Microtubule-Mediated NLRP3 Inflammasome Activation Is Independent of Microtubule-Associated Innate Immune Factor GEF-H1 in Murine Macrophages



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
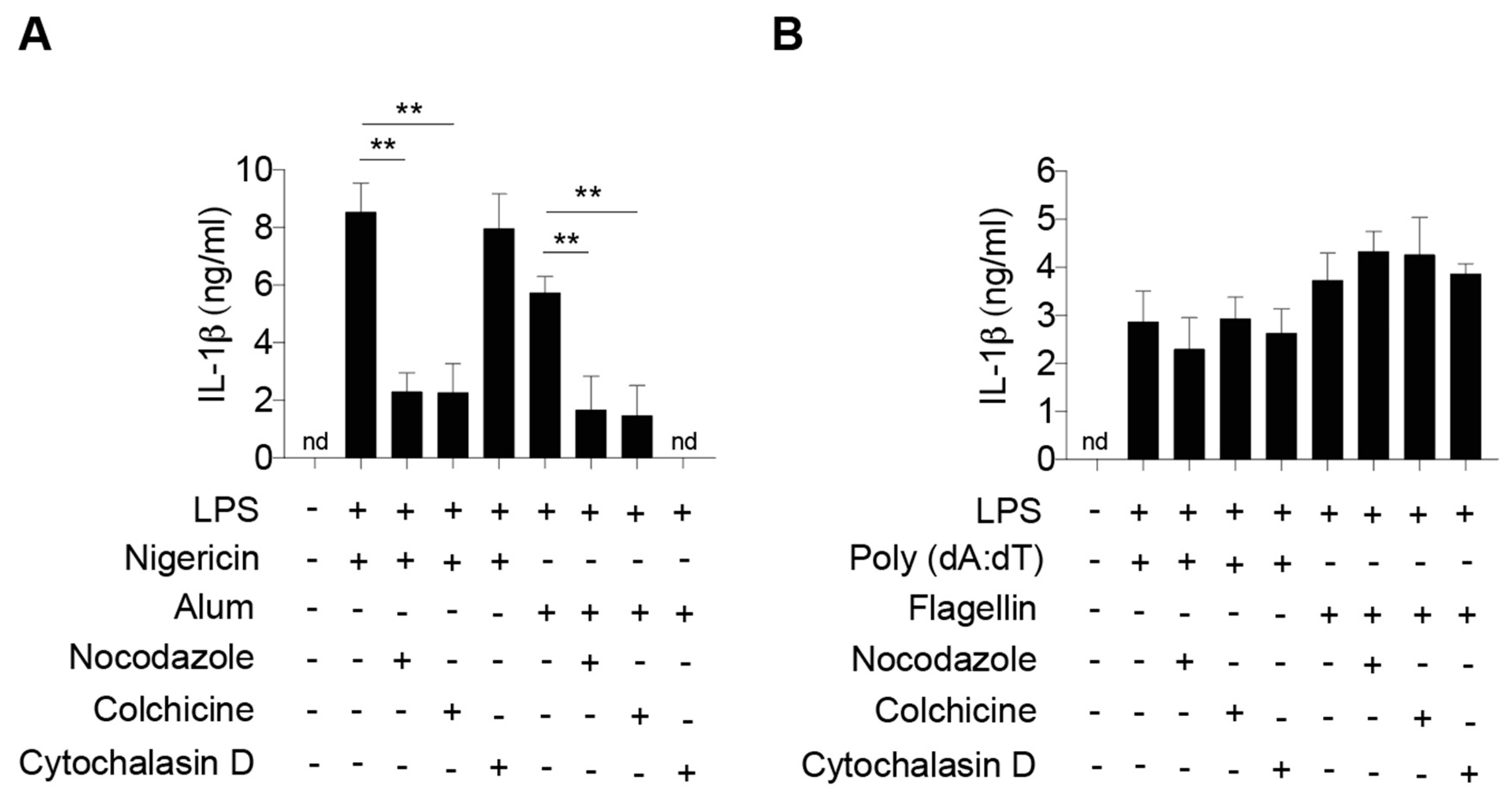
2.1. Dynamic Microtubule Network Controls NLRP3 Inflammasome-Mediated IL-1β Production
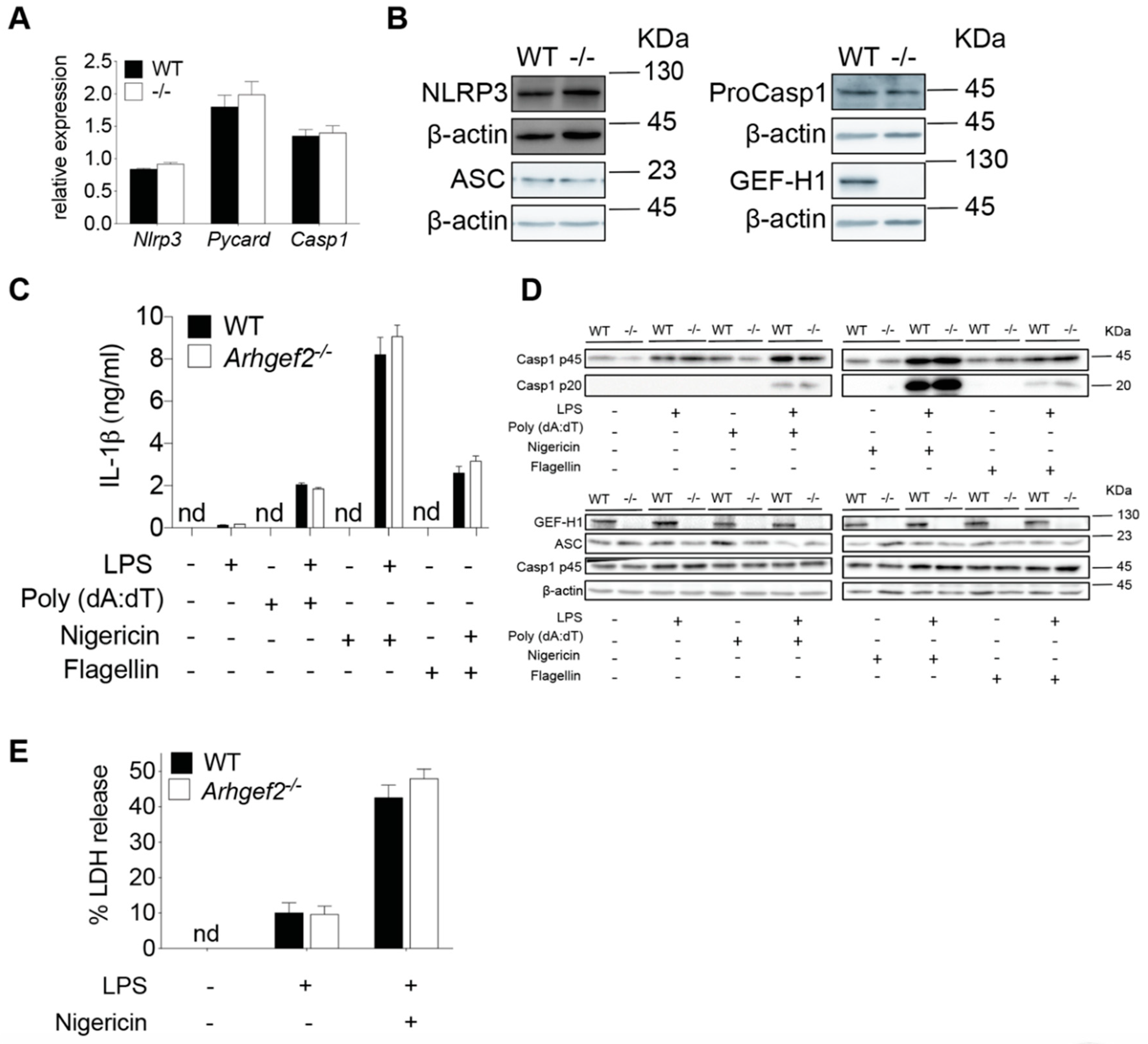
2.2. Deficiency of GEF-H1 Does Not Affect the Secretion of IL-1β in Response to NLRP3 Inflammasome Inducers
2.3. GEF-H1 Is Not Required for α-Tubulin Acetylation and Mitochondria Redistribution in LPS-Primed Macrophages Incubated with NLRP3 Inducer
2.4. GEF-H1 Is Dispensable for the Immune Defense against Salmonella typhimurim Infection in Macrophages
3. Discussion
4. Materials and Methods
4.1. Mice
4.2. Bone Marrow-Derived Macrophages, COS-7 Cells, and Plasmid
4.3. Treatments and Measurement of Il-1β
4.4. Immunoblotting
4.5. Detection of Lactate Dehydrogenase (LDH) Release
4.6. Quantitative RT-PCR (qRT-PCR)
4.7. Confocal Microscopy
4.8. Bacterial Infection and Gentamycin Protection Assay
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| PRR | Pattern recognition receptor |
| NLRP3 | NLR family pyrin domain-containing protein 3 |
| NLRC4 | NLR family CARD domain-containing protein 4 |
| NLR | NOD-like receptor |
| CARD | Caspase activation and recruitment domain |
| NOD | Nucleotide-binding oligomerization domain |
| NOD1 | Nucleotide-binding oligomerization domain-containing protein 1 |
| NOD2 | Nucleotide-binding oligomerization domain-containing protein 2 |
| AIM2 | Absent in melanoma-2 |
| NEK7 | NIMA-related kinase 7 |
| NIMA | Never in mitosis gene a |
| MARK4 | Microtubule-affinity regulating kinase 4 |
| GEF-H1 | Guanine nucleotide exchange factor-H1 |
| WT | Wild-type |
| BMDM | Bone marrow-derived macrophage |
| LPS | Lipopolysaccharide |
| SPI-1 | Salmonella pathogenicity island 1 |
| T3SS | Type 3 secretion system |
| RLR | RIG-I-like receptor |
References
- Kawai, T.; Akira, S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity 2011, 34, 637–650. [Google Scholar] [CrossRef] [Green Version]
- Martinon, F.; Burns, K.; Tschopp, J. The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef]
- Dinarello, C.A. Immunological and inflammatory functions of the interleukin-1 family. Annu. Rev. Immunol. 2009, 27, 519–550. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Gao, W.; Shao, F. Pyroptosis: Gasdermin-Mediated Programmed Necrotic Cell Death. Trends Biochem. Sci. 2017, 42, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Swanson, K.V.; Deng, M.; Ting, J.P. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef] [PubMed]
- Broz, P.; Monack, D.M. Newly described pattern recognition receptors team up against intracellular pathogens. Nat. Rev. Immunol. 2013, 13, 551–565. [Google Scholar] [CrossRef] [PubMed]
- Hughes, M.M.; O’Neill, L.A.J. Metabolic regulation of NLRP3. Immunol. Rev. 2018, 281, 88–98. [Google Scholar] [CrossRef]
- Guo, H.; Callaway, J.B.; Ting, J.P. Inflammasomes: Mechanism of action, role in disease, and therapeutics. Nat. Med. 2015, 21, 677–687. [Google Scholar] [CrossRef] [Green Version]
- Mostowy, S.; Shenoy, A.R. The cytoskeleton in cell-autonomous immunity: Structural determinants of host defence. Nat. Rev. Immunol. 2015, 15, 559–573. [Google Scholar] [CrossRef] [Green Version]
- Martinon, F.; Petrilli, V.; Mayor, A.; Tardivel, A.; Tschopp, J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 2006, 440, 237–241. [Google Scholar] [CrossRef] [Green Version]
- Misawa, T.; Takahama, M.; Kozaki, T.; Lee, H.; Zou, J.; Saitoh, T.; Akira, S. Microtubule-driven spatial arrangement of mitochondria promotes activation of the NLRP3 inflammasome. Nat. Immunol. 2013, 14, 454–460. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Zeng, M.Y.; Yang, D.; Motro, B.; Nunez, G. NEK7 is an essential mediator of NLRP3 activation downstream of potassium efflux. Nature 2016, 530, 354–357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmid-Burgk, J.L.; Chauhan, D.; Schmidt, T.; Ebert, T.S.; Reinhardt, J.; Endl, E.; Hornung, V. A Genome-wide CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats) Screen Identifies NEK7 as an Essential Component of NLRP3 Inflammasome Activation. J. Biol. Chem. 2016, 291, 103–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, H.; Wang, Y.; Li, X.; Zhan, X.; Tang, M.; Fina, M.; Su, L.; Pratt, D.; Bu, C.H.; Hildebrand, S.; et al. NLRP3 activation and mitosis are mutually exclusive events coordinated by NEK7, a new inflammasome component. Nat. Immunol. 2016, 17, 250–258. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Thome, S.; Ma, X.; Amrute-Nayak, M.; Finigan, A.; Kitt, L.; Masters, L.; James, J.R.; Shi, Y.; Meng, G.; et al. MARK4 regulates NLRP3 positioning and inflammasome activation through a microtubule-dependent mechanism. Nat. Commun. 2017, 8, 15986. [Google Scholar] [CrossRef] [Green Version]
- Ren, Y.; Li, R.; Zheng, Y.; Busch, H. Cloning and characterization of GEF-H1, a microtubule-associated guanine nucleotide exchange factor for Rac and Rho GTPases. J. Biol. Chem. 1998, 273, 34954–34960. [Google Scholar] [CrossRef] [Green Version]
- Krendel, M.; Zenke, F.T.; Bokoch, G.M. Nucleotide exchange factor GEF-H1 mediates cross-talk between microtubules and the actin cytoskeleton. Nat. Cell Biol. 2002, 4, 294–301. [Google Scholar] [CrossRef]
- Birkenfeld, J.; Nalbant, P.; Yoon, S.H.; Bokoch, G.M. Cellular functions of GEF-H1, a microtubule-regulated Rho-GEF: Is altered GEF-H1 activity a crucial determinant of disease pathogenesis? Trends Cell Biol. 2008, 18, 210–219. [Google Scholar] [CrossRef]
- Kashyap, A.S.; Fernandez-Rodriguez, L.; Zhao, Y.; Monaco, G.; Trefny, M.P.; Yoshida, N.; Martin, K.; Sharma, A.; Olieric, N.; Shah, P.; et al. GEF-H1 Signaling upon Microtubule Destabilization Is Required for Dendritic Cell Activation and Specific Anti-tumor Responses. Cell Rep. 2019, 28, 3367–3380. [Google Scholar] [CrossRef] [Green Version]
- Fukazawa, A.; Alonso, C.; Kurachi, K.; Gupta, S.; Lesser, C.F.; McCormick, B.A.; Reinecker, H.C. GEF-H1 mediated control of NOD1 dependent NF-kappaB activation by Shigella effectors. PLoS Pathog. 2008, 4, e1000228. [Google Scholar] [CrossRef]
- Matsuzawa, T.; Kuwae, A.; Yoshida, S.; Sasakawa, C.; Abe, A. Enteropathogenic Escherichia coli activates the RhoA signaling pathway via the stimulation of GEF-H1. EMBO J. 2004, 23, 3570–3582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Alonso, C.; Ballester, I.; Song, J.H.; Chang, S.Y.; Guleng, B.; Arihiro, S.; Murray, P.J.; Xavier, R.; Kobayashi, K.S.; et al. Control of NOD2 and Rip2-dependent innate immune activation by GEF-H1. Inflamm. Bowel Dis. 2012, 18, 603–612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Zagani, R.; Park, S.M.; Yoshida, N.; Shah, P.; Reinecker, H.C. Microbial recognition by GEF-H1 controls IKKepsilon mediated activation of IRF5. Nat. Commun. 2019, 10, 1349. [Google Scholar] [CrossRef] [PubMed]
- Chiang, H.S.; Zhao, Y.; Song, J.H.; Liu, S.; Wang, N.; Terhorst, C.; Sharpe, A.H.; Basavappa, M.; Jeffrey, K.L.; Reinecker, H.C. GEF-H1 controls microtubule-dependent sensing of nucleic acids for antiviral host defenses. Nat. Immunol. 2014, 15, 63–71. [Google Scholar] [CrossRef] [Green Version]
- Eisenbarth, S.C.; Colegio, O.R.; O’Connor, W.; Sutterwala, F.S.; Flavell, R.A. Crucial role for the Nalp3 inflammasome in the immunostimulatory properties of aluminium adjuvants. Nature 2008, 453, 1122–1126. [Google Scholar] [CrossRef]
- Schlesinger, N.; Schumacher, R.; Catton, M.; Maxwell, L. Colchicine for acute gout. Cochrane Database Syst. Rev. 2006, 4, CD006190. [Google Scholar]
- Fu, M.M.; Holzbaur, E.L. Integrated regulation of motor-driven organelle transport by scaffolding proteins. Trends Cell Biol. 2014, 24, 564–574. [Google Scholar] [CrossRef] [Green Version]
- Reed, N.A.; Cai, D.; Blasius, T.L.; Jih, G.T.; Meyhofer, E.; Gaertig, J.; Verhey, K.J. Microtubule acetylation promotes kinesin-1 binding and transport. Curr. Biol. 2006, 16, 2166–2172. [Google Scholar] [CrossRef] [Green Version]
- Meiri, D.; Marshall, C.B.; Greeve, M.A.; Kim, B.; Balan, M.; Suarez, F.; Bakal, C.; Wu, C.; Larose, J.; Fine, N.; et al. Mechanistic insight into the microtubule and actin cytoskeleton coupling through dynein-dependent RhoGEF inhibition. Mol. Cell 2012, 45, 642–655. [Google Scholar] [CrossRef] [Green Version]
- Broz, P.; Newton, K.; Lamkanfi, M.; Mariathasan, S.; Dixit, V.M.; Monack, D.M. Redundant roles for inflammasome receptors NLRP3 and NLRC4 in host defense against Salmonella. J. Exp. Med. 2010, 207, 1745–1755. [Google Scholar] [CrossRef]
- Lundberg, U.; Vinatzer, U.; Berdnik, D.; von Gabain, A.; Baccarini, M. Growth phase-regulated induction of Salmonella-induced macrophage apoptosis correlates with transient expression of SPI-1 genes. J. Bacteriol. 1999, 181, 3433–3437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, Q.Z.; Yang, F.; Li, C.G.; Xu, L.H.; He, X.H.; Mai, F.Y.; Zeng, C.Y.; Zhang, C.C.; Zha, Q.B.; Ouyang, D.Y. Paclitaxel Enhances the Innate Immunity by Promoting NLRP3 Inflammasome Activation in Macrophages. Front. Immunol. 2019, 10, 72. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.H.; Wood, G.; Kastner, D.L.; Chae, J.J. Pyrin inflammasome activation and RhoA signaling in the autoinflammatory diseases FMF and HIDS. Nat. Immunol. 2016, 17, 914–921. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; Yang, J.; Liu, W.; Wang, Y.; Shao, F. Site-specific phosphorylation and microtubule dynamics control Pyrin inflammasome activation. Proc. Natl. Acad. Sci. USA 2016, 113, E4857–E4866. [Google Scholar] [CrossRef] [Green Version]
- Van Gorp, H.; Saavedra, P.H.; de Vasconcelos, N.M.; Van Opdenbosch, N.; Vande Walle, L.; Matusiak, M.; Prencipe, G.; Insalaco, A.; Van Hauwermeiren, F.; Demon, D.; et al. Familial Mediterranean fever mutations lift the obligatory requirement for microtubules in Pyrin inflammasome activation. Proc. Natl. Acad. Sci. USA 2016, 113, 14384–14389. [Google Scholar] [CrossRef] [Green Version]
- Schnappauf, O.; Chae, J.J.; Kastner, D.L.; Aksentijevich, I. The Pyrin Inflammasome in Health and Disease. Front. Immunol. 2019, 10, 1745. [Google Scholar] [CrossRef]
- Raupach, B.; Peuschel, S.K.; Monack, D.M.; Zychlinsky, A. Caspase-1-mediated activation of interleukin-1beta (IL-1beta) and IL-18 contributes to innate immune defenses against Salmonella enterica serovar Typhimurium infection. Infect. Immun. 2006, 74, 4922–4926. [Google Scholar] [CrossRef] [Green Version]
- Qu, Y.; Misaghi, S.; Newton, K.; Maltzman, A.; Izrael-Tomasevic, A.; Arnott, D.; Dixit, V.M. NLRP3 recruitment by NLRC4 during Salmonella infection. J. Exp. Med. 2016, 213, 877–885. [Google Scholar] [CrossRef]
- Franchi, L.; Amer, A.; Body-Malapel, M.; Kanneganti, T.D.; Ozoren, N.; Jagirdar, R.; Inohara, N.; Vandenabeele, P.; Bertin, J.; Coyle, A.; et al. Cytosolic flagellin requires Ipaf for activation of caspase-1 and interleukin 1beta in salmonella-infected macrophages. Nat. Immunol. 2006, 7, 576–582. [Google Scholar] [CrossRef]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lai, H.-J.; Hsu, Y.-H.; Lee, G.-Y.; Chiang, H.-S. Microtubule-Mediated NLRP3 Inflammasome Activation Is Independent of Microtubule-Associated Innate Immune Factor GEF-H1 in Murine Macrophages. Int. J. Mol. Sci. 2020, 21, 1302. https://doi.org/10.3390/ijms21041302
Lai H-J, Hsu Y-H, Lee G-Y, Chiang H-S. Microtubule-Mediated NLRP3 Inflammasome Activation Is Independent of Microtubule-Associated Innate Immune Factor GEF-H1 in Murine Macrophages. International Journal of Molecular Sciences. 2020; 21(4):1302. https://doi.org/10.3390/ijms21041302
Chicago/Turabian StyleLai, Hsuan-Ju, Yi-Hsuan Hsu, Guan-Ying Lee, and Hao-Sen Chiang. 2020. "Microtubule-Mediated NLRP3 Inflammasome Activation Is Independent of Microtubule-Associated Innate Immune Factor GEF-H1 in Murine Macrophages" International Journal of Molecular Sciences 21, no. 4: 1302. https://doi.org/10.3390/ijms21041302