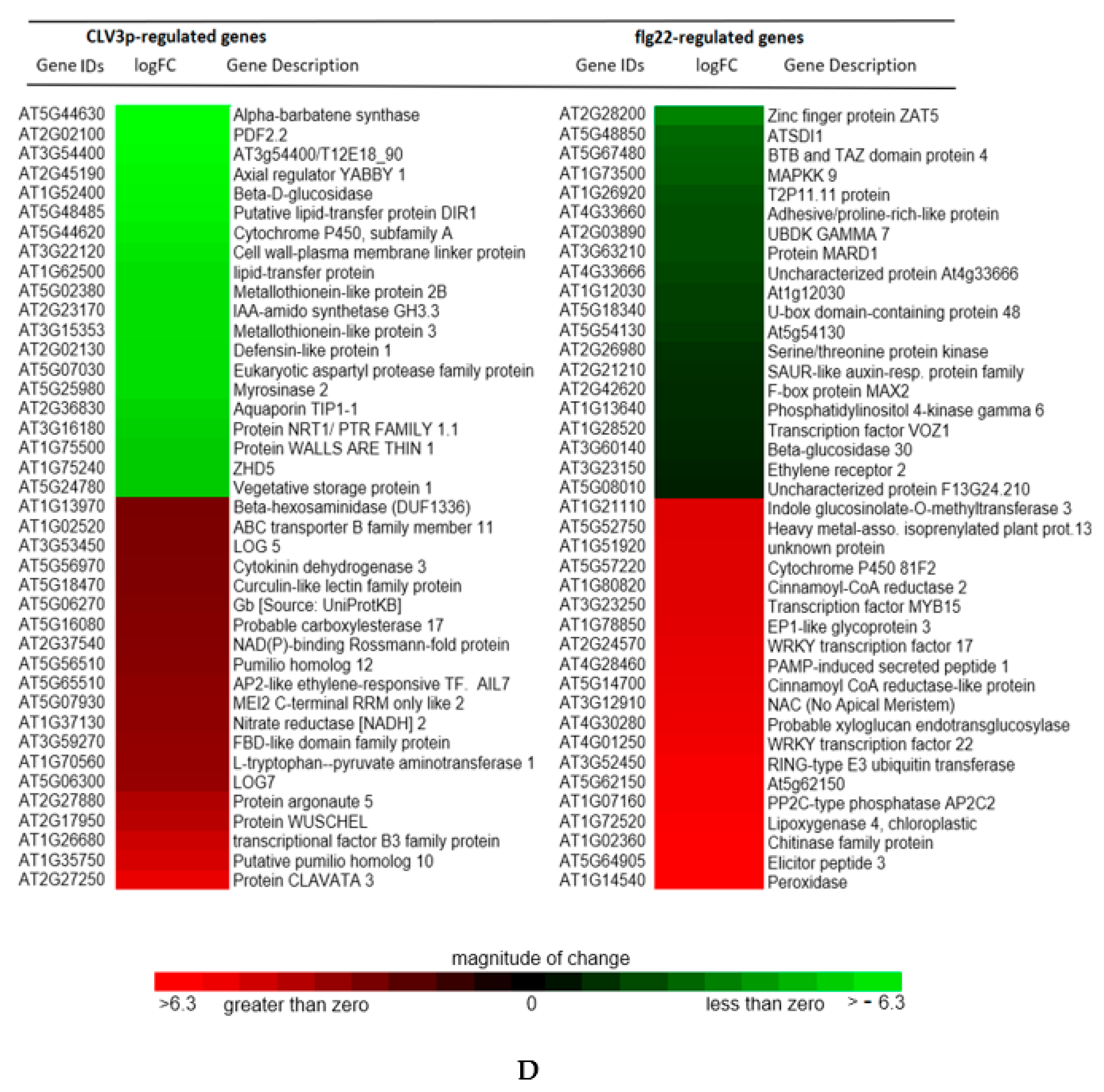{kind=link}
| Geo Series | Geo Sample | Cell Population | Cell Type | Source [Reference] |
|---|---|---|---|---|
| GSE13596 | GSM342135 | CLV3n | CLV3n cells lacks CLV3p cell type. | Yadav RK et al. 2009 [11] |
| GSE13596 | GSM342136 | CLV3n | CLV3n cells lacks CLV3p cell type. | Yadav RK et al. 2009 [11] |
| GSE13596 | GSM342137 | CLV3n | CLV3n cells lacks CLV3p cell type. | Yadav RK et al. 2009 [11] |
| GSE13596 | GSM342138 | CLV3p | central zone/stem cells | Yadav RK et al. 2009 [11] |
| GSE13596 | GSM342139 | CLV3p | central zone/stem cells | Yadav RK et al. 2009 [11] |
| GSE13596 | GSM342140 | CLV3p | central zone/stem cells | Yadav RK et al. 2009 [11] |
| GSE13596 | GSM342141 | FILp | organ primordia/peripheral zone cell type | Yadav RK et al. 2009 [11] |
| GSE13596 | GSM342142 | FILp | organ primordia/peripheral zone cell type | Yadav RK et al. 2009 [11] |
| GSE13596 | GSM342143 | FILp | organ primordia/peripheral zone cell type | Yadav RK et al. 2009 [11] |
| GSE13596 | GSM342144 | Prot_Ind | protoplast induced SAM | Yadav RK et al. 2009 [11] |
| GSE13596 | GSM342145 | Prot_Ind | protoplast induced SAM | Yadav RK et al. 2009 [11] |
| GSE13596 | GSM342146 | Prot_Unind | uninduced SAM | Yadav RK et al. 2009 [11] |
| GSE13596 | GSM342147 | Prot_Unind | uninduced SAM | Yadav RK et al. 2009 [11] |
| GSE13596 | GSM342148 | WUSp | L3 layer/rib zone cell type | Yadav RK et al. 2009 [11] |
| GSE13596 | GSM342149 | WUSp | L3 layer/rib zone cell type | Yadav RK et al. 2009 [11] |
| GSE16472 | GSM413942 | flg22cont | treated with mock solution | Boudsocq M et al. 2010 [22] |
| GSE16472 | GSM413943 | flg22 | treated with flg22 | Boudsocq M et al. 2010 [22] |
| GSE16472 | GSM413944 | flg22 | treated with flg22 | Boudsocq M et al. 2010 [22] |
| GSE16472 | GSM413945 | flg22cont | treated with mock solution | Boudsocq M et al. 2010 [22] |
| GSE16472 | GSM413946 | flg22 | treated with flg22 | Boudsocq M et al. 2010 [22] |
| GSE16472 | GSM413947 | flg22 | treated with flg22 | Boudsocq M et al. 2010 [22] |
| GSE28109 | GSM706470 | AtML1 | ubiquitous L1 layer/epidermal cell type | Yadav RK et al. 2014 [12] |
| GSE28109 | GSM706471 | AtML1 | ubiquitous L1 layer/epidermal cell type | Yadav RK et al. 2014 [12] |
| GSE28109 | GSM706472 | AtHB8 | shoot xylem/vasculature cell type | Yadav RK et al. 2014 [12] |
| GSE28109 | GSM706473 | AtHB8 | shoot xylem/vasculature cell type | Yadav RK et al. 2014 [12] |
| GSE28109 | GSM706474 | AtHB8 | shoot xylem/vasculature cell type | Yadav RK et al. 2014 [12] |
| GSE28109 | GSM706475 | HDG4 | L2 layer/subepidermal cell type | Yadav RK et al. 2014 [12] |
| GSE28109 | GSM706476 | HDG4 | L2 layer/subepidermal cell type | Yadav RK et al. 2014 [12] |
| GSE28109 | GSM706477 | HDG4 | L2 layer/subepidermal cell type | Yadav RK et al. 2014 [12] |
| GSE28109 | GSM706478 | HMG | meristematic L1 layer/epidermal cell type | Yadav RK et al. 2014 [12] |
| GSE28109 | GSM706479 | HMG | meristematic L1 layer/epidermal cell type | Yadav RK et al. 2014 [12] |
| GSE28109 | GSM706480 | HMG | meristematic L1 layer/epidermal cell type | Yadav RK et al. 2014 [12] |
| GSE28109 | GSM706481 | KAN1 | Abaxial organ boundaries/peripheral zone cell type | Yadav RK et al. 2014 [12] |
| GSE28109 | GSM706482 | KAN1 | Abaxial organ boundaries/peripheral zone cell type | Yadav RK et al. 2014 [12] |
| GSE28109 | GSM706483 | KAN1 | Abaxial organ boundaries/peripheral zone cell type | Yadav RK et al. 2014 [12] |
| GSE28109 | GSM706484 | LAS | Adaxial organ boundaries/peripheral zone cell type | Yadav RK et al. 2014 [12] |
| GSE28109 | GSM706485 | LAS | Adaxial organ boundaries/peripheral zone cell type | Yadav RK et al. 2014 [12] |
| GSE28109 | GSM706486 | S17 | shoot phloem/vasculature cell type | Yadav RK et al. 2014 [12] |
| GSE28109 | GSM706487 | S17 | shoot phloem/vasculature cell type | Yadav RK et al. 2014 [12] |
| GSE28109 | GSM706488 | S17 | shoot phloem/vasculature cell type | Yadav RK et al. 2014 [12] |
| Gene ID | Gene Description | Go Term | Cell Population |
|---|---|---|---|
| AT1G02450 | NIMIN1 | regulation of immune system(GO:0002682) | CLV3p |
| AT1G07000 | Exocyst subunit Exo70 family protein | regulation of defense response(GO:0031347) | CLV3p |
| AT1G37130 | Nitrate reductase [NADH] 2 | response to biotic stimulus(GO:0043207) | CLV3p |
| AT1G52400 | Beta-D-glucopyranosyl abscisate beta-glucosidase | defense response to fungus(GO:0050832) | CLV3p |
| AT2G27250 | CLAVATA 3 | innate immune response(GO:0045087) | CLV3p |
| AT2G35930 | E3 ubiquitin-protein ligase PUB23 | immune effector process(GO:0002252) | CLV3p |
| AT3G44300 | NIT2 | response to biotic stimulus(GO:0043207) | CLV3p |
| AT4G01610 | Cathepsin B-like protease 3 | defense response(GO:0006952) | CLV3p |
| AT4G24670 | Tryptophan aminotransferase-related protein 2 | defense response to bacterium(GO:0042742) | CLV3p |
| AT5G27420 | E3 ubiquitin-protein ligase ATL31 | innate immune response(GO:0045087) | CLV3p |
| AT1G31280 | Protein argonaute 2 | defense response to bacterium(GO:0042742) | HDG4 |
| AT1G74930 | Ethylene-responsive transcription factor ERF018 | defense response to organism(GO:0098542) | HDG4 |
| AT2G21660 | Glycine-rich RNA-binding protein 7 | innate immune response(GO:0045087) | HDG4 |
| AT4G16950 | Disease resistance protein RPP5 | defense response to fungus(GO:0050832) | HDG4 |
| AT4G34710 | Arginine decarboxylase 2 | response to jasmonic acid(GO:0009753) | HDG4 |
| AT4G39260 | GRP8 | innate immune response(GO:0045087) | HDG4 |
| AT5G15380 | DNA (cytosine-5)-methyltransferase DRM1 | defense response to fungus(GO:0050832) | HDG4 |
| AT5G65710 | LRR receptor-like serine/threonine-protein kinase | defense response to bacterium(GO:0042742) | HDG4 |
| AT5G66570 | Oxygen-evolving enhancer protein 1-1, chloroplastic | defense response to bacterium(GO:0042742) | HDG4 |
| AT1G11310 | MLO-like protein 2 | defense response to fungus(GO:0050832) | LAS |
| AT2G30750 | Cytochrome P450 71A12 | defense response to bacterium(GO:0042742) | LAS |
| AT2G46830 | Protein CCA1 | response to jasmonic acid(GO:0009753) | LAS |
| AT5G51630 | Disease resistance protein (TIR-NBS-LRR class) | defense response(GO:0006952) | LAS |
| Gene ID | Gene Description | Go Term | Cell Population |
|---|---|---|---|
| AT1G79090 | Protein PAT1 homolog | innate immune response(GO:0045087) | AtML1 |
| AT2G36890 | homeodomain-like superfamily protein | response to jasmonic acid(GO:0009753) | AtML1 |
| AT3G05710 | Syntaxin-43 | regulation of defense response(GO:0031347) | AtML1 |
| AT4G14720 | TIFY domain/Divergent CCT motif family protein | regulation of defense response(GO:0031347) | AtML1 |
| AT5G08280 | Porphobilinogen deaminase, chloroplastic | response to biotic stimulus(GO:0043207) | AtML1 |
| AT1G02360 | Chitinase family protein | response to biotic stimulus(GO:0043207) | HMG |
| AT1G55020 | Linoleate 9S-lipoxygenase 1 | response to jasmonic acid(GO:0009753) | HMG |
| AT1G71400 | Receptor-like protein 12 | defense response(GO:0006952) | HMG |
| AT2G24570 | WRKY transcription factor 17 | defense response to bacterium(GO:0042742) | HMG |
| AT2G26380 | Leucine-rich repeat (LRR) family protein | defense response(GO:0006952) | HMG |
| AT2G28790 | Pathogenesis-related thaumatin superfamily protein | response to biotic stimulus(GO:0043207) | HMG |
| AT4G32650 | KC1 | response to biotic stimulus(GO:0043207) | HMG |
| AT5G05190 | Protein ENHANCED DISEASE RESISTANCE 4 | response to biotic stimulus(GO:0002831) | HMG |
| AT5G23820 | MD-2-related lipid-recognition protein 3 | defense response(GO:0006952) | HMG |
| AT5G47910 | Respiratory burst oxidase homolog protein D | defense response to fungus(GO:0050832) | HMG |
| Gene ID | Gene Description | Go Term | Cell Population |
|---|---|---|---|
| AT1G02450 | NIMIN1 | regulation of immune process(GO:0002682) | FILp |
| AT1G20510 | 4-coumarate--CoA ligase-like 5 | jasmonic acid biosynthetic process(GO:0009695) | FILp |
| AT1G28480 | Glutaredoxin-C9 | response to jasmonic acid(GO:0009753) | FILp |
| AT1G73620 | Pathogenesis-related thaumatin superfamily protein | response to biotic stimulus(GO:0043207) | FILp |
| AT2G26440 | Probable pectinesterase/pectinesterase inhibitor 12 | response to biotic stimulus(GO:0043207) | FILp |
| AT2G41370 | Regulatory protein NPR5 | JA mediated signaling pathway(GO:0009864) | FILp |
| AT3G25250 | Serine/threonine-protein kinase OXI1 | defense response(GO:0006952) | FILp |
| AT3G57130 | Regulatory protein NPR6 | jasmonic acid mediated signaling (GO:0009864) | FILp |
| AT5G48485 | Putative lipid-transfer protein DIR1 | innate immune response(GO:0045087) | FILp |
| AT5G49520 | Probable WRKY transcription factor 48 | defense response to bacterium(GO:0042742) | FILp |
| AT1G02205 | Fatty acid hydroxylase superfamily | defense response to fungus(GO:0050832) | KAN1 |
| AT1G73620 | Pathogenesis-related thaumatin superfamily protein | response to biotic stimulus(GO:0043207) | KAN1 |
| AT1G75830 | Defensin-like protein 13 | defense response(GO:0006952) | KAN1 |
| AT3G01500 | Beta carbonic anhydrase 1, chloroplastic | defense response to bacterium(GO:0042742) | KAN1 |
| AT3G13790 | insoluble isoenzyme CWINV1 | defense response to fungus(GO:0050832) | KAN1 |
| AT3G26470 | Powdery mildew resistance protein containing protein | defense response to fungus(GO:0050832) | KAN1 |
| AT3G45140 | Lipoxygenase 2, chloroplastic | response to jasmonic acid(GO:0009753) | KAN1 |
| AT4G02410 | L-type lectin-domain containing receptor kinase IV.3 | defense response to bacterium(GO:0042742) | KAN1 |
| AT4G23670 | AT4G23670 protein | defense response to bacterium(GO:0042742) | KAN1 |
| AT5G14740 | Beta carbonic anhydrase 2, chloroplastic | defense response to bacterium(GO:0042742) | KAN1 |
| AT1G65390 | Protein PHLOEM PROTEIN 2-LIKE A5 | defense response(GO:0006952) | WUS |
| AT1G73805 | Protein SAR DEFICIENT 1 | response to biotic stimulus(GO:0002833) | WUS |
| AT3G29160 | Non-specific serine/threonine protein kinase | immune effector process(GO:0002697) | WUS |
| AT5G39580 | Peroxidase 62 | defense response to fungus(GO:0050832) | WUS |
| AT5G52450 | Protein DETOXIFICATION | response to biotic stimulus(GO:0043207) | WUS |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Naseem, M.; Osmanoğlu, Ö.; Kaltdorf, M.; Alblooshi, A.A.M.A.; Iqbal, J.; Howari, F.M.; Srivastava, M.; Dandekar, T. Integrated Framework of the Immune-Defense Transcriptional Signatures in the Arabidopsis Shoot Apical Meristem. Int. J. Mol. Sci. 2020, 21, 5745. https://doi.org/10.3390/ijms21165745
Naseem M, Osmanoğlu Ö, Kaltdorf M, Alblooshi AAMA, Iqbal J, Howari FM, Srivastava M, Dandekar T. Integrated Framework of the Immune-Defense Transcriptional Signatures in the Arabidopsis Shoot Apical Meristem. International Journal of Molecular Sciences. 2020; 21(16):5745. https://doi.org/10.3390/ijms21165745
Chicago/Turabian StyleNaseem, Muhammad, Özge Osmanoğlu, Martin Kaltdorf, Afnan Ali M. A. Alblooshi, Jibran Iqbal, Fares M. Howari, Mugdha Srivastava, and Thomas Dandekar. 2020. "Integrated Framework of the Immune-Defense Transcriptional Signatures in the Arabidopsis Shoot Apical Meristem" International Journal of Molecular Sciences 21, no. 16: 5745. https://doi.org/10.3390/ijms21165745