The Functions of ZIP8, ZIP14, and ZnT10 in the Regulation of Systemic Manganese Homeostasis


Department of Nutritional Sciences, The University of Arizona, Tucson, AZ 85721, USA
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2020, 21(9), 3304; https://doi.org/10.3390/ijms21093304
Submission received: 16 April 2020
/
Revised: 4 May 2020
/
Accepted: 5 May 2020
/
Published: 7 May 2020
(This article belongs to the Section Molecular Biology)
Abstract
:As an essential nutrient, manganese is required for the regulation of numerous cellular processes, including cell growth, neuronal health, immune cell function, and antioxidant defense. However, excess manganese in the body is toxic and produces symptoms of neurological and behavioral defects, clinically known as manganism. Therefore, manganese balance needs to be tightly controlled. In the past eight years, mutations of genes encoding metal transporters ZIP8 (SLC39A8), ZIP14 (SLC39A14), and ZnT10 (SLC30A10) have been identified to cause dysregulated manganese homeostasis in humans, highlighting the critical roles of these genes in manganese metabolism. This review focuses on the most recent advances in the understanding of physiological functions of these three identified manganese transporters and summarizes the molecular mechanisms underlying how the loss of functions in these genes leads to impaired manganese homeostasis and human diseases.
1. Introduction
Manganese is an abundant element in the Earth’s crust and is naturally found in a variety of minerals [1]. The most common natural form of manganese is manganese dioxide (MnO2), also known as pyrolusite, which contains about 60–63% manganese [2]. As a transition metal, manganese exists in different oxidation states, ranging from Mn3+ to Mn7+, with Mn2+ and Mn3+ being the most common forms found in biological species [3]. Manganese is widely used in modern industries, such as the production of steel, battery cathodes, soft ferrites for electronics, fertilizers, water treatment chemicals, colorant for automobile undercoating, bricks, frits, glass, textiles, and tiles [4,5,6].
As an essential nutrient, manganese is required for the function of numerous enzymes, including arginase, glycosyltransferases, manganese superoxide dismutase (MnSOD), phosphoenolpyruvate carboxykinase, prolidase, and pyruvate carboxylase [7,8,9,10]. Through these enzymes, manganese plays an important role in human health. Arginase is a manganese-containing enzyme that mediates the conversion of L-arginine to L-ornithine and urea in the last step of the urea cycle pathway [11,12]. The urea cycle is required for protection against the accumulation of excess ammonia. Glycosyltransferases require manganese as a cofactor and are involved in the synthesis of proteoglycans, including mucopolysaccharides, which are necessary for the production of cartilage and bone [13]. MnSOD is a manganese-dependent metalloenzyme that is located in the mitochondria of cells. The primary function of MnSOD is to facilitate the conversion of superoxide radicals to hydrogen peroxide, which is a vital antioxidant defense for nearly all cells [14]. Prolidase is a manganese-activated enzyme that recycles the amino acid proline for collagen synthesis and cell growth [15]. Collagen assembly is required in the process of wound healing. Phosphoenolpyruvate carboxykinase is vital in carbohydrate synthesis from pyruvate and plays an essential role in gluconeogenesis [16].
Deficiency in manganese has been associated with a number of health consequences, including impaired cognitive function, asthma, osteoporosis, and dyslipidemia. Since manganese is present in a broad range of foods and sufficient manganese can be obtained through diet under normal conditions, reports of dietary manganese deficiency are rare [17]. Manganese toxicity, on the other hand, has been observed in individuals drinking contaminated water, in drug addicts who use manganese-contaminated methcathinone, and in children or adults receiving prolonged parenteral nutrition [18]. Moreover, excessive occupational exposure may also occur, which can lead to manganese overload with severe side effects, clinically known as manganism. Individuals who have an increased risk of being exposed to higher than normal levels of manganese include workers in the production of iron and steel, dry cell batteries, fuel oil additives and antiknock agents, glasses, potassium permanganate, fungicides, and leather tanning [19,20,21]. The neurological symptoms of manganism consist of reduced response speed, irritability, intellectual deficits, mood changes, compulsive behaviors in the initial stages of the disorder, and more prominent and irreversible extrapyramidal dysfunction resembling Parkinson’s disease upon protracted exposure [22,23]. Classic clinical features include a mask-like face, limb rigidity, mild tremors, gait disturbance, cock-like walk, slurred speed, excessive salivation, sweating, and disruption in balance [24,25].
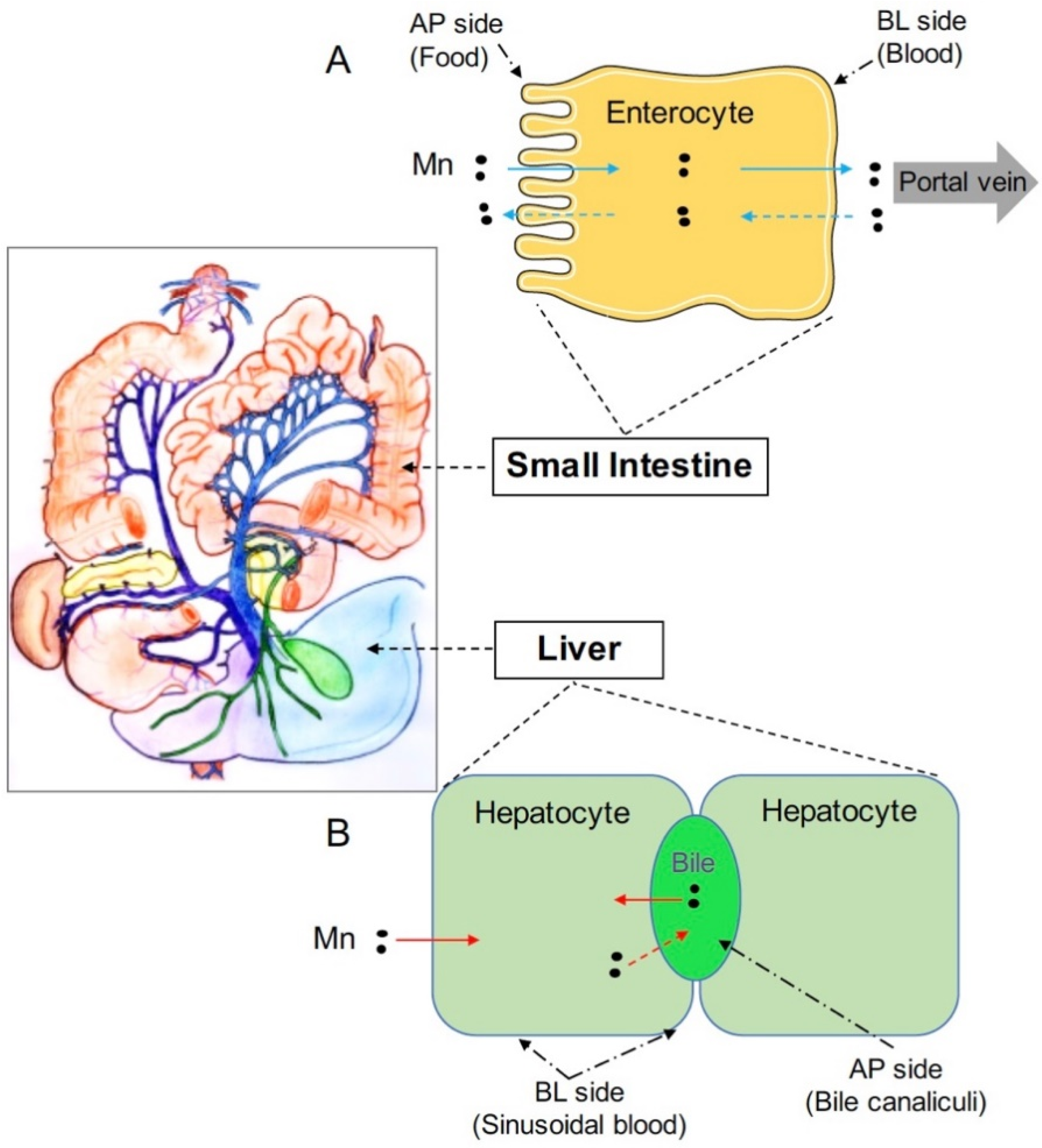
Tight homeostatic control is required to meet the dual challenge of avoiding manganese deficiency and preventing manganese overload. At the systemic level, this control is maintained mainly by the intestine and the liver [26,27,28,29]. The intestine regulates dietary manganese absorption; whereas, the liver clears manganese from the blood and secretes this metal as a bile conjugate for subsequent intestinal reabsorption or fecal excretion (Figure 1). At the cellular level, metal transporters responsible for manganese delivery into or out of the cells play important roles in this control. Within the past eight years, mutations of genes encoding metal transporters ZIP8 (SLC39A8), ZIP14 (SLC39A14), and ZnT10 (SLC30A10) have been reported to cause dysregulated manganese homeostasis [18,30,31,32,33], demonstrating that the primary physiological function of these metal transporters is to regulate body manganese metabolism. Among these three proteins, ZIP8 and ZIP14 are members of the Zrt- and Irt-like family proteins (ZIP), and ZnT10 is a member of the Zinc transporter (ZnT) family proteins. In general, the ZIP family proteins control the influx of metals from extracellular fluid or intracellular vesicles into the cytoplasm of the cell; whereas, the ZnT proteins are responsible for the efflux of metals from the cytosol into the extracellular fluid or intracellular vesicles [34]. In this review, we focus on the latest advances in the understanding of the physiological functions of these three manganese transporters and discuss the molecular mechanisms underlying dysregulated systemic manganese homeostasis caused by the deficiency of these genes.
2. ZIP8
2.1. ZIP8 Mutations and Manganese Deficiency
ZIP8 (also known as SLC39A8, solute carrier 39 family, member 8) is a member of the ZIP metal-ion transporter proteins. The ZIP family takes the name from zinc-regulated transporter (ZRT), iron-regulated transporter (IRT)-like proteins. The ZIP8 gene encodes a multi-transmembrane protein capable of transporting several divalent cations, including cadmium, zinc, iron, and manganese [35,36,37]. The identification of human diseases associated with ZIP8 mutations promoted the understanding of the physiological function of this transporter. In 2015, two clinical studies identified ZIP8 mutations in humans [30,31]. One study described recurrent homozygous ZIP8 mutations that led to manganese deficiency in a group of children [30]. The analysis of metal levels revealed non-detectable to extremely low levels of blood manganese in affected individuals. The clinical phenotypes of these children include intellectual disability, developmental delay, hypotonia, strabismus, cerebellar atrophy, and variable short stature. Another study identified compound heterozygous ZIP8 mutations in two unrelated patients with a congenital disorder of glycosylation (CDG) [31]. In this second study, manganese was not detectable in either the blood or urine samples from both patients. Because manganese is essential for glycosyltransferases, glycosylation profiles from both patients confirmed a severe defect in the glycosylation of transferrin, which was used as a marker for plasma proteins. Similar to the first report, clinical phenotypes of both patients include deformities in the skull, severe seizures, short limbs, psychomotor retardation, and hearing loss. In 2017, a homozygous point mutation of ZIP8 was identified in two siblings to cause severe manganese deficiency, presenting as CDG and Leigh-like syndrome, with standard features of manganese deficiency that included developmental delay, brain atrophy, hypotonia, and seizures [38].
So far, a majority of the identified ZIP8 mutations stem from consanguineous families, with both parents being heterozygous carriers of the same mutation as that identified in their children. The disease onset occurs at birth or very early during childhood. The detailed information about identified cases of human ZIP8 mutations are listed in Table 1.
2.2. The Molecular Mechanism Underlying ZIP8 Loss and Manganese Deficiency
ZIP8 is ubiquitously expressed throughout the body [39,40,41,42] and detected in almost all cell types with localization to the apical membrane of kidney epithelial cells [43], pulmonary alveolar epithelial cells [44,45], and hepatocytes [46]. In mouse fetal fibroblast cells (MFF cells), retrovirus-introduced ZIP8 functions maximally at pH 7.5 and transports both manganese and cadmium [37]. Overexpression of ZIP8 in HEK293 cells enhanced the cellular uptake of both iron and zinc from the culture media by 200% and 40%, respectively [47]. In Xenopus oocytes injected with ZIP8 cRNA, manganese serves as a strong inhibitor for Zn uptake, suggesting a primary role for ZIP8 in mediating manganese uptake into those cells [48]. These in vitro studies have suggested that ZIP8 can import a broad range of metals, including, zinc, iron, cadmium, and manganese.
Conventional whole-body knockout of Zip8 is embryonic lethal in mice [49]; therefore, to further examine ZIP8’s physiological function, tamoxifen-inducible global Zip8 knockout (Zip8-iKO) mice were generated, and metal concentrations in different organs of these mice were compared to that of wild-type (WT) animals. Zip8-iKO mice had markedly reduced manganese in multiple organs, including the liver, brain, kidney, and heart, and presented with decreased manganese-dependent arginase and β-1,4-galactosyltransferase activities [50], which is consistent with the observation that patients carrying ZIP8 mutations develop severe systemic manganese deficiency. Despite the ability of ZIP8 to transport iron and zinc in cell culture studies, the levels of tissue iron and zinc were not different in Zip8-iKO mice compared with that of the control animals, proving that the primary function of ZIP8 is to regulate manganese metabolism. Immunofluorescence analyses determined that ZIP8 is localized to the hepatocyte apical canalicular membrane, suggesting that hepatic ZIP8 may function to mediate manganese transport from the bile into hepatocytes.
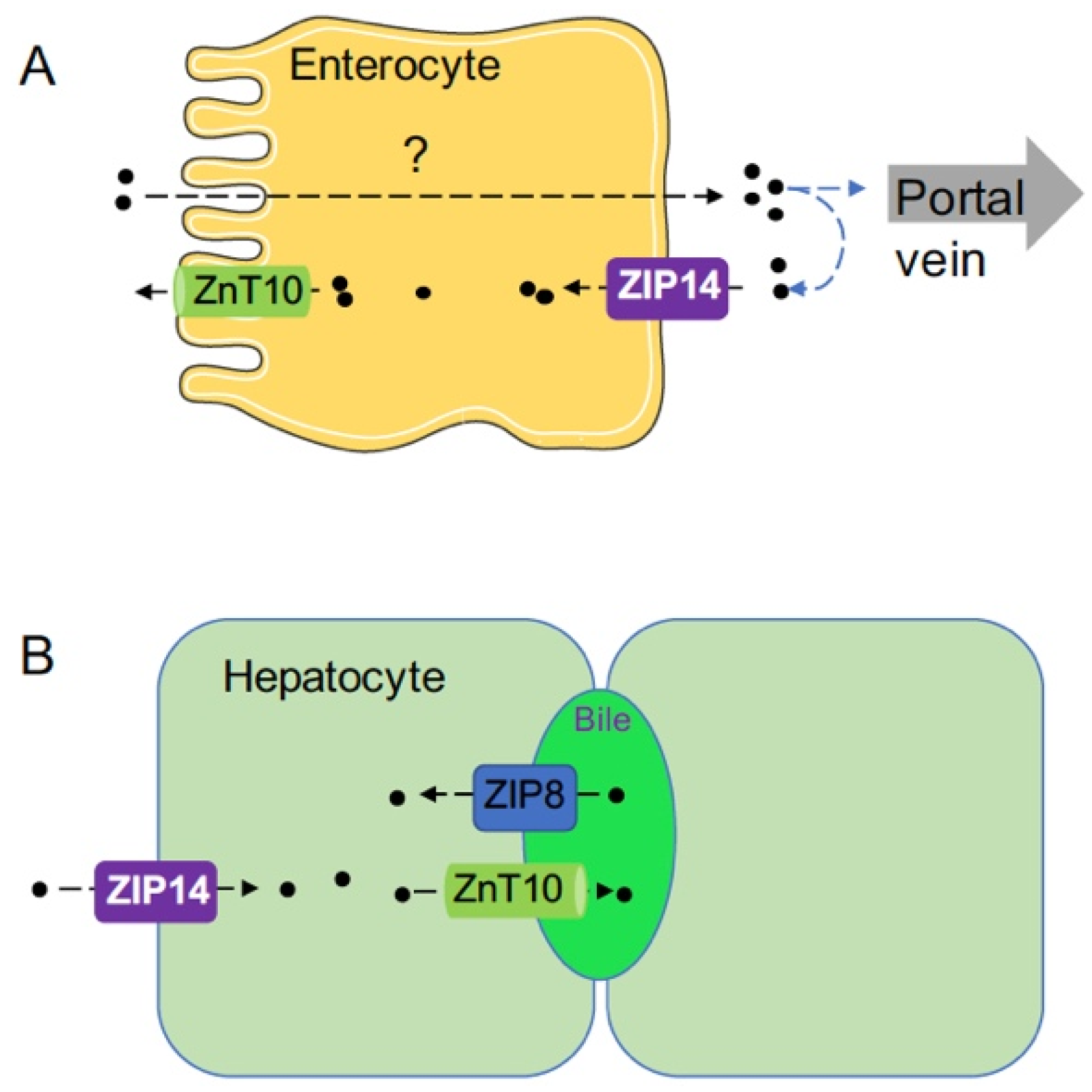
To determine the significance of hepatic ZIP8 in the regulation of whole-body manganese homeostasis, hepatocyte-specific Zip8 knockout (Zip8-L-KO) mice were created [50]. Similar to Zip8-iKO mice, Zip8-L-KO mice developed manganese deficiency in the liver, brain, kidney, and heart, suggesting that hepatic ZIP8 is required to regulate whole-body manganese homeostasis. Despite reduced whole-body manganese, Zip8-L-KO mice had a 55% increased level of manganese in the bile, supporting the role for ZIP8 in mediating transport of manganese from the bile into hepatocytes. Furthermore, liver-specific overexpression of ZIP8 through adeno-associated virus (AAV)-mediated gene delivery into WT mice resulted in a 76% decrease of bile manganese, an 87% increase of liver manganese, and a 94% increase of whole blood manganese, corroborating the view that higher than normal levels of ZIP8 will increase manganese accumulation and possibly lead to manganese overload, presumably through increased manganese reabsorption from the bile [50]. Taken together, these results from genetically modified mouse models demonstrate that ZIP8 localizes to the canalicular domain of hepatocytes to mediate transport of manganese from the bile into hepatocytes (Figure 2B). Thus, ZIP8 may allow reuptake of manganese from the bile when the liver senses deficiencies in manganese, providing one mechanism of manganese deficiency induced by the lack of ZIP8. Studies to examine ZIP8 expression under manganese deficiency and overload conditions are needed to further elucidate ZIP8’s function in regulating manganese homeostasis.
3. ZIP14
3.1. ZIP14 Mutations and Manganese Overload
ZIP14 (SLC39A14) was first identified as a gene responsible for cellular zinc influx based on the observation that overexpression of ZIP14 in Chinese hamster ovary (CHO) cells stimulates the uptake of zinc [51]. Later studies using ZIP14 overexpression models in HEK293 cells, Sf9 insect cells, and Xenopus oocytes determined that ZIP14 can mediate the cellular influx of a broad scope of metals, including cadmium, zinc, iron and manganese [52,53,54]. Northern blot analysis detected ubiquitous expressions of ZIP14, with the highest expression in the liver [55]. Human multiple tissue expression array analysis showed that the tissues with high levels of ZIP14 include the liver and small intestine [51]. A recent genome-wide transcriptomics analysis by RNA-sequencing of human samples from healthy individuals revealed that the greatest ZIP14 abundance was in the liver, followed by the small intestine [41]. In mice, under normal environmental and dietary conditions, the highest expression of ZIP14 was found in the small intestine, followed by the liver [56,57].
The physiological function of ZIP14 became clear with the identification of human mutations. Patients carrying ZIP14 mutations developed manganese toxicity and early-onset dystonia [32,58,59,60,61]. These patients did not accumulate manganese in the liver and had normal liver function. The levels of other essential metals, including iron and zinc, appeared to be within the normal range, suggesting that the primary physiological role of ZIP14 is to regulate manganese homeostasis. Like patients with ZIP8 mutations, a majority of ZIP14 mutation cases stem from consanguineous families. The onset of the disease occurs at infancy or during early childhood and are evident with loss of developmental milestones, progressive dystonia, bulbar dysfunction, spasticity, limb contractures, scoliosis, and loss of independent ambulation, with some showing parkinsonian features of hypomimia, tremor, and bradykinesia. The detailed information about identified human ZIP14 mutations are listed in Table 2.
3.2. The Molecular Mechanism Underlying ZIP14 Deficiency and Manganese Toxicity
Significant insights regarding the disease mechanisms underlying manganese toxicity induced by ZIP14 loss have been gained from animal models with ZIP14 deficiency. ZIP14-deficient zebrafish hyper-accumulated manganese in the brain, but not in the liver, and presented with reduced locomotor activities [32]; whole-body Zip14 knockout mice (Zip14−/−) had markedly increased manganese levels in the blood and brain [62,63,64,65], resulting in impaired locomotor behavior, but had decreased liver manganese [62,64,65]. These results clearly demonstrate an indispensable role for ZIP14 in controlling systemic manganese homeostasis and suggest a model where lack of ZIP14 impairs manganese delivery to the liver and subsequent clearance through biliary excretion, which in turn lead to manganese accumulation in the blood and extrahepatic tissues, including the brain [32,62,63,65].
Under normal dietary conditions, hepatocyte-specific Zip14 knockout mice (Zip14-L-KO) had significantly decreased manganese in the liver, confirming the essential function for ZIP14 to import manganese to hepatocytes; however, even with reduced manganese uptake into the liver, Zip14-L-KO mice had normal manganese levels in the blood and other tissues [63,66]. These results indicate that although hepatic ZIP14 is required for manganese delivery to the liver, the impaired hepatobiliary manganese excretion alone does not induce manganese hyper-accumulation in individuals lacking functional ZIP14, and suggest that ZIP14 in non-hepatic tissues acts as the primary control for systemic manganese homeostasis during normal physiological situations [63,66].
In addition to the liver, ZIP14 is highly expressed in the small intestine. To examine ZIP14’s function in enterocytes, ZIP14 knockout Caco-2 cells were created [66]. Caco-2 cells have many properties of absorptive enterocytes and have been widely used as a model to examine nutrient transport across the intestinal epithelium. Experiments using the Caco-2 Transwell system have indicated that ZIP14-deletion significantly increased apical-to-basolateral manganese transport. Mechanistically, it has been demonstrated that ZIP14 functions at the basolateral membrane of enterocytes to mediate the re-uptake of freshly released manganese, suggesting a role for ZIP14 in limiting manganese absorption and that a lack-of-ZIP14 in the intestine will increase the net manganese absorption to induce manganese overload (Figure 2A). To determine the physiological relevance of these findings, intestine-specific Zip14 knockout mice (Zip14-In-KO) were generated. In contrast to Zip14-L-KO mice that did not develop manganese overload in the body, Zip14-In-KO mice developed increased manganese in both the liver and brain under normal dietary conditions, verifying the importance of intestinal ZIP14 in maintaining systemic manganese homeostasis [66,67]. Based on the results from these most recent studies, we propose a new model for the function of ZIP14 in regulating systemic manganese homeostasis that includes the functions of intestinal ZIP14 to limit dietary manganese absorption and hepatic ZIP14 to clear manganese from the portal blood (Figure 2A,B). Future studies are needed to elucidate ZIP14’s role in other organs.
4. ZnT10
4.1. ZnT10 Mutations and Manganese Overload
ZnT10 (also known as SLC30A10, solute carrier 30 family, member 10) is a member of the Zinc transporter (ZnT) family proteins. In contrast to the function of ZIP members that increase cytosolic metal levels, ZnT proteins function to efflux metals from the cytoplasm either out of the cell or into intracellular organelles [68,69]. As the last member of the ZnT family, ZnT10 was identified by searching the human genome for homologous sequences of known ZnT proteins [70]. The amino acid sequence of ZnT10 was similar to that of ZnT1, which has been shown to principally mediate cellular zinc efflux [71,72]. Gene expression profiles indicated that ZnT10 was highly expressed in the small intestine and liver in both humans and mice [41,42,73]. In neuronal SH-SY5Y cells, Interleukin-6 treatment decreased ZnT10 expression and reduced manganese efflux [74]. In HeLa cells, ZnT10 was detected on the cell surface; and ZnT10 overexpression protected cells from manganese-induced cell toxicity by reducing intracellular manganese [75]. In lymphocyte-derived DT-40 cells, treated with high levels of zinc or manganese, ZnT10 had no effect on zinc export, but was determined to be the primary transporter mediating manganese efflux [76]. These cell culture studies suggest that ZnT10 functions as a manganese exporter to prevent manganese toxicity, which reflects the clinical status of patients carrying ZnT10 mutations.
In 2008, a clinical case report first described a 12-year old girl with severe dystonia and significantly elevated blood manganese [77]. No history of manganese exposure was noted, and her plasma levels of copper and zinc were within the normal range. This girl was born to healthy consanguineous parents. Her older brother died at 18 years of age and presented with the same disease phenotype prior to death, suggesting a genetic disorder of autosomal recessive inheritance within the family. However, the primary genetic variant and the pathophysiology of this disorder was not revealed at that time. The gene responsible for this disorder was later identified to be ZnT10 when two additional patients, presented with a similar clinical phenotype, were reported in 2012 [18]. At the same time, another research group also reported the identification of ZnT10 mutations as the causal gene defect in two consanguineous families with neurological disorders and juvenile-onset severe manganese overload [33]. Individuals with ZnT10 mutations are evident with phenotypical features of hypermanganesemia with dystonia. Genetic evaluations revealed that in the majority of identified cases, both parents of the affected individuals were heterozygous carriers of the same mutations found in their children. Neurological symptoms can occur during childhood (early-onset) or adulthood (adult-onset). In early-onset cases, children will experience symptoms that include dystonia in the arms or legs, involuntary trembling, unusually slow movement, and slurred speech. In adult-onset, symptoms include movement abnormalities, tremor, extraordinarily slow motion, muscle rigidity, and postural instability [33]. Additional symptoms may consist of polycythemia, low levels of iron in the body, enlarged liver from manganese accumulation, scarring in the liver, and irreversible liver cirrhosis [78,79,80]. Details about identified human ZnT10 mutations are listed in Table 3.
4.2. The Molecular Mechanism Underlying ZnT10’s Function in Manganese Metabolism
The identification of human ZnT10 mutations indicated that ZnT10 plays a pivotal role in regulating manganese homeostasis. While blood manganese concentrations are notably higher in individuals carrying homozygous ZnT10 mutations, zinc and iron concentrations in the blood either remain normal or show subtle changes compared to the standard values [18,33,81], further emphasizing the function of ZnT10 in manganese metabolism. Moreover, individuals affected by ZnT10 mutations also present with high levels of hepatic manganese [18]. Consistent with the human phenotype, ZnT10-deficient zebrafish had significantly increased body manganese, while levels of iron and zinc remained similar to the controls [84]; mice with constitutive whole-body Znt10 knockout (Znt10−/− mice) developed markedly elevated manganese in the liver, brain, and blood, with no changes or minor changes in body iron and zinc contents [64,85,86].
Because immunohistochemistry analysis of human liver tissues found that ZnT10 was abundant in hepatocytes and localized both intracellularly and on the plasma membrane facing the bile duct, it was initially postulated that the primary function of ZnT10 is to remove manganese from hepatocytes for biliary excretion and that the systemic manganese hyperaccumulation observed in individuals lacking ZnT10 is secondary to the defect of hepatic ZnT10 [33]. To determine ZnT10’s function in the liver, hepatocyte-specific Znt10 knockout mice (Znt10-L-KO) were generated [86,87]. After intra-orbital injection of manganese, Znt10-L-KO mice had almost no manganese excretion through the bile, confirming that ZnT10 is essential for biliary manganese excretion [86] (Figure 2B). However, in contrast to Znt10−/− mice that had 20–60 times more manganese in different tissues of the body, Znt10-L-KO mice only developed minimal excess of tissue manganese (about 2-fold increase compared to controls), indicating that ZnT10 in other organs may compensate for the loss of hepatic ZnT10 [86,87].
Besides the liver, the intestine also expresses high levels of ZnT10. To investigate the function of ZnT10 in enterocytes, polarized Caco-2 cells grown on Transwell inserts were used as a model to examine manganese transport. Immunofluorescence analysis and manganese transport assay indicated that ZnT10 localizes to the apical domain of Caco-2 cells and mediates the efflux of intracellular manganese at the apical membrane [87]. This function of ZnT10 may provide a compensatory mechanism to remove manganese into the intestinal lumen when hepatic ZnT10 is not present. To test the physiological significance of these findings, intestine-specific Znt10 knockout mice (Znt10-In-KO) and mice with Znt10 deletion in both hepatocytes and enterocytes (Znt10 double knockout (Znt10-DKO)) were created [86]. Similar to Znt10-L-KO mice, Znt10-In-KO mice only had moderate manganese accumulation in the body, despite reduced manganese export into the intestinal lumen. With a combined deletion of ZnT10 in hepatocytes and enterocytes, Znt10-DKO mice developed manganese overload that was more severe than that seen in Znt10-L-KO mice or Znt10-In-KO mice, but less severe than the manganese loading observed in the whole-body Znt10−/− mice. Together, these results demonstrated that hepatic and intestinal ZnT10 both contribute to the regulation of manganese homeostasis and that in addition to the liver and intestine, other organs with ZnT10 expression also contribute to the regulation of systemic manganese homeostasis. Future studies are required to determine ZnT10’s function at other sites.
5. Conclusions and Perspectives
Maintaining manganese balance within the body is essential for health. The discoveries of ZIP8, ZIP14, and ZnT10 as crucial manganese transporters have opened new paths towards a better understanding of manganese homeostatic regulation. Experiments using genetically modified animal models with global and tissue-specific knockouts of these genes have provided significant insights into the molecular basis of how deficiency in these genes leads to disorders of manganese metabolism.
The systemic manganese homeostasis is regulated mainly by intestinal absorption and hepatobiliary excretion. In enterocytes of the intestine, ZIP14 localizes to the basolateral membrane and imports extracellular manganese from the circulation into enterocytes, while ZnT10 functions at the apical membrane to export intracellular manganese into the lumen of the intestine. These two transporters function in the secretory direction to restrict the amount of manganese being absorbed into the blood (Figure 2A). With intestine-specific knockout of ZIP14 or ZnT10, mice develop manganese overload due to increased manganese absorption, suggesting that intestinal control of manganese absorption serves as a primary regulation for systemic manganese homeostasis. Future studies are required to address mechanisms for dietary manganese uptake at the apical membrane and manganese export at the basolateral membrane.
In hepatocytes of the liver, ZIP14 is expressed at the basolateral membrane to import circulating manganese into hepatocytes, while ZnT10 localizes at the apical canalicular membrane to export manganese into the bile. Both transporters work in the same direction to facilitate manganese removal through biliary excretion (Figure 2B). As a transporter functioning in the opposite direction, ZIP8 reclaims manganese from the bile to increase hepatic manganese storage. Continued studies are needed to explore the functions of these three manganese transporters in other organs to further advance our knowledge of manganese biology.
Author Contributions
Conceptualization, J.W.W.W. and N.Z.; writing—original draft preparation, J.W.W.W.; writing—review and editing, J.W.W.W., K.H.L., and N.Z.; funding acquisition, N.Z. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the National Institutes of Health Grant R00DK104066 (to N.Z.).
Acknowledgments
We thank Shannon E. Morgan for critical reading of the manuscript.
Conflicts of Interest
The authors declare no conflict of interest. The funder had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- Post, J.E. Manganese oxide minerals: Crystal structures and economic and environmental significance. Proc. Natl. Acad. Sci. USA 1999, 96, 3447–3454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nordberg, G. Handbook on the Toxicology of Metals, 3rd ed.; Academic Press: Cambridge, MA, USA, 2007; p. 975. [Google Scholar]
- Reaney, S.H.; Kwik-Uribe, C.L.; Smith, D.R. Manganese oxidation state and its implications for toxicity. Chem. Res. Toxicol. 2002, 15, 1119–1126. [Google Scholar] [CrossRef] [PubMed]
- Lankford, W.T. United States Steel Corporation; Association of Iron and Steel Engineers. In The Making, Shaping, and Treating of Steel, 10th ed.; Association of Iron and Steel Engineers: Pittsburgh, PA, USA, 1985; p. 1572. [Google Scholar]
- Simonelli, L.; Sorrentino, A.; Marini, C.; Ramanan, N.; Heinis, D.; Olszewski, W.; Mullaliu, A.; Birrozzi, A.; Laszczynski, N.; Giorgetti, M.; et al. Role of manganese in lithium- and manganese-rich layered oxides cathodes. J. Phys. Chem. Lett. 2019, 10, 3359–3368. [Google Scholar] [CrossRef]
- Hagelstein, K. Globally sustainable manganese metal production and use. J. Environ. Manag. 2009, 90, 3736–3740. [Google Scholar] [CrossRef] [PubMed]
- Aschner, M.; Guilarte, T.R.; Schneider, J.S.; Zheng, W. Manganese: Recent advances in understanding its transport and neurotoxicity. Toxicol. Appl. Pharmacol. 2007, 221, 131–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crossgrove, J.; Zheng, W. Manganese toxicity upon overexposure. NMR Biomed. 2004, 17, 544–553. [Google Scholar] [CrossRef] [PubMed]
- Guilarte, T.R. Manganese and parkinson’s disease: A critical review and new findings. Environ. Health Perspect. 2010, 118, 1071–1080. [Google Scholar] [CrossRef] [Green Version]
- Horning, K.J.; Caito, S.W.; Tipps, K.G.; Bowman, A.B.; Aschner, M. Manganese is essential for neuronal health. Annu. Rev. Nutr. 2015, 35, 71–108. [Google Scholar] [CrossRef]
- Munder, M. Arginase: An emerging key player in the mammalian immune system. Br. J. Pharmacol. 2009, 158, 638–651. [Google Scholar] [CrossRef] [Green Version]
- Caldwell, R.B.; Toque, H.A.; Narayanan, S.P.; Caldwell, R.W. Arginase: An old enzyme with new tricks. Trends Pharmacol. Sci. 2015, 36, 395–405. [Google Scholar] [CrossRef] [Green Version]
- Ram, B.P.; Munjal, D.D. Galactosyltransferases: Physical, chemical, and biological aspects. CRC Crit. Rev. Biochem. 1985, 17, 257–311. [Google Scholar] [CrossRef] [PubMed]
- Robinson, B.H. The role of manganese superoxide dismutase in health and disease. J. Inherit. Metab. Dis. 1998, 21, 598–603. [Google Scholar] [CrossRef] [PubMed]
- Besio, R.; Alleva, S.; Forlino, A.; Lupi, A.; Meneghini, C.; Minicozzi, V.; Profumo, A.; Stellato, F.; Tenni, R.; Morante, S. Identifying the structure of the active sites of human recombinant prolidase. Eur. Biophys. J. 2010, 39, 935–945. [Google Scholar] [CrossRef] [PubMed]
- Baly, D.L.; Keen, C.L.; Hurley, L.S. Pyruvate carboxylase and phosphoenolpyruvate carboxykinase activity in developing rats: Effect of manganese deficiency. J. Nutr. 1985, 115, 872–879. [Google Scholar] [CrossRef] [PubMed]
- Ross, A.C. Modern Nutrition in Health and Disease, 11th ed.; Wolters Kluwer Health/Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2014; p. 1616. [Google Scholar]
- Tuschl, K.; Clayton, P.T.; Gospe, S.M., Jr.; Gulab, S.; Ibrahim, S.; Singhi, P.; Aulakh, R.; Ribeiro, R.T.; Barsottini, O.G.; Zaki, M.S.; et al. Syndrome of hepatic cirrhosis, dystonia, polycythemia, and hypermanganesemia caused by mutations in slc30a10, a manganese transporter in man. Am. J. Hum. Genet. 2012, 90, 457–466. [Google Scholar] [CrossRef] [Green Version]
- Pesch, B.; Casjens, S.; Weiss, T.; Kendzia, B.; Arendt, M.; Eisele, L.; Behrens, T.; Ulrich, N.; Pundt, N.; Marr, A.; et al. Occupational exposure to manganese and fine motor skills in elderly men: Results from the heinz nixdorf recall study. Ann. Work Expo. Health 2017, 61, 1118–1131. [Google Scholar] [CrossRef]
- Kendzia, B.; Van Gelder, R.; Schwank, T.; Hagemann, C.; Zschiesche, W.; Behrens, T.; Weiss, T.; Bruning, T.; Pesch, B. Occupational exposure to inhalable manganese at german workplaces. Ann. Work Expo. Health 2017, 61, 1108–1117. [Google Scholar] [CrossRef]
- Clarke, C.; Upson, S. A global portrait of the manganese industry-a socioeconomic perspective. Neurotoxicology 2017, 58, 173–179. [Google Scholar] [CrossRef]
- Aschner, M. Manganese: Brain transport and emerging research needs. Environ. Health Perspect. 2000, 108 (Suppl. 3), 429–432. [Google Scholar]
- Huang, C.C.; Chu, N.S.; Lu, C.S.; Calne, D.B. Cock gait in manganese intoxication. Mov. Disord. 1997, 12, 807–808. [Google Scholar] [CrossRef]
- Roth, J.A. Homeostatic and toxic mechanisms regulating manganese uptake, retention, and elimination. Biol. Res. 2006, 39, 45–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukhopadhyay, S. Familial manganese-induced neurotoxicity due to mutations in slc30a10 or slc39a14. Neurotoxicology 2018, 64, 278–283. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Bornhorst, J.; Aschner, M. Manganese metabolism in humans. Front. Biosci. 2018, 23, 1655–1679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aschner, J.L.; Aschner, M. Nutritional aspects of manganese homeostasis. Mol. Asp. Med. 2005, 26, 353–362. [Google Scholar] [CrossRef] [PubMed]
- O’Neal, S.L.; Zheng, W. Manganese toxicity upon overexposure: A decade in review. Curr. Environ. Health Rep. 2015, 2, 315–328. [Google Scholar] [CrossRef] [Green Version]
- Avila, D.S.; Puntel, R.L.; Aschner, M. Manganese in health and disease. Met. Ions Life Sci. 2013, 13, 199–227. [Google Scholar]
- Boycott, K.M.; Beaulieu, C.L.; Kernohan, K.D.; Gebril, O.H.; Mhanni, A.; Chudley, A.E.; Redl, D.; Qin, W.; Hampson, S.; Kury, S.; et al. Autosomal-recessive intellectual disability with cerebellar atrophy syndrome caused by mutation of the manganese and zinc transporter gene slc39a8. Am. J. Hum. Genet. 2015, 97, 886–893. [Google Scholar] [CrossRef] [Green Version]
- Park, J.H.; Hogrebe, M.; Gruneberg, M.; DuChesne, I.; von der Heiden, A.L.; Reunert, J.; Schlingmann, K.P.; Boycott, K.M.; Beaulieu, C.L.; Mhanni, A.A.; et al. Slc39a8 deficiency: A disorder of manganese transport and glycosylation. Am. J. Hum. Genet. 2015, 97, 894–903. [Google Scholar] [CrossRef] [Green Version]
- Tuschl, K.; Meyer, E.; Valdivia, L.E.; Zhao, N.; Dadswell, C.; Abdul-Sada, A.; Hung, C.Y.; Simpson, M.A.; Chong, W.K.; Jacques, T.S.; et al. Mutations in slc39a14 disrupt manganese homeostasis and cause childhood-onset parkinsonism-dystonia. Nat. Commun. 2016, 7, 11601. [Google Scholar] [CrossRef] [Green Version]
- Quadri, M.; Federico, A.; Zhao, T.; Breedveld, G.J.; Battisti, C.; Delnooz, C.; Severijnen, L.A.; Di Toro Mammarella, L.; Mignarri, A.; Monti, L.; et al. Mutations in slc30a10 cause parkinsonism and dystonia with hypermanganesemia, polycythemia, and chronic liver disease. Am. J. Hum. Genet. 2012, 90, 467–477. [Google Scholar] [CrossRef] [Green Version]
- Hara, T.; Takeda, T.-A.; Takagishi, T.; Fukue, K.; Kambe, T.; Fukada, T. Physiological roles of zinc transporters: Molecular and genetic importance in zinc homeostasis. J. Physiol. Sci. 2017, 67, 283–301. [Google Scholar] [CrossRef] [PubMed]
- Galvez-Peralta, M.; He, L.; Jorge-Nebert, L.F.; Wang, B.; Miller, M.L.; Eppert, B.L.; Afton, S.; Nebert, D.W. Zip8 zinc transporter: Indispensable role for both multiple-organ organogenesis and hematopoiesis in utero. PLoS ONE 2012, 7, e36055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aydemir, T.B.; Liuzzi, J.P.; McClellan, S.; Cousins, R.J. Zinc transporter zip8 (slc39a8) and zinc influence ifn-gamma expression in activated human t cells. J. Leukoc. Biol. 2009, 86, 337–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, L.; Girijashanker, K.; Dalton, T.P.; Reed, J.; Li, H.; Soleimani, M.; Nebert, D.W. Zip8, member of the solute-carrier-39 (slc39) metal-transporter family: Characterization of transporter properties. Mol. Pharmacol. 2006, 70, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Riley, L.G.; Cowley, M.J.; Gayevskiy, V.; Roscioli, T.; Thorburn, D.R.; Prelog, K.; Bahlo, M.; Sue, C.M.; Balasubramaniam, S.; Christodoulou, J. A slc39a8 variant causes manganese deficiency, and glycosylation and mitochondrial disorders. J. Inherit. Metab. Dis. 2017, 40, 261–269. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Schneider, S.N.; Dragin, N.; Girijashanker, K.; Dalton, T.P.; He, L.; Miller, M.L.; Stringer, K.F.; Soleimani, M.; Richardson, D.D.; et al. Enhanced cadmium-induced testicular necrosis and renal proximal tubule damage caused by gene-dose increase in a slc39a8-transgenic mouse line. Am. J. Physiol. Cell Physiol. 2007, 292, C1523–C1535. [Google Scholar] [CrossRef] [Green Version]
- Nebert, D.W.; Liu, Z. Slc39a8 gene encoding a metal ion transporter: Discovery and bench to bedside. Hum. Genom. 2019, 13, 51. [Google Scholar] [CrossRef]
- Fagerberg, L.; Hallstrom, B.M.; Oksvold, P.; Kampf, C.; Djureinovic, D.; Odeberg, J.; Habuka, M.; Tahmasebpoor, S.; Danielsson, A.; Edlund, K.; et al. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol. Cell Proteom. 2014, 13, 397–406. [Google Scholar] [CrossRef] [Green Version]
- Yue, F.; Cheng, Y.; Breschi, A.; Vierstra, J.; Wu, W.; Ryba, T.; Sandstrom, R.; Ma, Z.; Davis, C.; Pope, B.D.; et al. A comparative encyclopedia of DNA elements in the mouse genome. Nature 2014, 515, 355–364. [Google Scholar] [CrossRef] [Green Version]
- Ajjimaporn, A.; Botsford, T.; Garrett, S.H.; Sens, M.A.; Zhou, X.D.; Dunlevy, J.R.; Sens, D.A.; Somji, S. Zip8 expression in human proximal tubule cells, human urothelial cells transformed by cd+2 and as+3 and in specimens of normal human urothelium and urothelial cancer. Cancer Cell Int. 2012, 12, 16. [Google Scholar] [CrossRef] [Green Version]
- Napolitano, J.R.; Liu, M.J.; Bao, S.; Crawford, M.; Nana-Sinkam, P.; Cormet-Boyaka, E.; Knoell, D.L. Cadmium-mediated toxicity of lung epithelia is enhanced through nf-kappab-mediated transcriptional activation of the human zinc transporter zip8. Am. J. Physiol.-Lung Cell Mol. Physiol. 2012, 302, L909–L918. [Google Scholar] [CrossRef] [PubMed]
- Scheiber, I.F.; Alarcon, N.O.; Zhao, N. Manganese uptake by a549 cells is mediated by both zip8 and zip14. Nutrients 2019, 11, 1473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, S.N.; Liu, Z.; Wang, B.; Miller, M.L.; Afton, S.E.; Soleimani, M.; Nebert, D.W. Oral cadmium in mice carrying 5 versus 2 copies of the slc39a8 gene: Comparison of uptake, distribution, metal content, and toxicity. Int. J. Toxicol. 2014, 33, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.Y.; Jenkitkasemwong, S.; Duarte, S.; Sparkman, B.K.; Shawki, A.; Mackenzie, B.; Knutson, M.D. Zip8 is an iron and zinc transporter whose cell-surface expression is up-regulated by cellular iron loading. J. Biol. Chem. 2012, 287, 34032–34043. [Google Scholar] [CrossRef] [Green Version]
- Nebert, D.W.; Galvez-Peralta, M.; Hay, E.B.; Li, H.; Johansson, E.; Yin, C.; Wang, B.; He, L.; Soleimani, M. Zip14 and zip8 zinc/bicarbonate symporters in xenopus oocytes: Characterization of metal uptake and inhibition. Metallomics 2012, 4, 1218–1225. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; He, L.; Dong, H.; Dalton, T.P.; Nebert, D.W. Generation of a slc39a8 hypomorph mouse: Markedly decreased zip8 zn (2) (+)/(hco(3)(-))(2) transporter expression. Biochem. Biophys. Res. Commun. 2011, 410, 289–294. [Google Scholar] [CrossRef] [Green Version]
- Lin, W.; Vann, D.R.; Doulias, P.T.; Wang, T.; Landesberg, G.; Li, X.; Ricciotti, E.; Scalia, R.; He, M.; Hand, N.J.; et al. Hepatic metal ion transporter zip8 regulates manganese homeostasis and manganese-dependent enzyme activity. J. Clin. Investig. 2017, 127, 2407–2417. [Google Scholar] [CrossRef] [Green Version]
- Taylor, K.M.; Morgan, H.E.; Johnson, A.; Nicholson, R.I. Structure-function analysis of a novel member of the liv-1 subfamily of zinc transporters, zip14. FEBS Lett. 2005, 579, 427–432. [Google Scholar] [CrossRef] [Green Version]
- Fujishiro, H.; Yano, Y.; Takada, Y.; Tanihara, M.; Himeno, S. Roles of zip8, zip14, and dmt1 in transport of cadmium and manganese in mouse kidney proximal tubule cells. Metallomics 2012, 4, 700–708. [Google Scholar] [CrossRef]
- Pinilla-Tenas, J.J.; Sparkman, B.K.; Shawki, A.; Illing, A.C.; Mitchell, C.J.; Zhao, N.; Liuzzi, J.P.; Cousins, R.J.; Knutson, M.D.; Mackenzie, B. Zip14 is a complex broad-scope metal-ion transporter whose functional properties support roles in the cellular uptake of zinc and nontransferrin-bound iron. Am. J. Physiol. Cell Physiol. 2011, 301, C862–C871. [Google Scholar] [CrossRef] [Green Version]
- Girijashanker, K.; He, L.; Soleimani, M.; Reed, J.M.; Li, H.; Liu, Z.; Wang, B.; Dalton, T.P.; Nebert, D.W. Slc39a14 gene encodes zip14, a metal/bicarbonate symporter: Similarities to the zip8 transporter. Mol. Pharmacol. 2008, 73, 1413–1423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nomura, N.; Nagase, T.; Miyajima, N.; Sazuka, T.; Tanaka, A.; Sato, S.; Seki, N.; Kawarabayasi, Y.; Ishikawa, K.; Tabata, S. Prediction of the coding sequences of unidentified human genes. Ii. The coding sequences of 40 new genes (kiaa0041-kiaa0080) deduced by analysis of cdna clones from human cell line kg-1. DNA Res. 1994, 1, 223–229. [Google Scholar] [CrossRef] [PubMed]
- Liuzzi, J.P.; Aydemir, F.; Nam, H.; Knutson, M.D.; Cousins, R.J. Zip14 (slc39a14) mediates non-transferrin-bound iron uptake into cells. Proc. Natl. Acad. Sci. USA 2006, 103, 13612–13617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Troche, C.; Aydemir, T.B.; Cousins, R.J. Zinc transporter slc39a14 regulates inflammatory signaling associated with hypertrophic adiposity. Am. J. Physiol. Endocrinol. Metab. 2016, 310, E258–E268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeglam, A.; Abugrara, A.; Kabuka, M. Autosomal-recessive iron deficiency anemia, dystonia and hypermanganesemia caused by new variant mutation of the manganese transporter gene slc39a14. Acta Neurol. Belg. 2018, 119, 379–384. [Google Scholar] [CrossRef]
- Rodan, L.H.; Hauptman, M.; D’Gama, A.M.; Qualls, A.E.; Cao, S.; Tuschl, K.; Al-Jasmi, F.; Hertecant, J.; Hayflick, S.J.; Wessling-Resnick, M.; et al. Novel founder intronic variant in slc39a14 in two families causing manganism and potential treatment strategies. Mol. Genet. Metab. 2018, 124, 161–167. [Google Scholar] [CrossRef]
- Marti-Sanchez, L.; Ortigoza-Escobar, J.D.; Darling, A.; Villaronga, M.; Baide, H.; Molero-Luis, M.; Batllori, M.; Vanegas, M.I.; Muchart, J.; Aquino, L.; et al. Hypermanganesemia due to mutations in slc39a14: Further insights into mn deposition in the central nervous system. Orphanet J. Rare Dis. 2018, 13, 28. [Google Scholar] [CrossRef]
- Juneja, M.; Shamim, U.; Joshi, A.; Mathur, A.; Uppili, B.; Sairam, S.; Ambawat, S.; Dixit, R.; Faruq, M. A novel mutation in slc39a14 causing hypermanganesemia associated with infantile onset dystonia. J. Gene Med. 2018, 20, e3012. [Google Scholar] [CrossRef]
- Aydemir, T.B.; Kim, M.H.; Kim, J.; Colon-Perez, L.M.; Banan, G.; Mareci, T.H.; Febo, M.; Cousins, R.J. Metal transporter zip14 (slc39a14) deletion in mice increases manganese deposition and produces neurotoxic signatures and diminished motor activity. J. Neurosci. 2017, 37, 5996–6006. [Google Scholar] [CrossRef] [Green Version]
- Xin, Y.; Gao, H.; Wang, J.; Qiang, Y.; Imam, M.U.; Li, Y.; Wang, J.; Zhang, R.; Zhang, H.; Yu, Y.; et al. Manganese transporter slc39a14 deficiency revealed its key role in maintaining manganese homeostasis in mice. Cell Discov. 2017, 3, 17025. [Google Scholar] [CrossRef]
- Liu, C.; Hutchens, S.; Jursa, T.; Shawlot, W.; Polishchuk, E.V.; Polishchuk, R.S.; Dray, B.K.; Gore, A.C.; Aschner, M.; Smith, D.R.; et al. Hypothyroidism induced by loss of the manganese efflux transporter slc30a10 may be explained by reduced thyroxine production. J. Biol. Chem. 2017, 292, 16605–16615. [Google Scholar] [CrossRef] [Green Version]
- Jenkitkasemwong, S.; Akinyode, A.; Paulus, E.; Weiskirchen, R.; Hojyo, S.; Fukada, T.; Giraldo, G.; Schrier, J.; Garcia, A.; Janus, C.; et al. Slc39a14 deficiency alters manganese homeostasis and excretion resulting in brain manganese accumulation and motor deficits in mice. Proc. Natl. Acad. Sci. USA 2018, 115, E1769–E1778. [Google Scholar] [CrossRef] [Green Version]
- Scheiber, I.F.; Wu, Y.; Morgan, S.E.; Zhao, N. The intestinal metal transporter zip14 maintains systemic manganese homeostasis. J. Biol. Chem. 2019, 294, 9147–9160. [Google Scholar] [CrossRef]
- Aydemir, T.B.; Thorn, T.L.; Ruggiero, C.H.; Pompilus, M.; Febo, M.; Cousins, R.J. Intestine-specific deletion of metal transporter zip14 (slc39a14) causes brain manganese overload and locomotor defects of manganism. Am. J. Physiol. Gastrointest. Liver Physiol. 2020, 318, G673–G681. [Google Scholar] [CrossRef]
- Cousins, R.J.; Liuzzi, J.P.; Lichten, L.A. Mammalian zinc transport, trafficking, and signals. J. Biol. Chem. 2006, 281, 24085–24089. [Google Scholar] [CrossRef] [Green Version]
- Kimura, T.; Kambe, T. The functions of metallothionein and zip and znt transporters: An overview and perspective. Int. J. Mol. Sci. 2016, 17, 336. [Google Scholar] [CrossRef] [Green Version]
- Seve, M.; Chimienti, F.; Devergnas, S.; Favier, A. In silico identification and expression of slc30 family genes: An expressed sequence tag data mining strategy for the characterization of zinc transporters’ tissue expression. BMC Genom. 2004, 5, 32. [Google Scholar] [CrossRef]
- Kambe, T. Molecular architecture and function of znt transporters. Curr Top. Membr 2012, 69, 199–220. [Google Scholar]
- Kambe, T.; Suzuki, T.; Nagao, M.; Yamaguchi-Iwai, Y. Sequence similarity and functional relationship among eukaryotic zip and cdf transporters. Genom. Proteom. Bioinform. 2006, 4, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Sreedharan, S.; Stephansson, O.; Schioth, H.B.; Fredriksson, R. Long evolutionary conservation and considerable tissue specificity of several atypical solute carrier transporters. Gene 2011, 478, 11–18. [Google Scholar] [CrossRef]
- Fujishiro, H.; Yoshida, M.; Nakano, Y.; Himeno, S. Interleukin-6 enhances manganese accumulation in sh-sy5y cells: Implications of the up-regulation of zip14 and the down-regulation of znt10. Metallomics 2014, 6, 944–949. [Google Scholar] [CrossRef] [Green Version]
- Leyva-Illades, D.; Chen, P.; Zogzas, C.E.; Hutchens, S.; Mercado, J.M.; Swaim, C.D.; Morrisett, R.A.; Bowman, A.B.; Aschner, M.; Mukhopadhyay, S. Slc30a10 is a cell surface-localized manganese efflux transporter, and parkinsonism-causing mutations block its intracellular trafficking and efflux activity. J. Neurosci. 2014, 34, 14079–14095. [Google Scholar] [CrossRef]
- Nishito, Y.; Tsuji, N.; Fujishiro, H.; Takeda, T.A.; Yamazaki, T.; Teranishi, F.; Okazaki, F.; Matsunaga, A.; Tuschl, K.; Rao, R.; et al. Direct comparison of manganese detoxification/efflux proteins and molecular characterization of znt10 protein as a manganese transporter. J. Biol. Chem. 2016, 291, 14773–14787. [Google Scholar] [CrossRef] [Green Version]
- Tuschl, K.; Mills, P.B.; Parsons, H.; Malone, M.; Fowler, D.; Bitner-Glindzicz, M.; Clayton, P.T. Hepatic cirrhosis, dystonia, polycythaemia and hypermanganesaemia—A new metabolic disorder. J. Inherit. Metab. Dis. 2008, 31, 151–163. [Google Scholar] [CrossRef]
- Zaki, M.S.; Issa, M.Y.; Elbendary, H.M.; El-Karaksy, H.; Hosny, H.; Ghobrial, C.; El Safty, A.; El-Hennawy, A.; Oraby, A.; Selim, L.; et al. Hypermanganesemia with dystonia, polycythemia and cirrhosis in 10 patients: Six novel slc30a10 mutations and further phenotype delineation. Clin. Genet. 2018, 93, 905–912. [Google Scholar] [CrossRef]
- Gulab, S.; Kayyali, H.R.; Al-Said, Y. Atypical neurologic phenotype and novel slc30a10 mutation in two brothers with hereditary hypermanganesemia. Neuropediatrics 2018, 49, 72–75. [Google Scholar] [CrossRef]
- Mukhtiar, K.; Ibrahim, S.; Tuschl, K.; Mills, P. Hypermanganesemia with dystonia, polycythemia and cirrhosis (hmdpc) due to mutation in the slc30a10 gene. Brain Dev. 2016, 38, 862–865. [Google Scholar] [CrossRef]
- Quadri, M.; Kamate, M.; Sharma, S.; Olgiati, S.; Graafland, J.; Breedveld, G.J.; Kori, I.; Hattiholi, V.; Jain, P.; Aneja, S.; et al. Manganese transport disorder: Novel slc30a10 mutations and early phenotypes. Mov. Disord. 2015, 30, 996–1001. [Google Scholar] [CrossRef]
- Tavasoli, A.; Arjmandi Rafsanjani, K.; Hemmati, S.; Mojbafan, M.; Zarei, E.; Hosseini, S. A case of dystonia with polycythemia and hypermanganesemia caused by slc30a10 mutation: A treatable inborn error of manganese metabolism. BMC Pediatr. 2019, 19, 229. [Google Scholar] [CrossRef]
- Yapici, Z.; Tuschl, K.; Eraksoy, M. Hypermanganesemia with dystonia 1: A novel mutation and response to iron supplementation. Mov. Disord. Clin. Pract. 2020, 7, 94–96. [Google Scholar] [CrossRef]
- Xia, Z.; Wei, J.; Li, Y.; Wang, J.; Li, W.; Wang, K.; Hong, X.; Zhao, L.; Chen, C.; Min, J.; et al. Zebrafish slc30a10 deficiency revealed a novel compensatory mechanism of atp2c1 in maintaining manganese homeostasis. PLoS Genet. 2017, 13, e1006892. [Google Scholar] [CrossRef]
- Hutchens, S.; Liu, C.; Jursa, T.; Shawlot, W.; Chaffee, B.K.; Yin, W.; Gore, A.C.; Aschner, M.; Smith, D.R.; Mukhopadhyay, S. Deficiency in the manganese efflux transporter slc30a10 induces severe hypothyroidism in mice. J. Biol. Chem. 2017, 292, 9760–9773. [Google Scholar] [CrossRef] [Green Version]
- Mercadante, C.J.; Prajapati, M.; Conboy, H.L.; Dash, M.E.; Herrera, C.; Pettiglio, M.A.; Cintron-Rivera, L.; Salesky, M.A.; Rao, D.B.; Bartnikas, T.B. Manganese transporter slc30a10 controls physiological manganese excretion and toxicity. J. Clin. Investig. 2019, 129, 5442–5461. [Google Scholar] [CrossRef]
- Taylor, C.A.; Hutchens, S.; Liu, C.; Jursa, T.; Shawlot, W.; Aschner, M.; Smith, D.R.; Mukhopadhyay, S. Slc30a10 transporter in the digestive system regulates brain manganese under basal conditions while brain slc30a10 protects against neurotoxicity. J. Biol. Chem. 2019, 294, 1860–1876. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
Both the intestine and liver play important roles in regulating systemic manganese metabolism. (A) Enterocytes are absorptive cells lining the lumen of intestine. Digested manganese (Mn) in the intestinal lumen can be transported across the apical (AP) membrane into enterocytes and exported across the basolateral (BL) membrane into the blood (solid blue arrows). Absorbed Mn will then be delivered to the liver through the portal vein. Mn in the blood can also be transported into enterocytes and exported back into the intestinal lumen (dashed blue arrows); (B) The liver clears Mn from the blood and secretes Mn into the bile for intestinal reabsorption or fecal excretion. Hepatocytes are polarized and represent the major cell type in the liver. The basal domain of hepatocytes faces the sinusoid blood; whereas, the apical domain forms the bile canalicular network. Hepatocytes express polarized, specific transporters that mediate manganese fluxes into or out of these cells (solid red arrows: manganese transport into hepatocytes from the blood or the bile; dashed red arrow: manganese export from hepatocytes into the bile).
Figure 1.
Both the intestine and liver play important roles in regulating systemic manganese metabolism. (A) Enterocytes are absorptive cells lining the lumen of intestine. Digested manganese (Mn) in the intestinal lumen can be transported across the apical (AP) membrane into enterocytes and exported across the basolateral (BL) membrane into the blood (solid blue arrows). Absorbed Mn will then be delivered to the liver through the portal vein. Mn in the blood can also be transported into enterocytes and exported back into the intestinal lumen (dashed blue arrows); (B) The liver clears Mn from the blood and secretes Mn into the bile for intestinal reabsorption or fecal excretion. Hepatocytes are polarized and represent the major cell type in the liver. The basal domain of hepatocytes faces the sinusoid blood; whereas, the apical domain forms the bile canalicular network. Hepatocytes express polarized, specific transporters that mediate manganese fluxes into or out of these cells (solid red arrows: manganese transport into hepatocytes from the blood or the bile; dashed red arrow: manganese export from hepatocytes into the bile).

Figure 2.
Schematic diagram illustrating the functions of ZIP8, ZIP14, and ZnT10 in enterocytes and hepatocytes. (A) The functions of ZIP14 and ZnT10 in enterocytes. How manganese is absorbed from the apical to the basolateral side is not well understood. ZIP14 plays a role in mediating the re-uptake of freshly released manganese at the basolateral membrane. After being reclaimed by enterocytes, manganese can then be transported back into the intestinal lumen by ZnT10 or remains in the intestine epithelium until it is sloughed. Therefore, loss of ZIP14 will decrease manganese re-uptake and increase the net manganese absorption to induce manganese overload. (B) The functions of ZIP14, ZnT10, and ZIP8 in hepatocytes. ZIP14 imports circulating manganese into hepatocytes at the basolateral membrane, while ZnT10 exports manganese into the bile at the apical canalicular membrane. ZIP8 reclaims manganese from the bile to increase hepatic manganese storage.
Figure 2.
Schematic diagram illustrating the functions of ZIP8, ZIP14, and ZnT10 in enterocytes and hepatocytes. (A) The functions of ZIP14 and ZnT10 in enterocytes. How manganese is absorbed from the apical to the basolateral side is not well understood. ZIP14 plays a role in mediating the re-uptake of freshly released manganese at the basolateral membrane. After being reclaimed by enterocytes, manganese can then be transported back into the intestinal lumen by ZnT10 or remains in the intestine epithelium until it is sloughed. Therefore, loss of ZIP14 will decrease manganese re-uptake and increase the net manganese absorption to induce manganese overload. (B) The functions of ZIP14, ZnT10, and ZIP8 in hepatocytes. ZIP14 imports circulating manganese into hepatocytes at the basolateral membrane, while ZnT10 exports manganese into the bile at the apical canalicular membrane. ZIP8 reclaims manganese from the bile to increase hepatic manganese storage.


{kind=link}
{kind=link}
Table 1.
Identified human cases of ZIP8 mutation.
| Subject and Reference | ZIP8 Mutation | Amino Acid Change | Gender | Age of Onset | Blood Manganese |
|---|---|---|---|---|---|
| A-1 ƒ § Ç [30] | c.[112G>C];[112G>C] | p.[Gly38Arg] | F | Birth | ND |
| B-1 ƒ § Ç [30] | c.[112G>C];[112G>C] | p.[Gly38Arg] | M | Birth | 20 nmol/L (Erythrocyte) (NR 273–728) |
| C-1 ƒ § Ç [30] | c.[112G>C];[112G>C] | p.[Gly38Arg] | M | Birth | 20 nmol/L (NR 78–289) |
| D-1 ƒ § Ç [30] | c.[112G>C];[112G>C] | p.[Gly38Arg] | F | Birth | 14.2 nmol/L (NR 5.3–40.8) |
| D-2 ƒ § Ç [30] | c.[112G>C];[112G>C] | p.[Gly38Arg] | F | Birth | 5.5 nmol/L (NR 5.3–40.8) |
| E-1 ƒ § Ç [30] | c.[112G>C];[112G>C] | p.[Gly38Arg] | M | Birth | 18.4 nmol/L (NR 5.3–40.8) |
| F-1 ƒ § Ç [30] | c.[112G>C];[112G>C] | p.[Gly38Arg] | F | Birth | 1.1 mcg/L (NR 5–12.4) |
| F-2 ƒ § Ç [30] | c.[112G>C];[112G>C] | p.[Gly38Arg] | M | Birth | 1.1 mcg/L (NR 5–12.4) |
| F-3 ƒ § Ç [30] | † | † | N/A | Birth | N/A |
| G-1 £ [31] | c.[112G>C];[1019T>A] | p.[Gly38Arg];[Ile340Asn] | F | <4 months | ND |
| H-2 £ [31] | c.[97G>A;1004G>C];[610G>T] | p.[Val33Met; Ser335Thr]; [Gly204Cys] | F | <1 year | ND |
| I-1 § Ç [38] | c.[338G>C];[338G>C] | p.[Cys113Ser] | F | 4 months | ND |
| I-2 § Ç [38] | c.[338G>C];[338G>C] | p.[Cys113Ser] | F | 3 months | ND |
Individual families are letters A–I with affected sibling listed. Abbreviations and symbols: Not detectable (ND); normal range (NR); male (M); female (F); †: DNA was not available for testing from the third affected member due to death at 7 months. The individual may have been affected by the same disorder and genetic mutation; ƒ: the mutation was present in two different families; £: expressed compound heterozygous mutations; §: both parents are heterozygous carriers of the identified mutation; Ç: patients from consanguineous families.
Table 2.
Identified human cases of ZIP14 mutation.
| Subject and Reference | ZIP14 Mutation | Amino Acid Change | Gender | Age of Onset | Blood Manganese |
|---|---|---|---|---|---|
| A-1 § Ç [32] | c.[292T>G];[292T>G] | p.[Phe98Val] | F | 7 Months | 2887 nmol/L (NR 73–325) |
| A-2 § Ç [32] | c.[292T>G];[292T>G] | p.[Phe98Val] | F | 6 Months | N/A |
| B-1 § Ç [32] | c.[313G>T];[313G>T] | p.[Glu105*] | F | 7 months | 8101 nmol/L (NR 73–325) |
| B-2 § Ç [32] | † | † | F | 7 months | N/A |
| C-1 § Ç [32] | c.[477_478del];[477_478del] | p.[S160Cysfs*5] | F | 3 years | 963 nmol/L (NR 73–325) |
| D-1 Ç [32] | c.[1147G>A];[1147G>A] | p.[Gly383Arg] | M | 10 months | 965 nmol/L (NR 145.6) |
| E-1 § Ç [32] | c.[1407C>G];[1407C>G] | p.[Asn469Lys] | F | 2 years | 2280 nmol/L (NR 73–325) |
| E-2 § Ç [32] | c.[1407C>G];[1407C>G] | p.[Asn469Lys] | F | 2 years | 3830 nmol/L (NR 73–325) |
| E-3 § Ç [32] | c.[1407C>G];[1407C>G] | p.[Asn469Lys] | M | 2 years | 1260 nmol/L (NR 73–325) |
| F-1 § Ç [60] | c.[311G>T];[311G>T] | p.[Ser104Ile] | M | 11 months | 10.5 mcg/L (Plasma) (NR 0.4–0.9) |
| F-2 § Ç [60] | ‡ | ‡ | M | 10 months | N/A |
| G-1 § [61] | c.[382C>T];[382C>T] | p.[Arg128Trp] | F | 2 months | 3640 nmol/L (NR 73–375) |
| H-1 ƒ § Ç [59] | c.[751-9C>G];[751-9C>G] | p.[His251Profs*26] | F | 8 months | 64.2 mcg/L (Serum) (NR 4–16.5) |
| I-1 ƒ [59] | c.[751-9C>G];[751-9C>G] | p.[His251Profs*26] | F | 18 months | 78 mcg/L (Serum) (NR 4–16.5) |
| J-1 § Ç [58] | c.[1136C>T];[1136C>T] | p.[Pro379Leu] | F | 15 months | 150 nmol/L (NR <10) |
Individual families are letters A–J with affected sibling listed. Abbreviations and symbols: Not detectable (ND); normal range (NR); male (M); female (F); †: DNA was not available for testing from the third affected member. Her clinical phenotype was similar to her siblings, suggesting that she may have been affected by the same disorder and genetic mutation; ‡: DNA of F-2 was not available for testing from the second affected member. His clinical phenotype was similar to his sibling (F-1), suggesting that he may have been affected by the same disorder and genetic mutation; ƒ: the mutation was present in two different families; §: both parents are heterozygous carriers of the identified mutation; Ç: patients from consanguineous families.
Table 3.
Identified human cases of ZnT10 mutation.
| Subject and Reference | ZnT10 Mutation | Amino Acid Change | Gender | Age of Onset | Blood Manganese |
|---|---|---|---|---|---|
| A-1 § Ç [18] | Deletion of exons 1 and 2 | N/A | F | 3 years | 6180 nmol/L (NR <320) |
| A-2 § Ç [18] | Deletion of exons 1 and 2 | N/A | F | 3 years | 3767 nmol/L (NR <320) |
| A-3 § Ç [18] | Deletion of exons 1 and 2 | N/A | M | 5 years | 5096 nmol/L (NR <320) |
| A-4 § Ç [18] | Deletion of exons 1 and 2 | N/A | M | 5 years | 6370 nmol/L (NR <320) |
| B-1 Ç Ð [33] | c.[507delG];[500T>C] | p.[Pro170Leufs*22] | M | 2 years | 231.6 nmol/L (NR <32.8) |
| B-2 Ç Ð [33] | c.[507delG];[500T>C] | p.[Pro170Leufs*22] | M | 14 years | 2626 nmol/L (NR 183–352) |
| B-3 Ç Ð [33] | † | † | F | 10 years | N/A |
| C-1 Ç Ð [33] | c.[1235delA];[1235delA] | p.[Gln412Argfs*26] | M | 47 years | 104 mcg/L (NR 3–8) |
| C-2 Ç Ð [33] | c.[1235delA];[1235delA] | p.[Gln412Argfs*26] | M | 57 years | 106 mcg/L (NR 3–8) |
| D-1 § Ç [18] | c.[266T>C];[266T>C] | p.[Leu89Pro] | F | 2 years | 2109 nmol/L (NR <320) |
| D-2 § Ç [18] | c.[266T>C];[266T>C] | p.[Leu89Pro] | F | 2 years | 1636 nmol/L (NR <320) |
| D-3 § Ç [18] | c.[266T>C];[266T>C] | p.[Leu89Pro] | F | 2 years | 1600 nmol/L (NR <320) |
| E-1 § [18] | c.[292_402del];[292_402del] | p.[Val98_Phe134del] | F | 4 years | 1145 nmol/L (NR <320) |
| F-1 § Ç [18] | c.[314_322del];[314_322del] | p.[Ala105_Pro107del] | M | 2 years | N/A |
| F-2 § Ç [18] | c.[314_322del];[314_322del] | p.[Ala105_Pro107del] | F | 11 years | 3285 nmol/L (NR <320) |
| G-1 § [18] | c.[585del];[585del] | p.[Thr196Profs*17] | M | 14 years | 3480 nmol/L (NR <320) |
| H-1 § [18] | c.[765_767del];[765_767del] | p.[Val256del] | F | 11 years | 3272 nmol/L (NR <320) |
| I-1 § Ç [18] | c.[922C>T];[922C>T] | p.[Gln308*] | M | 2 years | NA |
| I-2 § Ç [18] | c.[922C>T];[922C>T] | p.[Gln308*] | F | 3 years | 3114 nmol/L (NR <320) |
| J-1 § [18] | c.[1046T>C];[1046T>C] | p.[Leu349Pro] | F | 5 years | 2366 nmol/L (NR <320) |
| K-1 § Ç [81] | c.[496_553del58];[496_553del58] | p.[Ala166Glnfs*7] | F | 3 years | 9.8 mcg/L (serum) (NR 0.3–1.8) |
| L-1 § Ç [81] | c.[492delC];[492delC] | p.[Gly165Alafs*27] | M | 3 years | 19.5 mcg/L (serum) (NR 0.3–1.8) |
| L-2 § Ç [81] | c.[492delC];[492delC] | p.[Gly165Alafs*27] | F | 1 year | 23.9 mcg/L (serum) (NR 0.3–1.8) |
| L-3 § Ç [81] | c.[492delC];[492delC] | p.[Gly165Alafs*27] | F | 3 years | 29.5 mcg/L (serum) (NR 0.3–1.8) |
| M-1 § Ç [81] | c.[460C>T];[460C>T] | p.[Gln154*] | F | 4.5 years | 42 mcg/L (serum) (NR 0.3–1.8) |
| N-1 Ç € [80] | c.[1006C>T] | p.[His336Tyr] | M | 10 years | 14,972 nmol/L (NR <320) |
| N-2 Ç € [80] | c.[1006C>T] | p.[His336Tyr] | M | 8 years | 1511 nmol/L (NR <320) |
| N-3 Ç € [80] | c.[1006C>T] | p.[His336Tyr] | M | 6 years | 539 nmol/L (NR <320) |
| O-1 § Ç [79] | c.[359G>A] | p.[Gly120Asp] | M | 4 years | 2808 nmol/L (NR 100–260) |
| O-2 § Ç [79] | c.[359G>A] | p.[Gly120Asp] | M | 6 years | 2056 nmol/L (NR 100–260) |
| P-1 § Ç [78] | c.[957 + 1G>C] (Splice site mutation) | N/A | M | 10 years | 2900 nmol/L (NR <320) |
| P-2 § Ç [78] | c.[957 + 1G>C] (Splice site mutation) | N/A | M | 2 years | 3340 nmol/L (NR <320) |
| Q-1 § Ç [78] | c.[119A>C] | p.[Asp40A] | M | 1 year 6 months | 3200 nmol/L (NR <320) |
| R-1 § Ç [78] | c.[122_124delCCT] | p.[Ser41del] | M | 1 year 6 months | 3310 nmol/L (NR <320) |
| S-1 § Ç [78] | c.[90C>G] | p.[Tyr30*] | M | 1 year 3 months | 2980 nmol/L (NR <320) |
| T-1 § Ç [78] | c.[780_782delCAT] | p.[Iso260del] | F | 1 year 6 months | 3125 nmol/L (NR <320) |
| T-2 § Ç [78] | c.[780_782delCAT] | p.[Iso260del] | F | 1 year 3 months | 3300 nmol/L (NR <320) |
| U-1 ƒ § Ç [78] | c.[77T>C] | p.[Leu26Pro] | M | 2 year | 3245 nmol/L (NR <320) |
| U-2 ƒ § Ç [78] | c.[77T>C] | p.[Leu26Pro] | F | 1 year 6 months | 3120 nmol/L (NR <320) |
| V-1 ƒ § Ç [78] | c.[77T>C] | p.[Leu26Pro] | F | 4 years | 2750 nmol/L (NR <320) |
| W-1 ƒ § Ç [82] | c.[1006C>T] | p.[His336Tyr] | M | 10 years | 3000 nmol/L (NR <320) |
| X-1 § Ç [83] | c.[1188dup] | p.[Leu397Thrfs*15] | F | 1 year 6 months | 1946 nmol/L (at age of 7) (NR <273) |
Individual families are letters A–X with affected sibling listed. Abbreviations and symbols: Not detectable (ND); normal range (NR); male (M); female (F);†: DNA was not available for testing from the third affected member. Her clinical phenotype was similar to her siblings, suggesting that she may have been affected by the same disorder and genetic mutation; ƒ: the mutation was present in two different families; §: both parents are heterozygous carriers of the identified mutation; Ç: patients from consanguineous families; Ð: DNA from parents was not available for testing; €: father and one unaffected sibling were heterozygous for the mutation.
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Winslow, J.W.W.; Limesand, K.H.; Zhao, N. The Functions of ZIP8, ZIP14, and ZnT10 in the Regulation of Systemic Manganese Homeostasis. Int. J. Mol. Sci. 2020, 21, 3304. https://doi.org/10.3390/ijms21093304
AMA Style
Winslow JWW, Limesand KH, Zhao N. The Functions of ZIP8, ZIP14, and ZnT10 in the Regulation of Systemic Manganese Homeostasis. International Journal of Molecular Sciences. 2020; 21(9):3304. https://doi.org/10.3390/ijms21093304
Chicago/Turabian StyleWinslow, James W.W., Kirsten H. Limesand, and Ningning Zhao. 2020. "The Functions of ZIP8, ZIP14, and ZnT10 in the Regulation of Systemic Manganese Homeostasis" International Journal of Molecular Sciences 21, no. 9: 3304. https://doi.org/10.3390/ijms21093304
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.