Estradiol Metabolism: Crossroads in Pulmonary Arterial Hypertension

1
Heart, Lung, Blood and Vascular Medicine Institute, University of Pittsburgh, BST E1240, 200 Lothrop Street, Pittsburgh, PA 15261, USA
2
Department of Pharmacology and Chemical Biology University of Pittsburgh School of Medicine, 100 Technology Drive, PA 15219, USA
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2020, 21(1), 116; https://doi.org/10.3390/ijms21010116
Submission received: 2 December 2019
/
Accepted: 17 December 2019
/
Published: 23 December 2019
(This article belongs to the Special Issue Molecular Research on Pulmonary Hypertension 2.0)
Abstract
:Pulmonary arterial hypertension (PAH) is a debilitating and progressive disease that predominantly develops in women. Over the past 15 years, cumulating evidence has pointed toward dysregulated metabolism of sex hormones in animal models and patients with PAH. 17β-estradiol (E2) is metabolized at positions C2, C4, and C16, which leads to the formation of metabolites with different biological/estrogenic activity. Since the first report that 2-methoxyestradiol, a major non-estrogenic metabolite of E2, attenuates the development and progression of experimental pulmonary hypertension (PH), it has become increasingly clear that E2, E2 precursors, and E2 metabolites exhibit both protective and detrimental effects in PH. Furthermore, both experimental and clinical data suggest that E2 has divergent effects in the pulmonary vasculature versus right ventricle (estrogen paradox in PAH). The estrogen paradox is of significant clinical relevance for understanding the development, progression, and prognosis of PAH. This review updates experimental and clinical findings and provides insights into: (1) the potential impacts that pathways of estradiol metabolism (EMet) may have in PAH; (2) the beneficial and adverse effects of estrogens and their precursors/metabolites in experimental PH and human PAH; (3) the co-morbidities and pathological conditions that may alter EMet and influence the development/progression of PAH; (4) the relevance of the intracrinology of sex hormones to vascular remodeling in PAH; and (5) the advantages/disadvantages of different approaches to modulate EMet in PAH. Finally, we propose the three-tier-estrogen effects in PAH concept, which may offer reconciliation of the opposing effects of E2 in PAH and may provide a better understanding of the complex mechanisms by which EMet affects the pulmonary circulation–right ventricular interaction in PAH.
1. Introduction
Pulmonary arterial hypertension (PAH), a debilitating progressive cardiopulmonary disease in which increased pulmonary arterial pressure leads to right ventricle (RV) failure and premature death, predominantly develops in women. In the past two decades, data coming from various registries worldwide have confirmed the early observations of female preponderance of PAH made in the 1950s by Doctors Drexler and Wood [1,2]. The female-to-male (F:M) ratio in PAH ranges from 2:1 to 4:1, and even higher (4:1 to 9:1) in PAH associated with connective-tissue diseases (CTD) [3,4,5,6]. In elderly PAH patients, the gender bias disappears (F:M—1.2:1.0) [7], suggesting that female sex hormones are a risk factor for developing PAH. Yet, despite the very early recognition of female preponderance in PAH, for decades, estrogens were held to be protective in PAH. Consequently, the potential pathogenic roles of estrogens in PAH were ignored.
Several factors may have contributed to this lack of interest in the pathogenic roles of estrogens in PAH. First, estrogens are protective in classic rodent models of monocrotaline (MCT)-induced and chronic hypoxia (CH)-induced pulmonary hypertension [8]. We coined the term “estrogen paradox” to describe the discrepancy between female preponderance in humans and the protective effects of estrogens in classic models of pulmonary hypertension [8]. The limitations of classic models of pulmonary hypertension have been reviewed elsewhere [9,10,11]. Studying the role of sex hormones in these models may be of limited value and even misleading [8,12]. Therefore, we refer to the disease in rodents as pulmonary hypertension (PH). In classic models, there is minor involvement of endothelium and absence of occlusive and plexiform lesions (PXL), the hallmarks of vascular remodeling in human PAH. Media hypertrophy, which dominates in these models and is inhibited by estrogens, eventually disappears with the passage of time (MCT-PH) or upon re-exposure to normoxia (CH-PH). This is in contrast to the progressive character of disease in humans. Second, similar to their effects on systemic circulation, estrogens may be protective in the intact pulmonary vasculature, yet in injured endothelium that has been exposed to multiple unknown hits, estrogens may stimulate angioproliferation, promote inflammation, and augment the development of occlusive lesions. Finally, similar to the left heart, female sex hormones may protect the right ventricle (RV) when it is exposed to increased afterload. Recent data in animals, healthy people, and PAH patients support this notion [13,14,15,16]. Recently, the estrogen paradox concept was expanded to encompass the beneficial effects of estrogens concerning RV function and prognosis in PAH [17]. Collectively, we proposed the three-tier effects of estrogens in PAH concept [18], which may help in translating animal data to humans and in elucidating the role of sex hormones in the development, progression, and prognosis of PAH in both women and men.
17β-Estradiol (E2) metabolism occurs primarily by oxidation at the C2, C4, and C16 positions of E2 and leads to the formation of metabolites with different biological and estrogenic activity (Figure 1). Since the first report that 2-methoxyestradiol (2ME, a major non-estrogenic metabolite of E2) attenuates the development and progression of MCT-induced PH [12], it has become increasingly clear that E2 and its precursors and metabolites exhibit both protective and detrimental effects in PH [8,19]. Accumulating evidence indicates that, at least in part, both the protective and adverse effects of estradiol in experimental PH are mediated by these metabolites [20,21,22]. In the last decade, evidence has accumulated pointing toward dysregulated metabolism of sex hormones in animal models and patients with PAH. Importantly, these findings suggest the use of estrogen precursors, inhibitors of estrogen production and signaling, and non-estrogenic E2 metabolites for the treatment of PAH.
Previous reviews have focused on the effects of estrogens and their metabolites on pulmonary vascular pathobiology and the development of experimental PH [8,19]. The goal of this review is to provide updated insights into the following: (1) the potential impact that pathways of estradiol metabolism (EMet) may have on PAH; (2) the beneficial and adverse effects of estrogens and their precursors/metabolites in experimental PH and human PAH; (3) the co-morbidities and pathological conditions that, by altering EMet, may influence the development and progression of PAH; (3) the relevance of the intracrinology of sex hormones to vascular remodeling in PAH; and (5) the advantages and disadvantages of different approaches to modulate EMet in PAH. Estrogen signaling in PAH will be only briefly addressed in the context of their metabolism. For a comprehensive review of estrogen signaling in PAH readers are referred to review articles by Lahm and colleagues [23,24].
2. Sulfotransferase (SULT)–Sulfatase (STS) Pathway
The synthesis and breakdown of estrogens are complex (Figure 1). Dehydroepiandrosterone (DHEA) serves as pivotal precursor for E2 biosynthesis. DHEA is converted to DHEA sulfate (DHEA-S) by sulfotransferase (SULT), and DHEA-S can be converted back to DHEA by steroid sulfatase (STS). The balanced effects of STS and SULT [25] control the ratio of DHEA to DHEA-S. Until recently, sulfated steroids including DHEA-S were considered inert circulating metabolic products that were incapable of binding to and activating nuclear receptors. Circulating sulfated steroids are hydrophilic and they are readily available for renal excretion; to produce local/cellular effects they would require organic anion transporters (OATPs) for active transport into cells. Notably, OATPs are expressed in multiple cells and tissues, including endothelial and inflammatory cells and lungs [26,27]. Besides DHEA-S, other important hormone substrates for STS include estrone sulfate (E1-S) and estradiol sulfate (E2-S). Therefore, the sulfatase pathway represents a major intracrine route for regenerating biologically active estrogens (Figure 1). On the other hand, in target cells, the cytosolic enzyme SULT, by conjugating a sulfonate group to estrogens, inhibits E2 binding to and activity at estrogen receptors (ER) and augments the renal excretion of estrogens [28]. Thereby, SULT and STS are negative and positive regulators of the estrogen activity, respectively [29]. Sulfotransferase also catalyzes sulfation of 2ME, and increased SULT activity correlates with diminished anti-mitogenic effects of 2ME in various cancer cell lines [30]. Hence, SULT may modulate E2 signaling, regulate E2 intracrinology and inactivate biologically active E2 metabolites [30].
DHEA is synthesized in both postmenopausal women and men by the adrenal cortex and by peripheral conversion from circulating DHEA-S, and by ovaries and placenta in premenopausal women [31,32]. Adrenal production of DHEA takes place only in humans and primates, whereas rodents and other species have very low levels of DHEA that are produced by other organs. DHEA and its downstream metabolite androstenedione are the most abundant circulating steroids. The conversion of DHEA to androstenedione is facilitated by 3β-HSD, and in peripheral tissues, circulating androstenedione can be converted by aromatase to estrone [33,34].
2.1. SULT–STS Pathway in PAH
Although the roles of sulfatase, SULT, 3β-HSD, and androstenedione in PAH are unknown, a growing body of evidence supports important roles for DHEA in PAH (Table 1). At the cellular level, DHEA reduces hypoxia-induced accumulation of hypoxia-inducible factor 1α (HIF)-1α in human pulmonary artery endothelial cells (hPAECs) [35] and stimulates NO release in bovine endothelial cells (ECs) [36]. In animals, preventive and/or rescue treatments with DHEA or DHEA-S are effective in CH-PH [37,38,39], MCT-PH [40,41], fetal pulmonary circulation in sheep [42], the rat model of PH in infants [43], and in Sugen 5416 + hypoxia (Su-Hx)-induced PH [44]. DHEA also attenuates PH in patients with chronic obstructive pulmonary disease [45].
Recently, Ventetuolo et al. [46] reported that lower DHEA-S, greater E2 levels, and increased E2- to-testosterone ratios correlated with shorter 6-min walking distances (6MWD) and were associated with a greater risk of PAH in men. Furthermore, DHEA-S levels were inversely associated with right atrial pressures and pulmonary vascular resistances. Similar changes in sex hormones were associated with worse hemodynamics, functional class, shorter 6MWD, and greater risk of death in post-menopausal women with idiopathic-, CTD-, or heart disease-associated PAH [47]. Altogether, the reported pattern of hormonal changes in men and postmenopausal women with PAH points toward increased activity of sulfatase and increased downstream aromatization of androgens.
2.2. Future Directions and Clinical Implications
The above data suggest potential therapeutic effects of DHEA in PAH. However, although increased estrogen levels by pharmacological doses of DHEA may be beneficial with respect to the RV, they may exacerbate pulmonary angioproliferative remodeling and may increase the risk of sex hormone-dependent cancer in women with PAH. Although DHEA is an over-the-counter supplement with no major acute side effects, the long-term safety of DHEA in patients with PAH is unknown. Therefore, the long-term safety of high doses of DHEA (and the effects of subsequently elevated circulating estrogens) should be further investigated, at least in the Su+Hx PH model.
3. Aromatase Pathway
The aromatase pathway is a major biochemical system for the production of E2. Aromatase is encoded by the CYP19A1 gene and converts testosterone to E2 and androstenedione to E1. This conversion takes place in the ovaries and in peripheral tissues, and the aromatase pathway is a major source of extra gonadal estrogen production in postmenopausal women and men [66]. Compared to other CYP450 encoding genes, the CYP19A1 gene is unique, as it contains a number of untranslated exons that occur in aromatase transcripts in a tissue-specific fashion, due to differential splicing that gives rise to tissue-specific promoters [67,68]. Therefore, the regulation of aromatase via alternatively used promoters can differ in various tissues/organs in health and disease, and might be activated/inhibited by a host of local factors (including estrogens and their metabolites) [66,67]. Relevant to PAH, human endothelium expresses an entire aromatase–estrogen–E2 receptor system.
3.1. Aromatase in PAH
Early studies reported 2–3-fold greater aromatase activity in male than in female neuroendocrine tissues [67,69], whereas the most recent ones suggest increased aromatase expression in female human pulmonary artery smooth muscle cells (hPASMCs) and healthy and Sugen 5416 + Hypoxia treated female murine lung [55], but decreased lung aromatase expression in female MCT-PH rats [48]. These reported discrepancies may reflect tissue/disease specific regulation of aromatase. Similar to the increased aromatization of androgens in postmenopausal women and men with PAH [46,47], in patients with portopulmonary PAH, irrespective of sex, elevated aromatase activity and plasma E2 levels are associated with increased risk of PAH [58]. Significantly, E2 augments aromatase activity, and by increasing aromatization of androgens may augment its own production [70]. In contrast, 2-ME inhibits both basal- and TNFα-stimulated aromatase activity [71], and thereby may reduce extra-gonadal synthesis of estrogens.
In humans, the third-generation aromatase inhibitor (ARO-I) anastrozole reduces not only E2, but also E1 and E1S levels, suggesting indirect inhibition of local E2 production via 17β–HSD and sulfatase pathways. In hypoxic mice and Su-Hx rats, anastrozole reduces E2 levels and attenuates PH in female, but not in male animals [55]. Moreover, in combination with fulvestrant (selective estrogen receptor degrader) anastrozole reverses PH in BMPR2 mutant mice [57]. Finally, metformin, a first line anti-diabetic drug, inhibits aromatase expression [72] and has beneficial effects in angioproliferative PH in female rats [56]. Collectively, the experimental and observational data point toward a potential use of ARO-I in PH. Indeed, in a recent pilot phase 2 clinical trial in postmenopausal women and men with PAH, Kawut et al. reported that 12-week treatment with anastrozole reduced serum E2 levels by 40% and E1 levels by 70%, and significantly increased 6MWD [59].
3.2. Aromatase Inhibition and RV Function in PAH
Although experimental and initial clinical data with anastrazole are encouraging, the benefits and potential harm of long-term inhibition of E2 synthesis are unknown. ARO-I is well tolerated by cancer patients, yet reduced E2 production by ARO-I could deprive the RV of the protective effects of estrogens, in particular in PAH, where the RV is exposed to increased workload. In this regard, both exogenous and endogenous E2 protect RV function and structure in experimental PH [15,62]. Data from the Multi-Ethnic Study of Atherosclerosis (MESA study) clearly indicate that high E2 levels benefit RV function [14], and in ovariectomized (OVX) Su-Hx rats, exogenous E2 protects RV function by preserving mitochondrial content and oxidative capacity [60]. Preliminary data in female Su-Hx rats (published in abstract form [54]) suggest that despite reducing PH and the number of occlusive lesions, anastrozole does not reduce RV hypertrophy, as would be expected from reduced occlusive remodeling and reduced afterload. Finally, although anastrazole on average had no effects on echocardiographic parameters and biomarkers of RV function in the proof-of-concept study on PAH, anastrazole had varying effects on individual patients’ RV function, with some experiencing worsening in RV function [59].
3.3. Future Directions and Clinical Implications
The ongoing large phase 2 study (NCT03229499) should provide data on the short-term efficacy/safety of anastrazole in PAH. Yet, the long-term effects of anastrozole on the progression of disease, RV function, and mortality will remain unknown. Studies in the Su+Hx model focused on the interaction between anastrozole and progression of RV dysfunction could help address the potential long-term negative impact of anastrazole on RV function. Based on our preliminary data (published abstract [73]), we suggest studies of anastrazole in female rats exposed to escalating doses of SU5416 (20-50-100 mg/kg) that rapidly (within five to six weeks) produce an increasing level of RV dysfunction/failure.
4. 2-Hydroxylation/2–Methylation Pathway
Once formed, E2 can be converted to biologically active metabolites via the sequential steps of hydroxylation mediated by multiple CYP450 enzymes (Figure 1 and Figure 2), followed by methylation of hydroxyl groups catalyzed by catechol-O-methyl transferase (COMT). The 2-hydroxylation/2-methylation pathway is a major metabolic process that, by some estimates, accounts for ~50% of E2 metabolism. This process largely takes place in the liver, where E2 is mainly metabolized by CYP1A1/1A2/1B1 and CYP3A4 to 2-hydroxyestradiol (2HE) and, to a lesser degree (~5%), by Cyp1B1 to 4-hydroxyestradiol (4HE). Catechol estrogens have various levels of estrogenic activity and are converted by COMT to methoxyestradiols. In addition to hepatocytes and cancer cells, the conversion of E2 to downstream 2HE and 2ME also takes place in cardiovascular compartments [65]. The 2-hydroxylaton/methylation pathway produces non-estrogenic, anti-proliferative, anti-angiogenic, and anti-inflammatory metabolites (2HE and 2ME) [74]. Notably, the inhibition of CYP1A1 or COMT attenuates or blocks the antimitotic effects of E2 [74]. Furthermore, the inhibition of vascular remodeling in MCT-OVX rats by E2 and inhibition of carotid artery remodeling by 2ME are associated with reduced p-Akt and increased COX-2 expression [48,75].
4.1. 2-Hydroxylation/2-Methylation Pathway in PAH
Currently, the impact of this metabolic pathway in PAH are unknown. Yet, the above findings suggest that in vivo, the anti-proliferative effects of E2 in the vascular compartment may be at least in part mediated by the metabolism of E2 to 2ME (Figure 2). Indeed, as summarized in Table 2, 2ME not only reverses MCT-PH in male rats, but also, at least in part, mediates the beneficial effects of E2 in OVX rats with MCT- and bleomycin-induced PH and lung fibrosis [12,20,21]. 2ME is also protective in hypoxia-induced PH [76,77,78] and 2ME has synergistic therapeutic effects with sildenafil and bosentan to ameliorate MCT-induced PH, inflammation, and vascular remodeling [79]. Compared to E2, in ECs 2ME is a more potent modulator of NO, prostacyclin, and endothelin synthesis [8,74,80], three major pharmacological targets in PAH. The 2ME-induced NO release in ECs is mediated via activation of the PPARγ/PI3K/Akt pathway and results in vasodilation [81]. 2ME could be viewed as a mediator of E2′s beneficial effects in PAH. The high proliferative state of human leiomyoma cells (hLCs) is characterized by doubled ERα signaling, which is inherently regulated by microtubule dynamics [82]. Notably, COMT over expression or treatment with 2ME stabilizes microtubules, attenuates E2-induced proliferation, inhibits ERα signaling, and reduces HIF-1α and aromatase expression in hLCs [83,84]. That COMT and 2ME may induce similar changes in highly proliferative hPASMCs overexpressing aromatase and pulmonary artery endothelial cells (PAECs) overexpressing HIF-1α [55] is an attractive possibility that needs to be examined.
4.2. Other Effects of the 2-Methylation Pathway Relevant to PAH
There are several aspects of the 2-methylation metabolic pathway that are relevant to PAH. In this regard, cumulating evidence suggests that the renin–angiotensin system (RAS) contributes to the development of PAH [85,86,87]. Remarkably, COMT deficiency is associated with increased sensitivity to angiotensin II (Ang II), and in COMT -/- and CYP1B1 -/- mice that have reduced 2ME production, 2ME treatment abolishes hypersensitivity to and vascular injury induced by Ang II [88,89]. Of note is that 2ME via activation of the G-protein-coupled estrogen receptor (GPER) downregulates Ang II type 1 receptor [81,82,90,91]. Of relevance to PAH, 2ME attenuates Ang-II induced vascular and cardiac remodeling and fibrosis and isoproterenol-induced (Ang II-mediated) RV and LV hypertrophy and fibrosis [92].
Another aspect of the 2-methylation metabolic pathway relevant to PAH is its effects on obesity and the metabolic syndrome (MS), two established risk factors for PAH [93,94,95,96,97,98]. Experimental and clinical data link reduced COMT activity and 2ME levels to the development of obesity and insulin resistance [99,100]. Moreover, a low-activity variant of COMT (val/met 158) may contribute to MS [99], whereas the high-activity form of COMT (rs4680) is associated with lower HbA1c and protection from type 2 diabetes [101]. Also, both genetic and pharmacologically-induced deficiency of COMT exacerbates high-fat diet (HFD)-induced insulin resistance in mice [100]. In COMT-deficient mice on HFD, 2ME, which shares structural similarity with PPARγ ligands and acts as a PPARγ agonist [75,89], increases glucose tolerance, insulin secretion, and adenosine monophosphate kinase (AMPK) phosphorylation in the liver and islet cells [100].
Yet another aspect of the 2-methylation pathway potentially relevant to PAH is the fact that E2 regulates COMT activity and possibly its downstream conversion to non-estrogenic 2ME. On this subject, COMT is highly expressed in human and rat lungs [102,103]; men have higher hepatic COMT activity than women [104]; in vitro E2 decreases COMT transcription, activity, and protein levels [105,106]; exposure to E2 reduces hepatic COMT activity in rats [107]; and by antagonizing E2, tamoxifen increases COMT activity in peripheral tissues [108].
Additional biological effects of 2ME that may be of relevance to PAH are summarized in Figure 2. It should be reemphasized that in cardiovascular and renal cells, 2ME does not interact with classical ERα and ERβ nuclear receptors [74]. Cardiac and vascular protection of E2 has been linked to the activation of GPER located on the cell membrane [109]. More importantly, the activation of GPER reverses PH-related cardiopulmonary dysfunction and exercise intolerance in both male and female MCT-PH rats [110,111]. In this regard, 2ME binds the membrane GPER and, via GPER-mediated transactivation of EGFR and ERK1/2 phosphorylation, downregulates AT1 receptor expression [90,91]. Yet, the antimitogenic effects of 2ME in microglia cells and vascular smooth muscle cells (VSMCs) are independent of GPER or nuclear E2 receptors [112]. Whether cardiovascular protective effects of 2ME are mediated by GPER remains to be determined. Inhibition of the HIFα–VEGF axis is one of the most prominent effects of 2ME. This action is highly relevant to PAH and is discussed further in Section 7.
4.3. Future Directions and Clinical Implications
The role of COMT and the effects of 2ME in patients with PAH are unknown. Nonetheless, a growing body of evidence strongly supports further investigation of the E2–COMT–2ME interrelationships in experimental angioproliferative PH and PAH patients. This evidence includes: (1) the modulation of RAS activity by COMT and 2ME; (2) the role of COMT and 2ME in insulin resistance and the metabolic syndrome; (3) the effects E2 on COMT activity; and (4) the opposite effects of 2ME and E2 on angiogenesis (see Section 7).
5. 4- and 16α-Hydroxylation Pathways
The oxidation of estradiol at C4 by CYP1B1 leads to the production of 4-hydroxyestradiol (4HE), a catechol estradiol with reactive oxygen species (ROS)-dependent, but ER-independent, carcinogenic effects. 4HE can undergo methylation by COMT to 4-methoxyestradiol (4ME), oxidation to quinones, or dehydrogenation (by 17β–HSD) to 4-hydroxyestrone (4HE1; Figure 1). The oxidation of E2 and E1 at C16 (by CYP1A1, CYP1A2, CYP2C8, and CYP3A) produces 16α-hydroxylestradiol (16αHE; estriol) and 16α-hydroxyestrone (16αHE1), respectively. Estriol is a weak estrogen produced in abundance during pregnancy and is further oxidized by 17β–HSD2 to 16αHE1, a highly estrogenic metabolite that covalently binds to ERs and induces prolonged ER activation [121].
5.1. Role of CYP1B1 in PAH
New data obtained over the last decade suggest a major pathogenic role for 16αHE1 and Cyp1B1 in PAH. Both human and animal data coming largely from Vanderbilt [120,122,123,124], Glasgow [119,125,126], and Penn State Universities [14,127] clearly point toward a pathogenic role for CYP1B1 with regard to the risk of PAH in humans and toward a pathogenic 16αHE1–BMPR2 interaction in experimental PH. What is less clear is: (1) whether the 16α-hydroxylation pathway (i.e., 16αHE1) is a major pathogenic factor in human PAH; (2) whether CYP1B1 activity is directly linked to significant production of 16αHE1; and (3) whether the reported effects are related directly to CYP1B1 modulation of EMet versus the effects of CYP1B1 unrelated to EMet. What has been overlooked is: (1) differences in CYP1B1 activity in humans versus rodents; (2) the promiscuity of CYP1B1 for various non-estrogenic substrates (and thereby the effects of CYP1B1 inhibitors unrelated to estrogen metabolism); (3) the lack of direct/causative evidence about CYP1B1 effects on 16αHE1 production; and (4) limitations of interpreting urinary estrogen data (2HE1/16α-HE1 ratio) to draw associations between CYP1B1 activity, 16αHE1 production, and their pathogenic roles in PAH.
5.2. Divergent Effects on E2 and 2ME on CYP1B1 Activity
In humans, CYP1B1 is constitutively expressed in the lung and other tissues, including VSMCs and ECs [128], but not hepatocytes. Initial data indicated that human CYP1A1 predominantly controls 2HE and CYP1B1 4HE formation [129]. Importantly, in contrast to human CYP1B1, which favors a four-fold greater production of 4HE versus 2HE, rat CYP1B1 favors a two-fold production of 2HE compared to 4HE [130]. Notably, E2 and 2ME have divergent effects on CPY1B1 activity. Estradiol is not only a substrate for CYP1B1, but also (via ERα) is a transcriptional activator of CYP1B1 [131]. In contrast, in vitro 2ME exerts feedback inhibition on CYP1B1 activity [132] and inhibits aryl hydrocarbon receptor-mediated induction of CYP1B1 and downstream production of reactive metabolites. In vivo 2ME significantly inhibits CYP1B1 expression, reduces mid-chain hydroxyeicosatetraenoic acid (HETE) production, and attenuates pressure overload-induced cardiac remodeling [133].
5.3. Role of CYP1B1 and Estrogens in Arachidonic Acid Metabolism
In addition to the oxidation of xenobiotic and production of catechol estrogens, CYP1B1 is also involved in the metabolism of arachidonic acid (AA), melatonin, and retinoid. Of particular relevance to PAH is CYP1B1 lipoxygenase-like activity, which facilitates AA metabolism into mid-chain hydroxyeicosatetraenoic acids (HETEs) and epoxyeicosatrienoic acids (EETs; Figure 3) [134]. For example, CYP1B1 metabolizes AA into 12- and 20-HETEs, which stimulate VSMC growth [135]. HETEs also promote hypoxic pulmonary vasoconstriction and vascular remodeling and inhibit apoptosis of PAVSMCs [136,137,138]. In vivo inhibition of CYP1B1 reduces HETE formation and doxorubicin-induced cardiac fibrosis/failure [139]. In PAVSMCs and pulmonary artery ECs (PAECs), both EETs and HETEs have inflammatory, mitogenic/angiogenic, and vasoconstrictive effects and are implicated in the development of hypoxic PH [136,140]. Furthermore, in PAH patients, there is increased production of HETEs that correlates with poor prognosis [141].
What has been overlooked is the opposite effects that E2 and 2ME may have on HETE and EET production/metabolism. In this regard, E2 via ERα stimulates CYP1B1 activity and may increase HETE and EET production [142,143]. In contrast, 2ME inhibits CYP1B1 activity and reduces HETE formation and cardiac remodeling in rats with pressure overload-induced cardiac hypertrophy [114]. Moreover, E2 inhibits the expression/activity of soluble epoxide hydrolase (sEH), a key enzyme in EET metabolism [144]. Several studies linked low sEH activity to the pathophysiology of PAH: (1) E2-, genetic-, and pharmacologically-induced downregulation of sEH (and increased pulmonary EET) potentiates hypoxic pulmonary vasoconstriction [145,146,147]; (2) lungs from PH patients express no/little sEH; (3) hypoxia decreases the expression of sEH; and (4) when exposed to hypoxia, sEH KO mice exhibit exacerbated pulmonary vascular remodeling [147]. It seems that E2 plays a role in female-specific (physiological) downregulation of sEH, as suggested by the much higher sEH activity in male and OVX mice compared to intact female mice [148,149]. The E2 regulated sexually dimorphic expression of sEH and bioavailability of pathogenic EET and HETE in the pulmonary circulation could explain the female preponderance of PAH [142].
5.4. Future Directions and Clinical Implications
The fact that E2 in a sex-specific manner may regulate the bioavailability of pathogenic EET and HETE in the pulmonary circulation suggests an intriguing explanation for the female preponderance of PAH. However, this hypothesis requires further investigation. Thus far, the roles of CYP1B1 and estrogens in AA metabolism in PAH have not been studied. Any further investigation and discussion regarding the pathogenic roles of CYP1B1 in PAH should address the effects of E2 on CYP1B1 and sEH activity and related EET and HETE production and metabolism. The latter is also relevant to the assessment of the claimed “major role” of CYP1B1 in production of 16αHE1, a “major” pathogenic E2 metabolite in PAH. The dual-metabolic activity of CYP1B1, proinflammatory AA metabolites, and inflammation-instigated feedforward mechanism of E2 production (Figure 3) may shift E2 metabolism toward the production of pathogenic 16αHE1 and significantly reduce the 2HE1/16α-HE1 ratio (processes documented in patients with autoimmune inflammatory diseases). Accordingly, the 2HE1/16α-HE1 ratio should not be used to draw associations between CYP1B1 activity, 16αHE1 production, and their pathogenic role in PAH.
6. 17β-Hydroxysteroid Dehydrogenase Pathway
17β-Hydroxysteroid dehydrogenases (17β-HSDs) catalyze stereospecific oxido-reduction reactions at position 17 of estrogens and androgens and play a key role in the last step of activation and first step of degradation of estrogens and androgens (Figure 4). Type-1 17β–HSD (17βHSD-1) catalyzes the reductive transformation of less estrogenic estrone (E1) to E2 and this process takes place not only in cancer cells, but also in vascular smooth muscle cells [150,151]. The oxidative type-2 17β-HSD (17βHSD-2) converts E2 to E1. Additionally, 17β-HSD types 7 and 12 catalyze the production of E2, types 3 and 5 contribute to testosterone production, and types 4, 8, and 10 oxidize E2 and testosterone to less active forms of these steroids [152].
6.1. 17β-HSD Pathway and 2ME Disposition
The impact of the intracrinology and cellular disposition of estrogens and their metabolite in the pulmonary vasculature and PAH is unknown. However, it seems that the 17βHSD pathway plays a critical role in 2ME inactivation. 2ME is extensively metabolized by 17βHSD-2 to 2-methoxyestrone (2ME1), a metabolite largely considered to be biologically inactive. 2ME exhibits highly antimitogenic effects in both ER+/ER- human breast cancer cell lines with low 17βHSD-2 activity, whereas cells with high 17βHSD-2 activity are selectively resistant to the antimitotic effects of 2ME [138,153]. Related to 2ME disposition, the very fast uptake contributing to high cellular concentrations (>15 μmol/L) of 2ME takes place in highly proliferative and E2 sensitive MCF7 cells [154], and similar fast uptake and high cellular concentrations should be expected for more lipophilic 2ME1. Furthermore, in rats with MCT-induced PH, increases in the dose of 2ME do not additionally reduce PH and RV hypertrophy, yet additionally inhibit media remodeling and inflammation [113]. Finally, continuous use of large oral doses of 2ME in patients with various types of cancer results in minimal 2ME urinary excretion, but yields high plasma levels of 2ME1 (10–20-fold higher than 2ME) [155,156,157]. The above data indicate that 2ME–2ME1 interconversion takes place in humans and suggest that the cellular disposition of 2ME and 2ME1 may play a significant role in the biological and pharmacological effects of 2ME.
6.2. 17β–HSD Pathway in Experimental PH
The above discussion and data in female cancer patients point toward a key role of the 17β–HSD pathway in intracrinology. Thus, 17β–HSD likely modulates the biological effects of estrogens and their metabolites by altering their cellular disposition. Our preliminary data published in abstract form [115,116,117] (manuscript in preparation) support the notion that this may be also the case in PH. For example, in vitro, in contrast to 2ME, 2ME1 has only mild antimitogenic effects at high pharmacological concentrations (10 μM) [116]. However, in the presence of retinoic acid (RA, a 17βHSD-1 inducer), 2ME1 strongly inhibits the growth of human pulmonary artery smooth muscle cells (PASMCs) and lung fibroblasts, suggesting that the 2ME–2ME1 interconversion (2ME ↔ 2ME1) takes place in cells involved in vascular remodeling in PH [116]. In vivo, in MCT-induced PH in male rats, 2ME and RA have synergistic therapeutic effects; yet in vitro (in the absence of 2ME1), RA does not influence the marked antimitogenic effects of 2ME [115]. In vivo, in male MCT rats, rescue treatment with 2ME1 reduces PH, media remodeling, and inflammation (influx of ED1 cells) [116]. Finally, in intact and OVX female Su-Hx rats that exhibit severe angioproliferative PH and sporadically develop necrotizing arteritis, rescue treatment with 2ME1 reduces PH, RV dysfunction and remodeling, and the number of occlusive lesions and prevents the development of grade 6 lesions [117].
6.3. Future Directions and Clinical Implications
Currently, whether the 17β–HSD metabolic pathway plays a role in PAH is unknown. Yet, available data suggest that the 17β–HSD pathway controls the intracellular disposition of estrogens and their metabolites, and thereby determines the relative concentrations of steroids with low versus high estrogenic activity and “good” versus “bad” pharmacological effects. This warrants further investigation into the participation of 17β–HSD in PAH. Studying the effects of selective 17β–HSD1 inducers and/or 17β–HSD2 inhibitors on EMet (i.e., intracrinology of E2 and 2ME) and on development/progression of disease in angioproliferative PH should be part of this line of research.
7. Angiogenesis, Metabolic Reprograming, and Estradiol Metabolism in PAH
Vascular lesions in patients with severe PAH are characterized by the existence of two types of EC phenotypes: (1) normal quiescent apoptosis-sensitive ECs located in peripheral areas, which have a high expression of p27kip1 (a marker of low growth); and (2) highly proliferative apoptosis-resistant cells in the central core of the vascular lesion, which have low expression of p27kip1 and increased expression of HIF-1α, VEGF protein, and VEGF-2 receptor [158,159]. The hypoxia-activated HIF-1α–VEGF axis plays a key role in vascular reactivity/remodeling and angiogenesis [160]. In severe PAH in humans, HIF-1α is overexpressed in obliterative endothelial lesions [158] and in experimental PH, a time-dependent increase in HIF-1α levels/expression correlates with the development of disease and vascular and RV remodeling [161,162]. In the systemic circulation, E2 via the HIF-1α–VEGF axis prevents/reduces endothelial dysfunction, vascular inflammation, and neointima formation [163,164]. Relevant to PAH, following endothelial damage, E2 plays a key role in promoting endothelial healing and angiogenesis [165].
7.1. Opposing Effects of E2 and 2ME on Angiogenesis (Key Role of HIF-1α)
Data coming from basic and clinical research of female cancers indicate that E2 and 2ME have opposite effects on angiogenesis (Figure 2A; previously reviewed in references [8]). The angiogenic properties of E2 are in striking contrast to the strong antiangiogenic effects of its major non-estrogenic metabolite 2ME. The other biological effects of 2ME relevant to PAH (Figure 2) have been reviewed previously [166].
The regulation of HIF1α activity is mechanistically linked to microtubules, and disruption of the microtubule cytoskeleton downregulates the HIF1-α pathway and tumor angiogenesis [167,168]. Similar to other tubulin disruptors, 2ME binds to (or near) the colchicine-binding site, disrupts the microtubule cytoskeleton, downregulates HIF1-α, and inhibits angiogenesis ([168]; Figure 2B). Notably, the most consistently reported effect of 2ME is its ability to inhibit HIF-1α. Indeed, this effect is so reliable that numerous recent studies have used 2ME as a pharmacological tool to inhibit HIF-α expression and signaling [167,168]. Furthermore, the use of 2ME has been proposed to combat HIF-signaling and related glycolytic shift in PAH [169]. Noteworthy, in vitro in swine granulosa cells, hypoxia stimulates 2ME production, which simultaneously inhibits hypoxia-driven angiogenesis [170], and in vivo, in a murine model of rheumatoid arthritis, 2ME reduces the expression of mRNA for the angiogenic cytokines and prevents neovascularization into the joint [171]. Thus, 2ME could be viewed as a local modulator of angiogenesis. In OVX CH-PH rats, 2ME inhibits oxidative stress-induced activation of the HIF-1α pathway [69,79], and similar effects are seen with E2. Whether this inhibitory effect of E2 on the HIF-1α pathway in CH-PH rats is mediated by E2′s downstream metabolite 2ME is presently unknown. Importantly, a recent study from the MacLean lab [76] confirmed the previously reported therapeutic effects of 2ME in hypoxic PH [77,78] suggested sex-dependent differences in HIF-1α signaling and supports the role of 2ME as an anti-angiogenic factor in PH [76]. In this study, basal HIF-1α protein expression was higher in female than male hPASMCs, and the antimitogenic effects of 2ME in hPASMCs were associated with reduced HIF-1α expression. Similarly, in vivo 2ME attenuated hypoxia-induced PH in male and female rats, while decreasing the protein expression of HIF-1α [76].
Presently, it is unclear whether the effects of E2 and 2ME on angiogenesis and vascular remodeling play a role in PAH. Whether, based on its angiogenic properties, E2 exhibits protective or injurious effects in the healthy or injured pulmonary vasculature is a subject of the ongoing debate regarding the estrogens paradox in angioproliferative PH. On this subject, the available data are limited to media remodeling and are contradictory. In the Su-Hx model, female sex is associated with reduced RV fibrosis and greater survival, but with greater media remodeling of the pulmonary arteries [49]. On the contrary, preventive E2 treatment in OVX Su + Hx rats attenuates media remodeling and prevents the development of PH [172]. Unfortunately, to date, no single study has analyzed the effects of E2 on occlusive and plexiform lesions in a Su-Hx model.
7.2. 2ME, HIF1α, and Endothelial-to-Mesenchymal Transition in PAH
Hypoxia and HIF-1α upregulation drive endothelial-to-mesenchymal transition (EndoMT), i.e., ECs transition into a mesenchymal or myofibroblast phenotype. The activation of the HIF-1α pathway and subsequent EndoMT has been implicated in MCT- and CH-induced vascular remodeling and PH [173,174], as well as in the vascular pathology of pulmonary fibrosis and PAH in humans [175,176,177]. 2ME inhibits hypoxia- and irradiation-induced EndoMT and lung fibrosis via downregulation of HIF1α-dependent Smad signaling [178,179]. Whether 2ME reduces vascular remodeling via HIF-1α inhibition, not only in hypoxia-induced PH [76,77,78], but also in angioproliferative PH, needs to be determined.
7.3. 2ME, HIF-1α, and Metabolic Reprograming in PAH
A shift from oxidative phosphorylation to glycolysis (the Warburg effect) takes place in vasculature in PAH patients [168,180], and this shift has been linked to pathologic angiogenesis and pulmonary vascular remodeling. The Warburg effect has been investigated as a druggable target in highly proliferative ECs to reduce angiogenesis [181]. In this regard, the HIF-1α is a key regulator responsible for the glycolytic shift in vascular cells in both cancer and PAH patients [181,182]. HIF-1α-induced pyruvate dehydrogenase kinase (PDK) inhibits pyruvate dehydrogenase (PDH) and thereby increases glycolysis [180]. The PDK inhibitor dichloroacetate not only reverses the Warburg effect, but also has therapeutic effects on experimental PH [183,184,185]. The effects of estrogens on metabolic changes in pulmonary vasculature are unknown. Nonetheless, E2, via ERα-mediated upregulation of glycolytic enzymes, not only stimulates pathologic angiogenesis in breast cancer [186], but also boosts a mitogenic response in highly proliferative human endometrial cells and umbilical vein endothelial cells [187,188]. In contrast, because 2ME is a strong HIF-1α inhibitor, 2ME would be expected to inhibit metabolic reprograming in PAH. Although the effects of 2ME on metabolic reprograming in PAH are unknown, by inhibiting HIF-1α and PDK, 2ME attenuates glycolysis and inhibits proliferation of apoptosis resistant melanoma cells [189]. Whether, similar to cancer cells, 2ME inhibits metabolic reprograming in highly proliferative, apoptosis resistant endothelium in PAH warrants further investigation.
7.4. Future Directions and Clinical Implications
In contrast to solid experimental and clinical evidence about its beneficial effects on RV function, the role of E2 on angioproliferation (formation of occlusive and complex/plexiform lesions) in PAH remains largely unexplored. Future studies on the Su+Hx model should focus on head-to-head comparison of endogenous versus exogenous E2 and preventative versus rescue treatments with E2 and 2ME on metabolic reprograming and the development of occlusive and plexiform lesions in male and intact and OVX female animals.
8. Inflammation, Immunity, and Estradiol Metabolism in PAH
A growing body of evidence has implicated inflammation and altered immunity in the pathogenesis of PAH [190,191,192]. In PAH patients, the size of perivascular inflammatory and immune cell infiltrates correlates with intima + media remodeling and pulmonary artery pressure [193], and abnormally elevated levels of circulating proinflammatory cytokines are predictive of worse outcomes [194,195]. Notably, proinflammatory cytokines strongly induce aromatase and sulfatase activity [196,197] and peripheral E2 synthesis [198]. Because E2 upregulates CYP1B1 and sulfatase activity, in an inflammatory environment, via a feed-forward mechanism, E2 may increase the bioavailability of its metabolic precursor DHEA, which would augment its own production. In addition, upregulated CYP1B1 may increase the production of 4-HE and 16α-HE1, which are very estrogenic metabolites with significant angiogenic, proinflammatory, and mitogenic properties (Figure 3). As discussed above, increased E2 tissue levels may also augment the production of proinflammatory metabolites of arachidonic acid.
Women have stronger immune responses compared to men [199,200,201]. This may initially be physiologically beneficial, but with aging, a more aggressive immune response may become detrimental and may increase the risk of PAH. For example, ERα signaling promotes T cell activation and proliferation and contributes to T cell-mediated autoimmune inflammation [202]. Women more frequently develop various autoimmune diseases (including systemic sclerosis, systemic lupus erythematosus, mixed connective tissue disease, and rheumatoid arthritis) that are associated with increased risk of PAH [5]. The autoimmune inflammation may have significant effects on EMet and E2 levels. Both lung macrophages and immune cells express steroidogenic enzymes (sulfatase and aromatase), and in women with rheumatoid arthritis or systemic lupus, measures of autoimmune inflammation strongly correlate with E2 levels [203,204]. Furthermore, irrespective of sex, autoimmune inflammation augments the 16-hydroxylation pathway [205] and produces changes in sex steroids and their precursors and metabolites, similar to those reported in PAH [46,47,58].
8.1. Anti-Inflammatory and Immunomodulatory Effects of 2ME
In contrast to E2, non-estrogenic 2ME exhibits significant anti-inflammatory and immunomodulatory effects. The anti-inflammatory effects of 2ME are in part mediated by the suppression of macrophage activation [206,207], with the inhibition of macrophages influx/activation by 2ME reported in several models of cardiovascular and renal injury [208,209,210]. Furthermore, in an autoimmune model of rheumatoid arthritis, 2ME inhibits the expression of mRNA for inflammatory cytokines (IL-1β, TNF-α, IL-6, and IL-17), reduces local inflammation and prevents neovascularization and disease progression [171]. Furthermore, in several models of autoimmune inflammatory disease, 2ME attenuates the progression of disease by inhibiting T cell activation, proliferation, and cytokine release [211,212,213]. Of significance to PH, 2ME, its metabolic precursor 2HE, its “inactive” metabolite 2ME1, and/or its synthetic analog 2-ethoxyestradiol reduce the influx/activation of macrophages (ED1+ cells) in MCT- and bleomycin-induced PH, which correlates with reduced right ventricular peak systolic pressure and vascular remodeling and fibrosis [12,20,21,22,79,113,114,115,116,117,118].
8.2. Future Directions and Clinical Implications
An inflammatory environment and altered immunity may have significant effects on E2 metabolism. Through multiple mutually non-exclusive mechanisms, autoinflammation may increase the bioavailability of E2, other proinflammatory “bad estrogens”, and proinflammatory AA metabolites. As discussed above, the reported hormonal changes in men and postmenopausal women with PAH are similar to changes seen in autoimmune inflammatory diseases (systemic sclerosis and lupus). This suggests that, irrespective of sex and type of immune disease (Th1 versus Th2), the autoinflammation is associated with increased activity of sulfatase, aromatase, and 17β–HSD. Future translational and clinical studies should assess the impact that inflammation and altered immunity have on key metabolizing enzymes and E2 and AA metabolism in PAH.
9. Concluding Remarks and Future Directions
The proposed three-tier concept of estrogen action in PAH (Figure 5) offers an explanation for the contradictory effects of estrogens reported in different models of PH and in men and women with PAH.
Estrogens could be viewed as protectors of RV function in PAH, yet they can be considered instigators and perpetuators of vascular injury in the pulmonary circulation, which lead to the development of PAH. Based on the reported cellular effects and limited animal data, we view 2ME as a biological antagonist of E2 at the level of pulmonary ECs and AA metabolism. 2ME, via a multitude of actions (Figure 2 and Figure 3), can not only provide pulmonary vascular and RV protection, but also oppose the potential detrimental effects of E2 due to redirection of its own metabolism and metabolism of AA in PAH. Thereby, 2ME could be considered as both mediator and corrector of E2′s action and a new disease modifier in PAH [166].
Recently applied combination therapies, attacking different pathogenic pathways in PAH, have brought additional benefits to PAH patients over mono therapies [214,215]. Currently, three ongoing clinical trials are assessing the therapeutic benefits of inhibition of E2 production (anastrazole) or E2 signaling (tamoxifen and fulvestrant) in PAH patients. Testing combinations of modulators of E2 metabolism in experimental PH may give additional insight into the role of E2 metabolism in PAH and open the venue for clinical testing of E2 metabolites and metabolism modulators in both women and men with PAH. Combination therapy of E2 metabolism modulators/metabolites may bring more subtle and targeted correction of E2 metabolism derangements in PAH and offer protective and therapeutic effects in both women and men with PAH.
A multitude of factors and conditions can affect E2 metabolism (inflammation, hypoxia/oxidative stress, obesity/insulin resistance/leptin, diet, drugs, and genetic polymorphisms of metabolizing enzymes), suggesting that PAH heterogeneity may be due, at least in part, to dysregulated E2 metabolism. Increased E2 and decreased 2ME production may adversely affect pulmonary vascular remodeling and the progression of disease. Therefore, in-depth investigation of the vascular and RV effects of more than a dozen biologically active metabolites in severe PAH is warranted. Future prospective, randomized clinical trials (using highly selective and sensitive mass spectrometry-based methods) should not only quantify multiple estrogens, their metabolites, and metabolic precursors, but also markers of inflammation and angiogenesis. The use of high-throughput molecular data and contemporary computational techniques focused on the E2 metabolome may identify different PAH phenotypes and foster the precision medicine approach in PAH. In PAH patients, an accurate assessment of the activity of the numerous enzymes involved in estrogen metabolism may be very difficult. Animal and cell culture studies focused on sex hormone intracrinology (i.e., intracellular disposition and intracellular/extracellular equilibrium) in pulmonary vascular and RV cells would augment our understanding of the role of E2 metabolism in the inflammation, angioproliferation, and RV dysfunction of PAH.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Dresdale, D.T.; Schultz, M.; Michtom, R.J. Primary pulmonary hypertension. I. Clinical and hemodynamic study. Am. J. Med. 1951, 11, 686–705. [Google Scholar] [CrossRef] [Green Version]
- Wood, P. Pulmonary hypertension. Br. Med. Bull. 1952, 8, 348–353. [Google Scholar] [CrossRef] [PubMed]
- Benza, R.L.; Miller, D.P.; Barst, R.J.; Badesch, D.B.; Frost, A.E.; McGoon, M.D. An evaluation of long-term survival from time of diagnosis in pulmonary arterial hypertension from the REVEAL Registry. Chest 2012, 142, 448–456. [Google Scholar] [CrossRef] [PubMed]
- Tamura, Y.; Kumamaru, H.; Satoh, T.; Miyata, H.; Ogawa, A.; Tanabe, N.; Hatano, M.; Yao, A.; Abe, K.; Tsujino, I.; et al. Effectiveness and Outcome of Pulmonary Arterial Hypertension-Specific Therapy in Japanese Patients With Pulmonary Arterial Hypertension. Circ. J. 2017, 82, 275–282. [Google Scholar] [CrossRef] [Green Version]
- Batton, K.A.; Austin, C.O.; Bruno, K.A.; Burger, C.D.; Shapiro, B.P.; Fairweather, D. Sex differences in pulmonary arterial hypertension: Role of infection and autoimmunity in the pathogenesis of disease. Biol. Sex Differ. 2018, 9, 15. [Google Scholar] [CrossRef]
- Chung, L.; Farber, H.W.; Benza, R.; Miller, D.P.; Parsons, L.; Hassoun, P.M.; McGoon, M.; Nicolls, M.R.; Zamanian, R.T. Unique predictors of mortality in patients with pulmonary arterial hypertension associated with systemic sclerosis in the REVEAL registry. Chest 2014, 146, 1494–1504. [Google Scholar] [CrossRef] [Green Version]
- Hoeper, M.M.; Huscher, D.; Ghofrani, H.A.; Delcroix, M.; Distler, O.; Schweiger, C.; Grunig, E.; Staehler, G.; Rosenkranz, S.; Halank, M.; et al. Elderly patients diagnosed with idiopathic pulmonary arterial hypertension: Results from the COMPERA registry. Int. J. Cardiol. 2013, 168, 871–880. [Google Scholar] [CrossRef]
- Tofovic, S.P. Estrogens and development of pulmonary hypertension: Interaction of estradiol metabolism and pulmonary vascular disease. J. Cardiovasc. Pharmacol. 2010, 56, 696–708. [Google Scholar] [CrossRef] [Green Version]
- Colvin, K.L.; Yeager, M.E. Animal Models of Pulmonary Hypertension: Matching Disease Mechanisms to Etiology of the Human Disease. J. Pulm. Respir. Med. 2014, 4. [Google Scholar] [CrossRef] [Green Version]
- Maarman, G.; Lecour, S.; Butrous, G.; Thienemann, F.; Sliwa, K. A comprehensive review: The evolution of animal models in pulmonary hypertension research; are we there yet? Pulm. Circ. 2013, 3, 739–756. [Google Scholar] [CrossRef] [Green Version]
- Stenmark, K.R.; Meyrick, B.; Galie, N.; Mooi, W.J.; McMurtry, I.F. Animal models of pulmonary arterial hypertension: The hope for etiological discovery and pharmacological cure. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 297, L1013–L1032. [Google Scholar] [CrossRef] [PubMed]
- Tofovic, S.P.; Salah, E.M.; Mady, H.H.; Jackson, E.K.; Melhem, M.F. Estradiol metabolites attenuate monocrotaline-induced pulmonary hypertension in rats. J. Cardiovasc. Pharmacol. 2005, 46, 430–437. [Google Scholar] [CrossRef] [PubMed]
- Lahm, T.; Frump, A.L.; Albrecht, M.E.; Fisher, A.J.; Cook, T.G.; Jones, T.J.; Yakubov, B.; Whitson, J.; Fuchs, R.K.; Liu, A.; et al. 17beta-Estradiol mediates superior adaptation of right ventricular function to acute strenuous exercise in female rats with severe pulmonary hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2016, 311, L375–L388. [Google Scholar] [CrossRef] [PubMed]
- Ventetuolo, C.E.; Ouyang, P.; Bluemke, D.A.; Tandri, H.; Barr, R.G.; Bagiella, E.; Cappola, A.R.; Bristow, M.R.; Johnson, C.; Kronmal, R.A.; et al. Sex hormones are associated with right ventricular structure and function: The MESA-right ventricle study. Am. J. Respir. Crit. Care Med. 2011, 183, 659–667. [Google Scholar] [CrossRef] [PubMed]
- Frump, A.L.; Goss, K.N.; Vayl, A.; Albrecht, M.; Fisher, A.; Tursunova, R.; Fierst, J.; Whitson, J.; Cucci, A.R.; Brown, M.B.; et al. Estradiol improves right ventricular function in rats with severe angioproliferative pulmonary hypertension: Effects of endogenous and exogenous sex hormones. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 308, L873–L890. [Google Scholar] [CrossRef]
- Lahm, T.; Douglas, I.S.; Archer, S.L.; Bogaard, H.J.; Chesler, N.C.; Haddad, F.; Hemnes, A.R.; Kawut, S.M.; Kline, J.A.; Kolb, T.M.; et al. Assessment of Right Ventricular Function in the Research Setting: Knowledge Gaps and Pathways Forward. An Official American Thoracic Society Research Statement. Am. J. Respir. Crit. Care Med. 2018, 198, e15–e43. [Google Scholar] [CrossRef] [Green Version]
- Badlam, J.B.; Austin, E.D. Beyond oestrogens: Towards a broader evaluation of the hormone profile in pulmonary arterial hypertension. Eur. Respir. J. 2018, 51. [Google Scholar] [CrossRef]
- Tofovic, S.P.; Jackson, E.K. Estrogens in Men: Another Layer of Complexity of Estradiol Metabolism in Pulmonary Hypertension. Am. J. Respir. Crit. Care Med. 2016, 193, 1087–1090. [Google Scholar] [CrossRef] [Green Version]
- Austin, E.D.; Lahm, T.; West, J.; Tofovic, S.P.; Johansen, A.K.; Maclean, M.R.; Alzoubi, A.; Oka, M. Gender, sex hormones and pulmonary hypertension. Pulm. Circ. 2013, 3, 294–314. [Google Scholar] [CrossRef] [Green Version]
- Tofovic, S.P.; Zhang, X.; Jackson, E.K.; Dacic, S.; Petrusevska, G. 2-Methoxyestradiol mediates the protective effects of estradiol in monocrotaline-induced pulmonary hypertension. Vascul. Pharmacol. 2006, 45, 358–367. [Google Scholar] [CrossRef]
- Tofovic, S.P.; Zhang, X.; Jackson, E.K.; Zhu, H.; Petrusevska, G. 2-methoxyestradiol attenuates bleomycin-induced pulmonary hypertension and fibrosis in estrogen-deficient rats. Vascul. Pharmacol. 2009, 51, 190–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tofovic, S.P.; Zhang, X.; Zhu, H.; Jackson, E.K.; Rafikova, O.; Petrusevska, G. 2-Ethoxyestradiol is antimitogenic and attenuates monocrotaline-induced pulmonary hypertension and vascular remodeling. Vascul. Pharmacol. 2008, 48, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Lahm, T.; Tuder, R.M.; Petrache, I. Progress in solving the sex hormone paradox in pulmonary hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 307, L7–L26. [Google Scholar] [CrossRef] [PubMed]
- Lahm, T.; Kawut, S.M. Inhibiting oestrogen signalling in pulmonary arterial hypertension: Sex, drugs and research. Eur. Respir. J. 2017, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mueller, J.W.; Gilligan, L.C.; Idkowiak, J.; Arlt, W.; Foster, P.A. The Regulation of Steroid Action by Sulfation and Desulfation. Endocr. Rev. 2015, 36, 526–563. [Google Scholar] [CrossRef]
- Obaidat, A.; Roth, M.; Hagenbuch, B. The expression and function of organic anion transporting polypeptides in normal tissues and in cancer. Annu. Rev. Pharmacol. Toxicol. 2012, 52, 135–151. [Google Scholar] [CrossRef] [Green Version]
- Radford, D.J.; Wang, K.; McNelis, J.C.; Taylor, A.E.; Hechenberger, G.; Hofmann, J.; Chahal, H.; Arlt, W.; Lord, J.M. Dehydroepiandrosterone sulfate directly activates protein kinase C-beta to increase human neutrophil superoxide generation. Mol. Endocrinol. 2010, 24, 813–821. [Google Scholar] [CrossRef] [Green Version]
- Strott, C.A. Steroid sulfotransferases. Endocr. Rev. 1996, 17, 670–697. [Google Scholar] [CrossRef]
- Garbacz, W.G.; Jiang, M.; Xie, W. Sex-Dependent Role of Estrogen Sulfotransferase and Steroid Sulfatase in Metabolic Homeostasis. Adv. Exp. Med. Biol. 2017, 1043, 455–469. [Google Scholar] [CrossRef]
- Spink, B.C.; Katz, B.H.; Hussain, M.M.; Pang, S.; Connor, S.P.; Aldous, K.M.; Gierthy, J.F.; Spink, D.C. SULT1A1 catalyzes 2-methoxyestradiol sulfonation in MCF-7 breast cancer cells. Carcinogenesis 2000, 21, 1947–1957. [Google Scholar] [CrossRef] [Green Version]
- Prough, R.A.; Clark, B.J.; Klinge, C.M. Novel mechanisms for DHEA action. J. Mol. Endocrinol. 2016, 56, R139–R155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turcu, A.; Smith, J.M.; Auchus, R.; Rainey, W.E. Adrenal androgens and androgen precursors-definition, synthesis, regulation and physiologic actions. Compr. Physiol. 2014, 4, 1369–1381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forney, J.P.; Milewich, L.; Chen, G.T.; Garlock, J.L.; Schwarz, B.E.; Edman, C.D.; MacDonald, P.C. Aromatization of androstenedione to estrone by human adipose tissue in vitro. Correlation with adipose tissue mass, age, and endometrial neoplasia. J. Clin. Endocrinol. Metab. 1981, 53, 192–199. [Google Scholar] [CrossRef]
- Kley, H.K.; Deselaers, T.; Peerenboom, H.; Kruskemper, H.L. Enhanced conversion of androstenedione to estrogens in obese males. J. Clin. Endocrinol. Metab. 1980, 51, 1128–1132. [Google Scholar] [CrossRef]
- Dessouroux, A.; Akwa, Y.; Baulieu, E.E. DHEA decreases HIF-1alpha accumulation under hypoxia in human pulmonary artery cells: Potential role in the treatment of pulmonary arterial hypertension. J. Steroid Biochem. Mol. Biol. 2008, 109, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Dillon, J.S. Dehydroepiandrosterone stimulates nitric oxide release in vascular endothelial cells: Evidence for a cell surface receptor. Steroids 2004, 69, 279–289. [Google Scholar] [CrossRef]
- Bonnet, S.; Dumas-de-La-Roque, E.; Begueret, H.; Marthan, R.; Fayon, M.; Santos, P.D.; Savineau, J.P.; Baulieu, E.E. Dehydroepiandrosterone (DHEA) prevents and reverses chronic hypoxic pulmonary hypertension. Proc. Natl. Acad. Sci. USA 2003, 100, 9488–9493. [Google Scholar] [CrossRef] [Green Version]
- Hampl, V.; Bibova, J.; Povysilova, V.; Herget, J. Dehydroepiandrosterone sulphate reduces chronic hypoxic pulmonary hypertension in rats. Eur. Respir. J. 2003, 21, 862–865. [Google Scholar] [CrossRef]
- Oka, M.; Karoor, V.; Homma, N.; Nagaoka, T.; Sakao, E.; Golembeski, S.M.; Limbird, J.; Imamura, M.; Gebb, S.A.; Fagan, K.A.; et al. Dehydroepiandrosterone upregulates soluble guanylate cyclase and inhibits hypoxic pulmonary hypertension. Cardiovasc. Res. 2007, 74, 377–387. [Google Scholar] [CrossRef] [Green Version]
- Homma, N.; Nagaoka, T.; Karoor, V.; Imamura, M.; Taraseviciene-Stewart, L.; Walker, L.A.; Fagan, K.A.; McMurtry, I.F.; Oka, M. Involvement of RhoA/Rho kinase signaling in protection against monocrotaline-induced pulmonary hypertension in pneumonectomized rats by dehydroepiandrosterone. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008, 295, L71–L78. [Google Scholar] [CrossRef] [Green Version]
- Paulin, R.; Meloche, J.; Jacob, M.H.; Bisserier, M.; Courboulin, A.; Bonnet, S. Dehydroepiandrosterone inhibits the Src/STAT3 constitutive activation in pulmonary arterial hypertension. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H1798–H1809. [Google Scholar] [CrossRef]
- Sharma, D.; Coridon, H.; Aubry, E.; Houeijeh, A.; Houfflin-Debarge, V.; Besson, R.; Deruelle, P.; Storme, L. Vasodilator effects of dehydroepiandrosterone (DHEA) on fetal pulmonary circulation: An experimental study in pregnant sheep. PLoS ONE 2018, 13, e0198778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De la Roque, E.D.; Quignard, J.F.; Ducret, T.; Dahan, D.; Courtois, A.; Begueret, H.; Marthan, R.; Savineau, J.P. Beneficial effect of dehydroepiandrosterone on pulmonary hypertension in a rodent model of pulmonary hypertension in infants. Pediatr. Res. 2013, 74, 163–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alzoubi, A.; Toba, M.; Abe, K.; O’Neill, K.D.; Rocic, P.; Fagan, K.A.; McMurtry, I.F.; Oka, M. Dehydroepiandrosterone restores right ventricular structure and function in rats with severe pulmonary arterial hypertension. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H1708–H1718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De la Roque, E.D.; Savineau, J.P.; Metivier, A.C.; Billes, M.A.; Kraemer, J.P.; Doutreleau, S.; Jougon, J.; Marthan, R.; Moore, N.; Fayon, M.; et al. Dehydroepiandrosterone (DHEA) improves pulmonary hypertension in chronic obstructive pulmonary disease (COPD): A pilot study. Ann. Endocrinol. 2012, 73, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Ventetuolo, C.E.; Baird, G.L.; Barr, R.G.; Bluemke, D.A.; Fritz, J.S.; Hill, N.S.; Klinger, J.R.; Lima, J.A.; Ouyang, P.; Palevsky, H.I.; et al. Higher Estradiol and Lower Dehydroepiandrosterone-Sulfate Levels Are Associated with Pulmonary Arterial Hypertension in Men. Am. J. Respir. Crit. Care Med. 2016, 193, 1168–1175. [Google Scholar] [CrossRef] [PubMed]
- Baird, G.L.; Archer-Chicko, C.; Barr, R.G.; Bluemke, D.A.; Foderaro, A.E.; Fritz, J.S.; Hill, N.S.; Kawut, S.M.; Klinger, J.R.; Lima, J.A.C.; et al. Lower DHEA-S levels predict disease and worse outcomes in post-menopausal women with idiopathic, connective tissue disease- and congenital heart disease-associated pulmonary arterial hypertension. Eur. Respir. J. 2018, 51, 1800467. [Google Scholar] [CrossRef] [Green Version]
- Yuan, P.; Wu, W.H.; Gao, L.; Zheng, Z.Q.; Liu, D.; Mei, H.Y.; Zhang, Z.L.; Jing, Z.C. Oestradiol ameliorates monocrotaline pulmonary hypertension via NO, prostacyclin and endothelin-1 pathways. Eur. Respir. J. 2013, 41, 1116–1125. [Google Scholar] [CrossRef] [Green Version]
- Rafikova, O.; Rafikov, R.; Meadows, M.L.; Kangath, A.; Jonigk, D.; Black, S.M. The sexual dimorphism associated with pulmonary hypertension corresponds to a fibrotic phenotype. Pulm. Circ. 2015, 5, 184–197. [Google Scholar] [CrossRef] [Green Version]
- De la Roque, E.D.; Bellance, N.; Rossignol, R.; Begueret, H.; Billaud, M.; Santos, P.D.; Ducret, T.; Marthan, R.; Dahan, D.; Ramos-Barbon, D.; et al. Dehydroepiandrosterone reverses chronic hypoxia/reoxygenation-induced right ventricular dysfunction in rats. Eur. Respir. J. 2012, 40, 1420–1429. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.T.; Xue, J.J.; Wang, Q.; Cheng, S.Y.; Chen, Z.C.; Li, H.Y.; Shan, J.J.; Cheng, K.L.; Zeng, W.J. Dehydroepiandrosterone attenuates pulmonary artery and right ventricular remodeling in a rat model of pulmonary hypertension due to left heart failure. Life Sci. 2019, 219, 82–89. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.H.; Yuan, P.; Zhang, S.J.; Jiang, X.; Wu, C.; Li, Y.; Liu, S.F.; Liu, Q.Q.; Li, J.H.; Pudasaini, B.; et al. Impact of Pituitary-Gonadal Axis Hormones on Pulmonary Arterial Hypertension in Men. Hypertension 2018, 72, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.X.; Wang, L.; Lu, W.Z.; Yuan, P.; Wu, W.H.; Zhou, Y.P.; Zhao, Q.H.; Zhang, S.J.; Li, Y.; Wu, T.; et al. Association between High FSH, Low Progesterone and Idiopathic Pulmonary Arterial Hypertension in Women of Reproductive Age. Am. J. Hypertens. 2019. [Google Scholar] [CrossRef] [PubMed]
- Tofovic, S.P.; Bilan, V.; Mi, Z.; Jackson, E.K.; Schneider, F. Aromatase inhibition attenuates and ovariectomy and 4-hydroxyestradiol have mixed effects on development of angioproliferative pulmonary hypertension in female rats. Am. J. Respir. Crit. Care Med. 2013, 187, A6099. [Google Scholar]
- Mair, K.M.; Wright, A.F.; Duggan, N.; Rowlands, D.J.; Hussey, M.J.; Roberts, S.; Fullerton, J.; Nilsen, M.; Loughlin, L.; Thomas, M.; et al. Sex-dependent influence of endogenous estrogen in pulmonary hypertension. Am. J. Respir. Crit. Care Med. 2014, 190, 456–467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dean, A.; Nilsen, M.; Loughlin, L.; Salt, I.P.; MacLean, M.R. Metformin Reverses Development of Pulmonary Hypertension via Aromatase Inhibition. Hypertension 2016, 68, 446–454. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Austin, E.D.; Talati, M.; Fesse, J.P.; Farber-Eger, E.H.; Brittain, E.L.; Hemnes, A.R.; Loyd, J.E.; West, E. Oestrogen inhibition reverses pulmonary arterial hypertension and associated metabolic defects. Eur. Respir. J. 2017, 50. [Google Scholar] [CrossRef] [Green Version]
- Roberts, K.E.; Fallon, M.B.; Krowka, M.J.; Brown, R.S.; Trotter, J.F.; Peter, I.; Tighiouart, H.; Knowles, J.A.; Rabinowitz, D.; Benza, R.L.; et al. Genetic risk factors for portopulmonary hypertension in patients with advanced liver disease. Am. J. Respir. Crit. Care Med. 2009, 179, 835–842. [Google Scholar] [CrossRef]
- Kawut, S.M.; Archer-Chicko, C.L.; DeMichele, A.; Fritz, J.S.; Klinger, J.R.; Ky, B.; Palevsky, H.I.; Palmisciano, A.J.; Patel, M.; Pinder, D.; et al. Anastrozole in Pulmonary Arterial Hypertension. A Randomized, Double-Blind, Placebo-controlled Trial. Am. J. Respir. Crit. Care Med. 2017, 195, 360–368. [Google Scholar] [CrossRef] [Green Version]
- Liu, A.; Philip, J.; Vinnakota, K.C.; van den Bergh, F.; Tabima, D.M.; Hacker, T.; Beard, D.A.; Chesler, N.C. Estrogen maintains mitochondrial content and function in the right ventricle of rats with pulmonary hypertension. Physiol. Rep. 2017, 5. [Google Scholar] [CrossRef]
- Liu, A.; Tian, L.; Golob, M.; Eickhoff, J.C.; Boston, M.; Chesler, N.C. 17beta-Estradiol Attenuates Conduit Pulmonary Artery Mechanical Property Changes With Pulmonary Arterial Hypertension. Hypertension 2015, 66, 1082–1088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, A.; Schreier, D.; Tian, L.; Eickhoff, J.C.; Wang, Z.; Hacker, T.A.; Chesler, N.C. Direct and indirect protection of right ventricular function by estrogen in an experimental model of pulmonary arterial hypertension. Am. J. Physiol. Heart Circ. Physiol. 2014, 307, H273–H283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Umar, S.; Partow-Navid, R.; Ruffenach, G.; Iorga, A.; Moazeni, S.; Eghbali, M. Severe pulmonary hypertension in aging female apolipoprotein E-deficient mice is rescued by estrogen replacement therapy. Biol. Sex Differ. 2017, 8, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, K.; Dempsie, Y.; Nilsen, M.; Wright, A.F.; Loughlin, L.; MacLean, M.R. The serotonin transporter, gender, and 17beta oestradiol in the development of pulmonary arterial hypertension. Cardiovasc. Res. 2011, 90, 373–382. [Google Scholar] [CrossRef] [PubMed]
- Tofovic, P.S.; Zhang, X.; Petrusevska, G. Progesterone inhibits vascular remodeling and attenuates monocrotaline-induced pulmonary hypertension in estrogen-deficient rats. Prilozi 2009, 30, 25–44. [Google Scholar] [PubMed]
- Blakemore, J.; Naftolin, F. Aromatase: Contributions to Physiology and Disease in Women and Men. Physiology 2016, 31, 258–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simpson, E.R.; Clyne, C.; Rubin, G.; Boon, W.C.; Robertson, K.; Britt, K.; Speed, C.; Jones, M. Aromatase—A brief overview. Annu. Rev. Physiol. 2002, 64, 93–127. [Google Scholar] [CrossRef]
- Simpson, E.R.; Zhao, Y.; Agarwal, V.R.; Michael, M.D.; Bulun, S.E.; Hinshelwood, M.M.; Graham-Lorence, S.; Sun, T.; Fisher, C.R.; Qin, K.; et al. Aromatase expression in health and disease. Recent Prog. Horm. Res. 1997, 52, 185–213. [Google Scholar]
- Naftolin, F.; Ryan, K.J.; Davies, I.J.; Reddy, V.V.; Flores, F.; Petro, Z.; Kuhn, M.; White, R.J.; Takaoka, Y.; Wolin, L. The formation of estrogens by central neuroendocrine tissues. Recent Prog. Horm. Res. 1975, 31, 295–319. [Google Scholar]
- Adashi, E.Y.; Hsueh, A.J. Estrogens augment the stimulation of ovarian aromatase activity by follicle-stimulating hormone in cultured rat granulosa cells. J. Biol. Chem. 1982, 257, 6077–6083. [Google Scholar]
- Purohit, A.; Singh, A.; Ghilchik, M.W.; Reed, M.J. Inhibition of tumor necrosis factor alpha-stimulated aromatase activity by microtubule-stabilizing agents, paclitaxel and 2-methoxyestradiol. Biochem. Biophys. Res. Commun. 1999, 261, 214–217. [Google Scholar] [CrossRef] [PubMed]
- Brown, K.A.; Hunger, N.I.; Docanto, M.; Simpson, E.R. Metformin inhibits aromatase expression in human breast adipose stromal cells via stimulation of AMP-activated protein kinase. Breast Cancer Res. Treat. 2010, 123, 591–596. [Google Scholar] [CrossRef] [PubMed]
- Tofovic, S.P.; Bilan, V.; Jackson, E.K.; Schneider, F. Sugen 5416 Dose-Hypoxia-Normoxia-Gender Interaction in Angioproliferative Pulmonary Hypertension in Rats. Am. J. Respir. Crit. Care Med. 2014, 189, A5566. [Google Scholar]
- Dubey, R.K.; Tofovic, S.P.; Jackson, E.K. Cardiovascular pharmacology of estradiol metabolites. J. Pharmacol. Exp. Ther. 2004, 308, 403–409. [Google Scholar] [CrossRef]
- Barchiesi, F.; Jackson, E.K.; Fingerle, J.; Gillespie, D.G.; Odermatt, B.; Dubey, R.K. 2-Methoxyestradiol, an estradiol metabolite, inhibits neointima formation and smooth muscle cell growth via double blockade of the cell cycle. Circ. Res. 2006, 99, 266–274. [Google Scholar] [CrossRef] [Green Version]
- Docherty, C.K.; Nilsen, M.; MacLean, M.R. Influence of 2-Methoxyestradiol and Sex on Hypoxia-Induced Pulmonary Hypertension and Hypoxia-Inducible Factor-1-alpha. J. Am. Heart Assoc. 2019, 8, e011628. [Google Scholar] [CrossRef] [Green Version]
- Hao, S.; Jiang, L.; Fu, C.; Wu, X.; Liu, Z.; Song, J.; Lu, H.; Wu, X.; Li, S. 2-Methoxyestradiol attenuates chronic-intermittent hypoxia-induced pulmonary hypertension through regulating microRNA-223. J. Cell Physiol. 2019, 234, 6324–6335. [Google Scholar] [CrossRef]
- Wang, L.; Zheng, Q.; Yuan, Y.; Li, Y.; Gong, X. Effects of 17beta-estradiol and 2-methoxyestradiol on the oxidative stress-hypoxia inducible factor-1 pathway in hypoxic pulmonary hypertensive rats. Exp. Ther. Med. 2017, 13, 2537–2543. [Google Scholar] [CrossRef] [Green Version]
- Tofovic, S.P.; Jones, T.J.; Bilan, V.P.; Jackson, E.K.; Petrusevska, G. Synergistic therapeutic effects of 2-methoxyestradiol with either sildenafil or bosentan on amelioration of monocrotaline-induced pulmonary hypertension and vascular remodeling. J. Cardiovasc. Pharmacol. 2010, 56, 475–483. [Google Scholar] [CrossRef]
- Fenoy, F.J.; Hernandez, M.E.; Hernandez, M.; Quesada, T.; Salom, M.G.; Hernandez, I. Acute effects of 2-methoxyestradiol on endothelial aortic No release in male and ovariectomized female rats. Nitric Oxide 2010, 23, 12–19. [Google Scholar] [CrossRef]
- Chen, W.; Cui, Y.; Zheng, S.; Huang, J.; Li, P.; Simoncini, T.; Zhang, Y.; Fu, X. 2-methoxyestradiol induces vasodilation by stimulating NO release via PPARgamma/PI3K/Akt pathway. PLoS ONE 2015, 10, e0118902. [Google Scholar] [CrossRef]
- Manavathi, B.; Acconcia, F.; Rayala, S.K.; Kumar, R. An inherent role of microtubule network in the action of nuclear receptor. Proc. Natl. Acad. Sci. USA 2006, 103, 15981–15986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salama, S.A.; Kamel, M.W.; Botting, S.; Salih, S.M.; Borahay, M.A.; Hamed, A.A.; Kilic, G.S.; Saeed, M.; Williams, M.Y.; Diaz-Arrastia, C.R. Catechol-o-methyltransferase expression and 2-methoxyestradiol affect microtubule dynamics and modify steroid receptor signaling in leiomyoma cells. PLoS ONE 2009, 4, e7356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salama, S.A.; Nasr, A.B.; Dubey, R.K.; Al-Hendy, A. Estrogen metabolite 2-methoxyestradiol induces apoptosis and inhibits cell proliferation and collagen production in rat and human leiomyoma cells: A potential medicinal treatment for uterine fibroids. J. Soc. Gynecol. Investig. 2006, 13, 542–550. [Google Scholar] [CrossRef]
- Morrell, N.W.; Stenmark, K.R. The renin-angiotensin system in pulmonary hypertension. Am. J. Respir. Crit. Care Med. 2013, 187, 1138–1139. [Google Scholar] [CrossRef] [Green Version]
- Maron, B.A.; Leopold, J.A. The role of the renin-angiotensin-aldosterone system in the pathobiology of pulmonary arterial hypertension (2013 Grover Conference series). Pulm. Circ. 2014, 4, 200–210. [Google Scholar] [CrossRef] [Green Version]
- Yuan, Y.M.; Luo, L.; Guo, Z.; Yang, M.; Ye, R.S.; Luo, C. Activation of renin-angiotensin-aldosterone system (RAAS) in the lung of smoking-induced pulmonary arterial hypertension (PAH) rats. J. Renin Angiotensin Aldosterone Syst. 2015, 16, 249–253. [Google Scholar] [CrossRef]
- Pingili, A.K.; Davidge, K.N.; Thirunavukkarasu, S.; Khan, N.S.; Katsurada, A.; Majid, D.S.A.; Gonzalez, F.J.; Navar, L.G.; Malik, K.U. 2-Methoxyestradiol Reduces Angiotensin II-Induced Hypertension and Renal Dysfunction in Ovariectomized Female and Intact Male Mice. Hypertension 2017, 69, 1104–1112. [Google Scholar] [CrossRef]
- Ueki, N.; Kanasaki, K.; Kanasaki, M.; Takeda, S.; Koya, D. Catechol-O-Methyltransferase Deficiency Leads to Hypersensitivity of the Pressor Response against Angiotensin II. Hypertension 2017, 69, 1156–1164. [Google Scholar] [CrossRef]
- Koganti, S.; Snyder, R.; Gumaste, U.; Karamyan, V.T.; Thekkumkara, T. 2-methoxyestradiol binding of GPR30 down-regulates angiotensin AT(1) receptor. Eur. J. Pharmacol. 2014, 723, 131–140. [Google Scholar] [CrossRef] [Green Version]
- Ogola, B.; Zhang, Y.; Iyer, L.; Thekkumkara, T. 2-Methoxyestradiol causes matrix metalloproteinase 9-mediated transactivation of epidermal growth factor receptor and angiotensin type 1 receptor downregulation in rat aortic smooth muscle cells. Am. J. Physiol. Cell Physiol. 2018, 314, C554–C568. [Google Scholar] [CrossRef] [PubMed]
- Salah, E.; Bastacky, S.I.; Jackson, E.K.; Tofovic, S.P. 2-Methoxyestradiol Attenuates Angiotensin II-Induced Hypertension, Cardiovascular Remodeling, and Renal Injury. J. Cardiovasc. Pharmacol. 2019, 73, 165–177. [Google Scholar] [CrossRef] [PubMed]
- Hansmann, G.; Wagner, R.A.; Schellong, S.; Perez, V.A.; Urashima, T.; Wang, L.; Sheikh, A.Y.; Suen, R.S.; Stewart, D.J.; Rabinovitch, M. Pulmonary arterial hypertension is linked to insulin resistance and reversed by peroxisome proliferator-activated receptor-gamma activation. Circulation 2007, 115, 1275–1284. [Google Scholar] [CrossRef] [PubMed]
- Assad, T.R.; Hemnes, A.R. Metabolic Dysfunction in Pulmonary Arterial Hypertension. Curr. Hypertens. Rep. 2015, 17, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Summer, R.; Walsh, K.; Medoff, B.D. Obesity and pulmonary arterial hypertension: Is adiponectin the molecular link between these conditions? Pulm. Circ. 2011, 1, 440–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zamanian, R.T.; Hansmann, G.; Snook, S.; Lilienfeld, D.; Rappaport, K.M.; Reaven, G.M.; Rabinovitch, M.; Doyle, R.L. Insulin resistance in pulmonary arterial hypertension. Eur. Respir. J. 2009, 33, 318–324. [Google Scholar] [CrossRef]
- Hemnes, A.R.; Luther, J.M.; Rhodes, C.J.; Burgess, J.P.; Carlson, J.; Fan, R.; Fessel, J.P.; Fortune, N.; Gerszten, R.E.; Halliday, S.J.; et al. Human PAH is characterized by a pattern of lipid-related insulin resistance. JCI Insight 2019, 4. [Google Scholar] [CrossRef] [Green Version]
- West, J.; Niswender, K.D.; Johnson, J.A.; Pugh, M.E.; Gleaves, L.; Fessel, J.P.; Hemnes, A.R. A potential role for insulin resistance in experimental pulmonary hypertension. Eur. Respir. J. 2013, 41, 861–871. [Google Scholar] [CrossRef] [Green Version]
- Annerbrink, K.; Westberg, L.; Nilsson, S.; Rosmond, R.; Holm, G.; Eriksson, E. Catechol O-methyltransferase val158-met polymorphism is associated with abdominal obesity and blood pressure in men. Metabolism 2008, 57, 708–711. [Google Scholar] [CrossRef]
- Kanasaki, M.; Srivastava, S.P.; Yang, F.; Xu, L.; Kudoh, S.; Kitada, M.; Ueki, N.; Kim, H.; Li, J.; Takeda, S.; et al. Deficiency in catechol-o-methyltransferase is linked to a disruption of glucose homeostasis in mice. Sci. Rep. 2017, 7, 7927. [Google Scholar] [CrossRef] [Green Version]
- Hall, K.T.; Jablonski, K.A.; Chen, L.; Harden, M.; Tolkin, B.R.; Kaptchuk, T.J.; Bray, G.A.; Ridker, P.M.; Florez, J.C.; Mukamal, K.J.; et al. Catechol-O-methyltransferase association with hemoglobin A1c. Metabolism 2016, 65, 961–967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bryan-Lluka, L.J. Evidence for saturation of catechol-O-methyltransferase by low concentrations of noradrenaline in perfused lungs of rats. Naunyn Schmiedebergs Arch. Pharmacol. 1995, 351, 408–416. [Google Scholar] [CrossRef] [PubMed]
- De Santi, C.; Giulianotti, P.C.; Pietrabissa, A.; Mosca, F.; Pacifici, G.M. Catechol-O-methyltransferase: Variation in enzyme activity and inhibition by entacapone and tolcapone. Eur. J. Clin. Pharmacol. 1998, 54, 215–219. [Google Scholar] [CrossRef] [PubMed]
- Boudikova, B.; Szumlanski, C.; Maidak, B.; Weinshilboum, R. Human liver catechol-O-methyltransferase pharmacogenetics. Clin. Pharmacol. Ther. 1990, 48, 381–389. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Xie, T.; Ramsden, D.B.; Ho, S.L. Human catechol-O-methyltransferase down-regulation by estradiol. Neuropharmacology 2003, 45, 1011–1018. [Google Scholar] [CrossRef]
- Xie, T.; Ho, S.L.; Ramsden, D. Characterization and implications of estrogenic down-regulation of human catechol-O-methyltransferase gene transcription. Mol. Pharmacol. 1999, 56, 31–38. [Google Scholar] [CrossRef]
- Parvez, S.; Parvez, S.H.; Youdim, M.B. Variation in activity of monoamine metabolizing enzymes in rat liver during pregnancy. Br. J. Pharmacol. 1975, 53, 241–246. [Google Scholar] [CrossRef] [Green Version]
- Schendzielorz, N.; Rysa, A.; Reenila, I.; Raasmaja, A.; Mannisto, P.T. Complex estrogenic regulation of catechol-O-methyltransferase (COMT) in rats. J. Physiol. Pharmacol. 2011, 62, 483–490. [Google Scholar]
- Meyer, M.R.; Fredette, N.C.; Daniel, C.; Sharma, G.; Amann, K.; Arterburn, J.B.; Barton, M.; Prossnitz, E.R. Obligatory role for GPER in cardiovascular aging and disease. Sci. Signal. 2016, 9, ra105. [Google Scholar] [CrossRef] [Green Version]
- Alencar, A.K.; Montes, G.C.; Montagnoli, T.; Silva, A.M.; Martinez, S.T.; Fraga, A.G.; Wang, H.; Groban, L.; Sudo, R.T.; Zapata-Sudo, G. Activation of GPER ameliorates experimental pulmonary hypertension in male rats. Eur. J. Pharm. Sci. 2017, 97, 208–217. [Google Scholar] [CrossRef] [Green Version]
- Alencar, A.K.; Montes, G.C.; Costa, D.G.; Mendes, L.V.; Silva, A.M.; Martinez, S.T.; Trachez, M.M.; Cunha, V.D.M.; Montagnoli, T.L.; Fraga, A.G.; et al. Cardioprotection Induced by Activation of GPER in Ovariectomized Rats With Pulmonary Hypertension. J. Gerontol. A Biol. Sci. Med. Sci. 2018, 73, 1158–1166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaufelberger, S.A.; Rosselli, M.; Barchiesi, F.; Gillespie, D.G.; Jackson, E.K.; Dubey, R.K. 2-Methoxyestradiol, an endogenous 17beta-estradiol metabolite, inhibits microglial proliferation and activation via an estrogen receptor-independent mechanism. Am. J. Physiol. Endocrinol. Metab. 2016, 310, E313–E322. [Google Scholar] [CrossRef] [PubMed]
- Tofovic, S.P.; Jones, T.; Petrusevska, G. Dose-dependent therapeutic effects of 2-Methoxyestradiol on Monocrotaline-Induced pulmonary hypertension and vascular remodeling. Prilozi 2010, 31, 279–295. [Google Scholar] [PubMed]
- Tofovic, S.P.; Zhang, X.C.; Jones, T.; Jackson, E.K.; Petrusevska, G. 2-Methoxyestradiol attenuates the development and retards the progression of chronic hypoxia-induced pulmonary hypertension in rats. Circulation 2005, 112, 98–99. [Google Scholar]
- Tofovic, S.P.; Jackson, E.K.; Rafikova, O. Synergistic Effects of 2-Methoxyestradiol and Retinoic Acid on Amelioration of Monocrotaline-Induced Pulmonary Hypertension. Am. J. Res. Crit. Care Med. 2008, 177, A592. [Google Scholar]
- Tofovic, S.P.; Zhu, H.; Jackson, E.K.; Rafikova, O. 2-Methoxyestrone Inhibits Vascular Remodeling and Attenuates Monocrotaline-Induced Pulmonary Hypertension. Eur. Res. J. 2008, 32 (Suppl. 52), 995. [Google Scholar]
- Hu, J.; Rafikova, O.; Schneider, F.; Jackson, E.K. Tofovic SP 2-Methoxyestrone Retards the Progression of Angioproliferative Pulmonary Hypertension in Rats—Role of 17beta-Hydroxysteroid Dehydrogenase Pathway. Circulation 2015, 132 (Suppl. 3), A18888. [Google Scholar]
- Tofovic, S.P.; Hu, J.; Jackson, E.K.; Schneider, F. 2-Hydroxyestradiol Attenuates Metabolic Syndrome-Induced Pulmonary Hypertension. Am. J. Respir. Crit. Care Med. 2015, 191, A4096. [Google Scholar]
- White, K.; Johansen, A.K.; Nilsen, M.; Ciuclan, L.; Wallace, E.; Paton, L.; Campbell, A.; Morecroft, I.; Loughlin, L.; McClure, J.D.; et al. Activity of the estrogen-metabolizing enzyme cytochrome P450 1B1 influences the development of pulmonary arterial hypertension. Circulation 2012, 126, 1087–1098. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Talati, M.; Fessel, J.P.; Hemnes, A.R.; Gladson, S.; French, J.; Shay, S.; Trammell, A.; Phillips, J.A.; Hamid, R.; et al. Estrogen Metabolite 16alpha-Hydroxyestrone Exacerbates Bone Morphogenetic Protein Receptor Type II-Associated Pulmonary Arterial Hypertension Through MicroRNA-29-Mediated Modulation of Cellular Metabolism. Circulation 2016, 133, 82–97. [Google Scholar] [CrossRef]
- Swaneck, G.E.; Fishman, J. Covalent binding of the endogenous estrogen 16 alpha-hydroxyestrone to estradiol receptor in human breast cancer cells: Characterization and intranuclear localization. Proc. Natl. Acad. Sci. USA 1988, 85, 7831–7835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Austin, E.D.; Cogan, J.D.; West, J.D.; Hedges, L.K.; Hamid, R.; Dawson, E.P.; Wheeler, L.A.; Parl, F.F.; Loyd, J.E.; Phillips, J. Alterations in oestrogen metabolism: Implications for higher penetrance of familial pulmonary arterial hypertension in females. Eur. Respir. J. 2009, 34, 1093–1099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loyd, J.E. Pulmonary arterial hypertension: Insights from genetic studies. Proc. Am. Thorac. Soc. 2011, 8, 154–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- West, J.; Cogan, J.; Geraci, M.; Robinson, L.; Newman, J.; Phillips, J.A.; Lane, K.; Meyrick, B.; Loyd, J. Gene expression in BMPR2 mutation carriers with and without evidence of pulmonary arterial hypertension suggests pathways relevant to disease penetrance. BMC Med. Genom. 2008, 1, 45. [Google Scholar] [CrossRef] [Green Version]
- Johansen, A.K.; Dean, A.; Morecroft, I.; Hood, K.; Nilsen, M.; Loughlin, L.; Anagnostopoulou, A.; Touyz, R.M.; White, K.; MacLean, M.R. The serotonin transporter promotes a pathological estrogen metabolic pathway in pulmonary hypertension via cytochrome P450 1B1. Pulm. Circ. 2016, 6, 82–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hood, K.Y.; Montezano, A.C.; Harvey, A.P.; Nilsen, M.; MacLean, M.R.; Touyz, R.M. Nicotinamide Adenine Dinucleotide Phosphate Oxidase-Mediated Redox Signaling and Vascular Remodeling by 16alpha-Hydroxyestrone in Human Pulmonary Artery Cells: Implications in Pulmonary Arterial Hypertension. Hypertension 2016, 68, 796–808. [Google Scholar] [CrossRef]
- Ventetuolo, C.E.; Mitra, N.; Wan, F.; Manichaikul, A.; Barr, R.G.; Johnson, C.; Bluemke, D.A.; Lima, J.A.; Tandri, H.; Ouyang, P.; et al. Oestradiol metabolism and androgen receptor genotypes are associated with right ventricular function. Eur. Respir. J. 2016, 47, 553–563. [Google Scholar] [CrossRef]
- Kerzee, J.K.; Ramos, K.S. Constitutive and inducible expression of Cyp1a1 and Cyp1b1 in vascular smooth muscle cells: Role of the Ahr bHLH/PAS transcription factor. Circ. Res. 2001, 89, 573–582. [Google Scholar] [CrossRef] [Green Version]
- Lee, A.J.; Cai, M.X.; Thomas, P.E.; Conney, A.H.; Zhu, B.T. Characterization of the oxidative metabolites of 17beta-estradiol and estrone formed by 15 selectively expressed human cytochrome p450 isoforms. Endocrinology 2003, 144, 3382–3398. [Google Scholar] [CrossRef]
- Rahman, M.; Sutter, C.H.; Emmert, G.L.; Sutter, T.R. Regioselective 2-hydroxylation of 17beta-estradiol by rat cytochrome P4501B1. Toxicol. Appl. Pharmacol. 2006, 216, 469–478. [Google Scholar] [CrossRef]
- Tsuchiya, Y.; Nakajima, M.; Yokoi, T. Cytochrome P450-mediated metabolism of estrogens and its regulation in human. Cancer Lett. 2005, 227, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Dawling, S.; Roodi, N.; Parl, F.F. Methoxyestrogens exert feedback inhibition on cytochrome P450 1A1 and 1B1. Cancer Res. 2003, 63, 3127–3132. [Google Scholar] [PubMed]
- Maayah, Z.H.; Levasseur, J.; Piragasam, R.S.; Abdelhamid, G.; Dyck, J.R.B.; Fahlman, R.P.; Siraki, A.G.; El-Kadi, A.O.S. 2-Methoxyestradiol protects against pressure overload-induced left ventricular hypertrophy. Sci. Rep. 2018, 8, 2780. [Google Scholar] [CrossRef] [Green Version]
- Choudhary, D.; Jansson, I.; Stoilov, I.; Sarfarazi, M.; Schenkman, J.B. Metabolism of retinoids and arachidonic acid by human and mouse cytochrome P450 1b1. Drug Metab. Dispos. 2004, 32, 840–847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, W.; Parrish, A.R.; Ramos, K.S. Constitutive and inducible expression of cytochrome P450IA1 and P450IB1 in human vascular endothelial and smooth muscle cells. In Vitro Cell. Dev. Biol. Anim. 1998, 34, 671–673. [Google Scholar] [CrossRef]
- Zhu, D.; Ran, Y. Role of 15-lipoxygenase/15-hydroxyeicosatetraenoic acid in hypoxia-induced pulmonary hypertension. J. Physiol. Sci. 2012, 62, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Wang, S.; Wang, Z.; Ma, J.; Liu, S.; Li, W.; Zhu, D. The role of ERK1/2 in 15-HETE-inhibited apoptosis in pulmonary arterial smooth muscle cells. J. Recept. Signal. Transduct. Res. 2011, 31, 45–52. [Google Scholar] [CrossRef]
- Sugumaran, P.K.; Wang, S.; Song, S.; Nie, X.; Zhang, L.; Feng, Y.; Ma, W.; Zhu, D. 15-oxo-Eicosatetraenoic acid prevents serum deprivation-induced apoptosis of pulmonary arterial smooth muscle cells by activating pro-survival pathway. Prostaglandins Leukot. Essent. Fat. Acids 2014, 90, 89–98. [Google Scholar] [CrossRef]
- Maayah, Z.H.; Althurwi, H.N.; Abdelhamid, G.; Lesyk, G.; Jurasz, P.; El-Kadi, A.O. CYP1B1 inhibition attenuates doxorubicin-induced cardiotoxicity through a mid-chain HETEs-dependent mechanism. Pharmacol. Res. 2016, 105, 28–43. [Google Scholar] [CrossRef]
- Ma, C.; Li, Y.; Ma, J.; Liu, Y.; Li, Q.; Niu, S.; Shen, Z.; Zhang, L.; Pan, Z.; Zhu, D. Key role of 15-lipoxygenase/15-hydroxyeicosatetraenoic acid in pulmonary vascular remodeling and vascular angiogenesis associated with hypoxic pulmonary hypertension. Hypertension 2011, 58, 679–688. [Google Scholar] [CrossRef] [Green Version]
- Al-Naamani, N.; Sagliani, K.D.; Dolnikowski, G.G.; Warburton, R.R.; Toksoz, D.; Kayyali, U.; Hill, N.S.; Fanburg, B.L.; Roberts, K.E.; Preston, I.R. Plasma 12- and 15-hydroxyeicosanoids are predictors of survival in pulmonary arterial hypertension. Pulm. Circ. 2016, 6, 224–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, A.; Sun, D. Sexually Dimorphic Regulation of EET Synthesis and Metabolism: Roles of Estrogen. Front. Pharmacol. 2018, 9, 1222. [Google Scholar] [CrossRef] [PubMed]
- Somjen, D.; Kohen, F.; Limor, R.; Sharon, O.; Knoll, E.; Many, A.; Stern, N. Estradiol-17beta increases 12- and 15-lipoxygenase (type2) expression and activity and reactive oxygen species in human umbilical vascular smooth muscle cells. J. Steroid Biochem. Mol. Biol. 2016, 163, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.M.; Sun, D.; Kandhi, S.; Froogh, G.; Zhuge, J.; Huang, W.; Hammock, B.D.; Huang, A. Estrogen-dependent epigenetic regulation of soluble epoxide hydrolase via DNA methylation. Proc. Natl. Acad. Sci. USA 2018, 115, 613–618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kandhi, S.; Qin, J.; Froogh, G.; Jiang, H.; Luo, M.; Wolin, M.S.; Huang, A.; Sun, D. EET-dependent potentiation of pulmonary arterial pressure: Sex-different regulation of soluble epoxide hydrolase. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 309, L1478–L1486. [Google Scholar] [CrossRef] [Green Version]
- Kandhi, S.; Zhang, B.; Froogh, G.; Qin, J.; Alruwaili, N.; Le, Y.; Yang, Y.M.; Hwang, S.H.; Hammock, B.D.; Wolin, M.S.; et al. EETs promote hypoxic pulmonary vasoconstriction via constrictor prostanoids. Am. J. Physiol. Lung Cell. Mol. Physiol. 2017, 313, L350–L359. [Google Scholar] [CrossRef]
- Keseru, B.; Barbosa-Sicard, E.; Schermuly, R.T.; Tanaka, H.; Hammock, B.D.; Weissmann, N.; Fisslthaler, B.; Fleming, I. Hypoxia-induced pulmonary hypertension: Comparison of soluble epoxide hydrolase deletion vs. inhibition. Cardiovasc. Res. 2010, 85, 232–240. [Google Scholar] [CrossRef] [Green Version]
- Denlinger, C.L.; Vesell, E.S. Hormonal regulation of the developmental pattern of epoxide hydrolases. Studies in rat liver. Biochem. Pharmacol. 1989, 38, 603–610. [Google Scholar] [CrossRef]
- Pinot, F.; Grant, D.F.; Spearow, J.L.; Parker, A.G.; Hammock, B.D. Differential regulation of soluble epoxide hydrolase by clofibrate and sexual hormones in the liver and kidneys of mice. Biochem. Pharmacol. 1995, 50, 501–508. [Google Scholar] [CrossRef]
- Bayard, F.; Clamens, S.; Meggetto, F.; Blaes, N.; Delsol, G.; Faye, J.C. Estrogen synthesis, estrogen metabolism, and functional estrogen receptors in rat arterial smooth muscle cells in culture. Endocrinology 1995, 136, 1523–1529. [Google Scholar] [CrossRef]
- Zhang, C.Y.; Chen, J.; Yin, D.C.; Lin, S.X. The contribution of 17beta-hydroxysteroid dehydrogenase type 1 to the estradiol-estrone ratio in estrogen-sensitive breast cancer cells. PLoS ONE 2012, 7, e29835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soubhye, J.; Alard, I.C.; van Antwerpen, P.; Dufrasne, F. Type 2 17-beta hydroxysteroid dehydrogenase as a novel target for the treatment of osteoporosis. Future Med. Chem. 2015, 7, 1431–1456. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.J.; Lee, W.J.; Zhu, B.T. Selective insensitivity of ZR-75-1 human breast cancer cells to 2-methoxyestradiol: Evidence for type II 17beta-hydroxysteroid dehydrogenase as the underlying cause. Cancer Res. 2005, 65, 5802–5811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamath, K.; Okouneva, T.; Larson, G.; Panda, D.; Wilson, L.; Jordan, M.A. 2-Methoxyestradiol suppresses microtubule dynamics and arrests mitosis without depolymerizing microtubules. Mol. Cancer Ther. 2006, 5, 2225–2233. [Google Scholar] [CrossRef] [Green Version]
- James, J.; Murry, D.J.; Treston, A.M.; Storniolo, A.M.; Sledge, G.W.; Sidor, C.; Miller, K.D. Phase I safety, pharmacokinetic and pharmacodynamic studies of 2-methoxyestradiol alone or in combination with docetaxel in patients with locally recurrent or metastatic breast cancer. Investig. New Drugs 2007, 25, 41–48. [Google Scholar] [CrossRef]
- Matei, D.; Schilder, J.; Sutton, G.; Perkins, S.; Breen, T.; Quon, C.; Sidor, C. Activity of 2 methoxyestradiol (Panzem NCD) in advanced, platinum-resistant ovarian cancer and primary peritoneal carcinomatosis: A Hoosier Oncology Group trial. Gynecol. Oncol. 2009, 115, 90–96. [Google Scholar] [CrossRef]
- Sweeney, C.; Liu, G.; Yiannoutsos, C.; Kolesar, J.; Horvath, D.; Staab, M.J.; Fife, K.; Armstrong, V.; Treston, A.; Sidor, C.; et al. A phase II multicenter, randomized, double-blind, safety trial assessing the pharmacokinetics, pharmacodynamics, and efficacy of oral 2-methoxyestradiol capsules in hormone-refractory prostate cancer. Clin. Cancer Res. 2005, 11, 6625–6633. [Google Scholar] [CrossRef] [Green Version]
- Cool, C.D.; Stewart, J.S.; Werahera, P.; Miller, G.J.; Williams, R.L.; Voelkel, N.F.; Tuder, R.M. Three-dimensional reconstruction of pulmonary arteries in plexiform pulmonary hypertension using cell-specific markers: Evidence for a dynamic and heterogeneous process of pulmonary endothelial cell growth. Am. J. Pathol. 1999, 155, 411–419. [Google Scholar] [CrossRef]
- Tuder, R.M.; Chacon, M.; Alger, L.; Wang, J.; Taraseviciene-Stewart, L.; Kasahara, Y.; Cool, C.D.; Bishop, A.E.; Geraci, M.; Semenza, G.L.; et al. Expression of angiogenesis-related molecules in plexiform lesions in severe pulmonary hypertension: Evidence for a process of disordered angiogenesis. J. Pathol. 2001, 195, 367–374. [Google Scholar] [CrossRef]
- Semenza, G.L. HIF-1: Mediator of physiological and pathophysiological responses to hypoxia. J. Appl. Physiol. 2000, 88, 1474–1480. [Google Scholar] [CrossRef] [Green Version]
- Lai, Y.L.; Law, T.C. Chronic hypoxia- and monocrotaline-induced elevation of hypoxia-inducible factor-1 alpha levels and pulmonary hypertension. J. Biomed. Sci. 2004, 11, 315–321. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Zhang, Y.; Lu, L.; Wang, L.; Chen, M.; Hu, T. Expression and Correlation of Hypoxia-Inducible Factor-1alpha (HIF-1alpha) with Pulmonary Artery Remodeling and Right Ventricular Hypertrophy in Experimental Pulmonary Embolism. Med. Sci. Monit. 2017, 23, 2083–2088. [Google Scholar] [CrossRef] [PubMed]
- Concina, P.; Sordello, S.; Barbacanne, M.A.; Elhage, R.; Pieraggi, M.T.; Fournial, G.; Plouet, J.; Bayard, F.; Arnal, J.F. The mitogenic effect of 17beta-estradiol on in vitro endothelial cell proliferation and on in vivo reendothelialization are both dependent on vascular endothelial growth factor. J. Vasc. Res. 2000, 37, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Morales, D.E.; McGowan, K.A.; Grant, D.S.; Maheshwari, S.; Bhartiya, D.; Cid, M.C.; Kleinman, H.K.; Schnaper, H.W. Estrogen promotes angiogenic activity in human umbilical vein endothelial cells in vitro and in a murine model. Circulation 1995, 91, 755–763. [Google Scholar] [CrossRef] [PubMed]
- Trenti, A.; Tedesco, S.; Boscaro, C.; Trevisi, L.; Bolego, C.; Cignarella, A. Estrogen, Angiogenesis, Immunity and Cell Metabolism: Solving the Puzzle. Int. J. Mol. Sci. 2018, 19, 859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tofovic, S.P.; Jackson, E.K. 2-Methoxyestradiol in Pulmonary Arterial Hypertension: A New Disease Modifier. In Interventional Pulmonology and Pulmonary Hypertension; Intec Open: London, UK, 2019. [Google Scholar] [CrossRef] [Green Version]
- Escuin, D.; Kline, E.R.; Giannakakou, P. Both microtubule-stabilizing and microtubule-destabilizing drugs inhibit hypoxia-inducible factor-1alpha accumulation and activity by disrupting microtubule function. Cancer Res. 2005, 65, 9021–9028. [Google Scholar] [CrossRef] [Green Version]
- Mabjeesh, N.J.; Escuin, D.; LaVallee, T.M.; Pribluda, V.S.; Swartz, G.M.; Johnson, M.S.; Willard, M.T.; Zhong, H.; Simons, J.W.; Giannakakou, P. 2ME2 inhibits tumor growth and angiogenesis by disrupting microtubules and dysregulating HIF. Cancer Cell 2003, 3, 363–375. [Google Scholar] [CrossRef] [Green Version]
- Fijalkowska, I.; Xu, W.; Comhair, S.A.; Janocha, A.J.; Mavrakis, L.A.; Krishnamachary, B.; Zhen, L.; Mao, T.; Richter, A.; Erzurum, S.C.; et al. Hypoxia inducible-factor1alpha regulates the metabolic shift of pulmonary hypertensive endothelial cells. Am. J. Pathol. 2010, 176, 1130–1138. [Google Scholar] [CrossRef] [Green Version]
- Basini, G.; Grasselli, F.; Bussolati, S.; Baioni, L.; Bianchi, F.; Musci, M.; Careri, M.; Mangia, A. Hypoxia stimulates the production of the angiogenesis inhibitor 2-methoxyestradiol by swine granulosa cells. Steroids 2011, 76, 1433–1436. [Google Scholar] [CrossRef]
- Plum, S.M.; Park, E.J.; Strawn, S.J.; Moore, E.G.; Sidor, C.F.; Fogler, W.E. Disease modifying and antiangiogenic activity of 2-methoxyestradiol in a murine model of rheumatoid arthritis. BMC Musculoskelet. Disord. 2009, 10, 46. [Google Scholar] [CrossRef] [Green Version]
- Philip, J.L.; Tabima, D.M.; Wolf, G.D.; Frump, A.L.; Cheng, T.C.; Schreier, D.A.; Hacker, T.A.; Lahm, T.; Chesler, N.C. Exogenous Estrogen Preserves Distal Pulmonary Arterial Mechanics and Prevents Pulmonary Hypertension in Rats. Am. J. Respir. Crit. Care Med. 2019. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Yuan, T.; Zhang, H.; Yan, Y.; Wang, D.; Fang, L.; Lu, Y.; Du, G. Activation of Nrf2 Attenuates Pulmonary Vascular Remodeling via Inhibiting Endothelial-to-Mesenchymal Transition: An Insight from a Plant Polyphenol. Int. J. Biol. Sci. 2017, 13, 1067–1081. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Niu, W.; Dong, H.Y.; Liu, M.L.; Luo, Y.; Li, Z.C. Hypoxia induces endothelialmesenchymal transition in pulmonary vascular remodeling. Int. J. Mol. Med. 2018, 42, 270–278. [Google Scholar] [PubMed] [Green Version]
- Hopper, R.K.; Moonen, J.R.; Diebold, I.; Cao, A.; Rhodes, C.J.; Tojais, N.F.; Hennigs, J.K.; Gu, M.; Wang, L.; Rabinovitch, M. In Pulmonary Arterial Hypertension, Reduced BMPR2 Promotes Endothelial-to-Mesenchymal Transition via HMGA1 and Its Target Slug. Circulation 2016, 133, 1783–1794. [Google Scholar] [CrossRef] [Green Version]
- Stenmark, K.R.; Frid, M.; Perros, F. Endothelial-to-Mesenchymal Transition: An Evolving Paradigm and a Promising Therapeutic Target in PAH. Circulation 2016, 133, 1734–1737. [Google Scholar] [CrossRef]
- Tang, H.; Babicheva, A.; McDermott, K.M.; Gu, Y.; Ayon, R.J.; Song, S.; Wang, Z.; Gupta, A.; Zhou, T.; Sun, X.; et al. Endothelial HIF-2alpha contributes to severe pulmonary hypertension due to endothelial-to-mesenchymal transition. Am. J. Physiol. Lung Cell. Mol. Physiol. 2018, 314, L256–L275. [Google Scholar] [CrossRef]
- Liu, C.; Zhou, X.; Lu, J.; Zhu, L.; Li, M. Autophagy mediates 2-methoxyestradiol-inhibited scleroderma collagen synthesis and endothelial-to-mesenchymal transition induced by hypoxia. Rheumatology 2019, 58, 1966–1975. [Google Scholar] [CrossRef]
- Choi, S.H.; Hong, Z.Y.; Nam, J.K.; Lee, H.J.; Jang, J.; Yoo, R.J.; Lee, Y.J.; Lee, C.Y.; Kim, K.H.; Park, S.; et al. A Hypoxia-Induced Vascular Endothelial-to-Mesenchymal Transition in Development of Radiation-Induced Pulmonary Fibrosis. Clin. Cancer Res. 2015, 21, 3716–3726. [Google Scholar] [CrossRef] [Green Version]
- Chan, S.Y.; Rubin, L.J. Metabolic dysfunction in pulmonary hypertension: From basic science to clinical practice. Eur. Respir. Rev. 2017, 26. [Google Scholar] [CrossRef] [Green Version]
- Fitzgerald, G.; Soro-Arnaiz, I.; de Bock, K. The Warburg Effect in Endothelial Cells and its Potential as an Anti-angiogenic Target in Cancer. Front. Cell Dev. Biol. 2018, 6, 100. [Google Scholar] [CrossRef] [Green Version]
- Tuder, R.M.; Davis, L.A.; Graham, B.B. Targeting energetic metabolism: A new frontier in the pathogenesis and treatment of pulmonary hypertension. Am. J. Respir. Crit. Care Med. 2012, 185, 260–266. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Yan, J.; Shen, Y.; Liu, Y.; Ma, Z. Dichloroacetate prevents but not reverses the formation of neointimal lesions in a rat model of severe pulmonary arterial hypertension. Mol. Med. Rep. 2014, 10, 2144–2152. [Google Scholar] [CrossRef] [PubMed]
- McMurtry, M.S.; Bonnet, S.; Wu, X.; Dyck, J.R.; Haromy, A.; Hashimoto, K.; Michelakis, E.D. Dichloroacetate prevents and reverses pulmonary hypertension by inducing pulmonary artery smooth muscle cell apoptosis. Circ. Res. 2004, 95, 830–840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michelakis, E.D.; McMurtry, M.S.; Wu, X.C.; Dyck, J.R.; Moudgil, R.; Hopkins, T.A.; Lopaschuk, G.D.; Puttagunta, L.; Waite, R.; Archer, S.L. Dichloroacetate, a metabolic modulator, prevents and reverses chronic hypoxic pulmonary hypertension in rats: Role of increased expression and activity of voltage-gated potassium channels. Circulation 2002, 105, 244–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gandhi, N.; Das, G.M. Metabolic Reprogramming in Breast Cancer and Its Therapeutic Implications. Cells 2019, 8, 89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salama, S.A.; Mohammad, M.A.; Diaz-Arrastia, C.R.; Kamel, M.W.; Kilic, G.S.; Ndofor, B.T.; Abdel-Baki, M.S.; Theiler, S.K. Estradiol-17beta upregulates pyruvate kinase M2 expression to coactivate estrogen receptor-alpha and to integrate metabolic reprogramming with the mitogenic response in endometrial cells. J. Clin. Endocrinol. Metab. 2014, 99, 3790–3799. [Google Scholar] [CrossRef] [Green Version]
- Trenti, A.; Tedesco, S.; Boscaro, C.; Ferri, N.; Cignarella, A.; Trevisi, L.; Bolego, C. The Glycolytic Enzyme PFKFB3 Is Involved in Estrogen-Mediated Angiogenesis via GPER1. J. Pharmacol. Exp. Ther. 2017, 361, 398–407. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Jiang, H.; Li, Z.; Zhuang, Y.; Liu, Y.; Zhou, S.; Xiao, Y.; Xie, C.; Zhou, F.; Zhou, Y. 2-Methoxyestradiol enhances radiosensitivity in radioresistant melanoma MDA-MB-435R cells by regulating glycolysis via HIF-1alpha/PDK1 axis. Int. J. Oncol. 2017, 50, 1531–1540. [Google Scholar] [CrossRef] [Green Version]
- Kuebler, W.M.; Bonnet, S.; Tabuchi, A. Inflammation and autoimmunity in pulmonary hypertension: Is there a role for endothelial adhesion molecules? (2017 Grover Conference Series). Pulm. Circ. 2018, 8, 2045893218757596. [Google Scholar] [CrossRef] [Green Version]
- Nicolls, M.R.; Voelkel, N.F. The Roles of Immunity in the Prevention and Evolution of Pulmonary Arterial Hypertension. Am. J. Respir. Crit. Care Med. 2017, 195, 1292–1299. [Google Scholar] [CrossRef] [Green Version]
- Rabinovitch, M.; Guignabert, C.; Humbert, M.; Nicolls, M.R. Inflammation and immunity in the pathogenesis of pulmonary arterial hypertension. Circ. Res. 2014, 115, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Stacher, E.; Graham, B.B.; Hunt, J.M.; Gandjeva, A.; Groshong, S.D.; McLaughlin, V.V.; Jessup, M.; Grizzle, W.E.; Aldred, M.A.; Cool, C.D.; et al. Modern age pathology of pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2012, 186, 261–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cracowski, J.L.; Chabot, F.; Labarere, J.; Faure, P.; Degano, B.; Schwebel, C.; Chaouat, A.; Reynaud-Gaubert, M.; Cracowski, C.; Sitbon, O.; et al. Proinflammatory cytokine levels are linked to death in pulmonary arterial hypertension. Eur. Respir. J. 2014, 43, 915–917. [Google Scholar] [CrossRef] [Green Version]
- Soon, E.; Holmes, A.M.; Treacy, C.M.; Doughty, N.J.; Southgate, L.; Machado, R.D.; Trembath, R.C.; Jennings, S.; Barker, L.; Nicklin, P.; et al. Elevated levels of inflammatory cytokines predict survival in idiopathic and familial pulmonary arterial hypertension. Circulation 2010, 122, 920–927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macdiarmid, F.; Wang, D.; Duncan, L.J.; Purohit, A.; Ghilchick, M.W.; Reed, M.J. Stimulation of aromatase activity in breast fibroblasts by tumor necrosis factor alpha. Mol. Cell. Endocrinol. 1994, 106, 17–21. [Google Scholar] [CrossRef]
- Seriolo, B.; Accardo, S.; Garnero, A.; Fasciolo, D.; Cutolo, M. Association between anticardiolipin antibody positivity and increased 17-beta-estradiol levels in premenopausal women with rheumatoid arthritis. Ann. N. Y. Acad. Sci. 1999, 876, 159–163. [Google Scholar] [CrossRef] [PubMed]
- Zuckerman, S.H.; Ahmari, S.E.; Bryan-Poole, N.; Evans, G.F.; Short, L.; Glasebrook, A.L. Estriol: A potent regulator of TNF and IL-6 expression in a murine model of endotoxemia. Inflammation 1996, 20, 581–597. [Google Scholar] [CrossRef]
- Foo, Y.Z.; Nakagawa, S.; Rhodes, G.; Simmons, L.W. The effects of sex hormones on immune function: A meta-analysis. Biol. Rev. 2017, 92, 551–571. [Google Scholar] [CrossRef] [Green Version]
- Roved, J.; Westerdahl, H.; Hasselquist, D. Sex differences in immune responses: Hormonal effects, antagonistic selection, and evolutionary consequences. Horm. Behav. 2017, 88, 95–105. [Google Scholar] [CrossRef]
- Osman, M.S.; Michelakis, E.D. Immunity Comes to Play in the “Sex Paradox” of Pulmonary Arterial Hypertension. Circ. Res. 2018, 122, 1635–1637. [Google Scholar] [CrossRef]
- Mohammad, I.; Starskaia, I.; Nagy, T.; Guo, J.; Yatkin, E.; Vaananen, K.; Watford, W.T.; Chen, Z. Estrogen receptor alpha contributes to T cell-mediated autoimmune inflammation by promoting T cell activation and proliferation. Sci. Signal. 2018, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Folomeev, M.; Dougados, M.; Beaune, J.; Kouyoumdjian, J.C.; Nahoul, K.; Amor, B.; Alekberova, Z. Plasma sex hormones and aromatase activity in tissues of patients with systemic lupus erythematosus. Lupus 1992, 1, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Tengstrand, B.; Carlstrom, K.; Fellander-Tsai, L.; Hafstrom, I. Abnormal levels of serum dehydroepiandrosterone, estrone, and estradiol in men with rheumatoid arthritis: High correlation between serum estradiol and current degree of inflammation. J. Rheumatol. 2003, 30, 2338–2343. [Google Scholar] [PubMed]
- Capellino, S.; Straub, R.H.; Cutolo, M. Aromatase and regulation of the estrogen-to-androgen ratio in synovial tissue inflammation: Common pathway in both sexes. Ann. N. Y. Acad. Sci. 2014, 1317, 24–31. [Google Scholar] [CrossRef]
- Shand, F.H.; Langenbach, S.Y.; Keenan, C.R.; Ma, S.P.; Wheaton, B.J.; Schuliga, M.J.; Ziogas, J.; Stewart, A.G. In vitro and in vivo evidence for anti-inflammatory properties of 2-methoxyestradiol. J. Pharmacol. Exp. Ther. 2011, 336, 962–972. [Google Scholar] [CrossRef] [Green Version]
- Sutherland, T.E.; Anderson, R.L.; Hughes, R.A.; Altmann, E.; Schuliga, M.; Ziogas, J.; Stewart, A.G. 2-Methoxyestradiol—A unique blend of activities generating a new class of anti-tumour/anti-inflammatory agents. Drug Discov. Today 2007, 12, 577–584. [Google Scholar] [CrossRef]
- Tofovic, S.P.; Dubey, R.K.; Jackson, E.K. 2-Hydroxyestradiol attenuates the development of obesity, the metabolic syndrome, and vascular and renal dysfunction in obese ZSF1 rats. J. Pharmacol. Exp. Ther. 2001, 299, 973–977. [Google Scholar]
- Tofovic, S.P.; Salah, E.M.; Dubey, R.K.; Melhem, M.F.; Jackson, E.K. Estradiol metabolites attenuate renal and cardiovascular injury induced by chronic nitric oxide synthase inhibition. J. Cardiovasc. Pharmacol. 2005, 46, 25–35. [Google Scholar] [CrossRef]
- Zhang, X.; Jia, Y.; Jackson, E.K.; Tofovic, S.P. 2-Methoxyestradiol and 2-ethoxyestradiol retard the progression of renal disease in aged, obese, diabetic ZSF1 rats. J. Cardiovasc. Pharmacol. 2007, 49, 56–63. [Google Scholar] [CrossRef]
- Duncan, G.S.; Brenner, D.; Tusche, M.W.; Brustle, A.; Knobbe, C.B.; Elia, A.J.; Mock, T.; Bray, M.R.; Krammer, P.H.; Mak, T.W. 2-Methoxyestradiol inhibits experimental autoimmune encephalomyelitis through suppression of immune cell activation. Proc. Natl. Acad. Sci. USA 2012, 109, 21034–21039. [Google Scholar] [CrossRef] [Green Version]
- Luc, J.G.; Paulin, R.; Zhao, J.Y.; Freed, D.H.; Michelakis, E.D.; Nagendran, J. 2-Methoxyestradiol: A Hormonal Metabolite Modulates Stimulated T-Cells Function and proliferation. Transplant. Proc. 2015, 47, 2057–2066. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Yang, T.; Su, S.; Wang, F. 2-Methoxyestradiol Alleviates Experimental Autoimmune Uveitis by Inhibiting Lymphocytes Proliferation and T cell Differentiation. BioMed Res. Int. 2016, 2016, 7948345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fox, B.D.; Shtraichman, O.; Langleben, D.; Shimony, A.; Kramer, M.R. Combination Therapy for Pulmonary Arterial Hypertension: A Systematic Review and Meta-analysis. Can. J. Cardiol. 2016, 32, 1520–1530. [Google Scholar] [CrossRef] [PubMed]
- Lajoie, A.C.; Bonnet, S.; Provencher, S. Combination therapy in pulmonary arterial hypertension: Recent accomplishments and future challenges. Pulm. Circ. 2017, 7, 312–325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
Metabolism of sex steroids. (1) The delicate balance between inactive sulfated sex steroids and sex steroids and their biologically active metabolites/precursors is controlled by the sulfotransferase (SULT)–sulfatase (STS) pathway. (2) Conversion of androgenic precursors to estrogens and intracrine production of estrogens is controlled by the aromatase pathway. (3) The 2-hydroxylation/methylation pathway of estrogen metabolism produces non-estrogenic metabolites with opposite effects to maternal estrogens. (4) Activation of the 4-Hydroxylation/16α-Hydroxylation pathway leads to the production of highly estrogenic metabolites with proliferative, proinflammatory, and angiogenic properties; (5) The 17β–hydroxysteroid dehydrogenase (17β–HSD) pathway controls the interconversion and delicate intracrine balance between estrogens with high and moderate estrogenic activity, as well as the conversion of biologically inactive 2-methoxyestrone (2ME1) to anti-angiogenic, anti-inflammatory, and pro-apoptotic 2-methoxyestradiol (2ME).
Figure 1.
Metabolism of sex steroids. (1) The delicate balance between inactive sulfated sex steroids and sex steroids and their biologically active metabolites/precursors is controlled by the sulfotransferase (SULT)–sulfatase (STS) pathway. (2) Conversion of androgenic precursors to estrogens and intracrine production of estrogens is controlled by the aromatase pathway. (3) The 2-hydroxylation/methylation pathway of estrogen metabolism produces non-estrogenic metabolites with opposite effects to maternal estrogens. (4) Activation of the 4-Hydroxylation/16α-Hydroxylation pathway leads to the production of highly estrogenic metabolites with proliferative, proinflammatory, and angiogenic properties; (5) The 17β–hydroxysteroid dehydrogenase (17β–HSD) pathway controls the interconversion and delicate intracrine balance between estrogens with high and moderate estrogenic activity, as well as the conversion of biologically inactive 2-methoxyestrone (2ME1) to anti-angiogenic, anti-inflammatory, and pro-apoptotic 2-methoxyestradiol (2ME).

Figure 2.
Putative mechanisms of action of 2ME relevant to PAH. 2ME could be viewed as a new disease modifier in PAH. (A) In injured endothelium in PAH, 2ME behaves as a biological antagonist of estradiol (E2). 2ME and E2 have opposite effects on key regulators of angioproliferation (p27Kip1, AKT, HIF1-α, VEGF) and 2ME is a more potent modulator of prostacyclin, endothelin, and nitric oxide synthesis/release than E2. (B) 2ME binds to the colchicine-binding site, disrupts the microtubule cytoskeleton, downregulates HIF1-α and inhibits angiogenesis and glycolysis. +++ vs. +, stronger effect of 2ME vs. E2; ↑ and ↓: E2 (red) and 2ME (blue) related increase/stimulation and decrease/inhibition, respectively. [B] black ↑ and ↓: subsequent and/or indirect effects of 2ME; blue arrows: binding of or stimulation by 2ME; blue T arrows: inhibition/down-regulation by 2ME.
Figure 2.
Putative mechanisms of action of 2ME relevant to PAH. 2ME could be viewed as a new disease modifier in PAH. (A) In injured endothelium in PAH, 2ME behaves as a biological antagonist of estradiol (E2). 2ME and E2 have opposite effects on key regulators of angioproliferation (p27Kip1, AKT, HIF1-α, VEGF) and 2ME is a more potent modulator of prostacyclin, endothelin, and nitric oxide synthesis/release than E2. (B) 2ME binds to the colchicine-binding site, disrupts the microtubule cytoskeleton, downregulates HIF1-α and inhibits angiogenesis and glycolysis. +++ vs. +, stronger effect of 2ME vs. E2; ↑ and ↓: E2 (red) and 2ME (blue) related increase/stimulation and decrease/inhibition, respectively. [B] black ↑ and ↓: subsequent and/or indirect effects of 2ME; blue arrows: binding of or stimulation by 2ME; blue T arrows: inhibition/down-regulation by 2ME.

Figure 3.
(A) Dual metabolic activity of CYP1B1 and an inflammation-instigated estradiol (E2)-feed forward mechanism that involves sulfatase, aromatase, E2, and BMPR2 and results in the development of an angioproliferative and inflammatory phenotype and increased risk of PAH in women. E2 and 2-methoxyestradiol (2ME) have opposite effects on CYP1B1 activity and E2 and arachidonic acid (AA) metabolism, which leads to the production of proinflammatory E2 and AA metabolites. Red arrows: estrogens related increase/stimulation or red T arrows reduction/inhibition of enzyme expression/activity, E2 and AA metabolite production and inflammation, and BMPR2 down-regulation. Magenta arrows: CYP1B1 activation and related AA metabolism result in the production of proinflammatory AA metabolites and up-regulation of E2 producing enzymes; Blue T arrows: 2ME inhibits CYP1B1 activity and the production of AA metabolites and proinflammatory cytokines induced by estrogen-producing enzymes. ↑ and ↓: estrogens (red) CYP1B1 (magenta) and 2ME (blue) related increase and decrease, respectively; BMPR2 = bone morphogenetic protein receptor type 2; COMT = catechol-O-methyltransferase; CYP = cytochrome p450 enzymes; EETs = epoxyeicosatrienoic acids; HETEs = hydroxyeicosatetraenoic acids; she = soluble epoxide hydrolase. (B) Compared to males, female SU + Hx PH rats exhibit (indicated by arrows) exacerbated angioproliferation, i.e. occlusive lesions (a) and perivascular inflammation (b,c), and sporadically develop necrotizing arteritis i.e., grade 6 lesions (d).
Figure 3.
(A) Dual metabolic activity of CYP1B1 and an inflammation-instigated estradiol (E2)-feed forward mechanism that involves sulfatase, aromatase, E2, and BMPR2 and results in the development of an angioproliferative and inflammatory phenotype and increased risk of PAH in women. E2 and 2-methoxyestradiol (2ME) have opposite effects on CYP1B1 activity and E2 and arachidonic acid (AA) metabolism, which leads to the production of proinflammatory E2 and AA metabolites. Red arrows: estrogens related increase/stimulation or red T arrows reduction/inhibition of enzyme expression/activity, E2 and AA metabolite production and inflammation, and BMPR2 down-regulation. Magenta arrows: CYP1B1 activation and related AA metabolism result in the production of proinflammatory AA metabolites and up-regulation of E2 producing enzymes; Blue T arrows: 2ME inhibits CYP1B1 activity and the production of AA metabolites and proinflammatory cytokines induced by estrogen-producing enzymes. ↑ and ↓: estrogens (red) CYP1B1 (magenta) and 2ME (blue) related increase and decrease, respectively; BMPR2 = bone morphogenetic protein receptor type 2; COMT = catechol-O-methyltransferase; CYP = cytochrome p450 enzymes; EETs = epoxyeicosatrienoic acids; HETEs = hydroxyeicosatetraenoic acids; she = soluble epoxide hydrolase. (B) Compared to males, female SU + Hx PH rats exhibit (indicated by arrows) exacerbated angioproliferation, i.e. occlusive lesions (a) and perivascular inflammation (b,c), and sporadically develop necrotizing arteritis i.e., grade 6 lesions (d).

Figure 4.
The 17β-HSD pathway controls the interconversion and delicate intracrine balance between estrogens with high and moderate estrogenic activity, as well as the conversion of biologically inactive 2-methoxyestrone (2ME1) to anti-angiogenic, anti-inflammatory, and pro-apoptotic 2-methoxyestradiol (2ME). RA = retinoic acid, an inducer of 17β-HSD1; hPASMCs = human pulmonary artery smooth muscle cells; MCT = monocrotaline; OVX = ovariectomy.
Figure 4.
The 17β-HSD pathway controls the interconversion and delicate intracrine balance between estrogens with high and moderate estrogenic activity, as well as the conversion of biologically inactive 2-methoxyestrone (2ME1) to anti-angiogenic, anti-inflammatory, and pro-apoptotic 2-methoxyestradiol (2ME). RA = retinoic acid, an inducer of 17β-HSD1; hPASMCs = human pulmonary artery smooth muscle cells; MCT = monocrotaline; OVX = ovariectomy.

Figure 5.
Three-tier effects of estradiol concept in PAH. The effects on vascular pathobiology, progression, and prognosis of PAH are shown. Estradiol could be viewed as: (1) the protector of heathy pulmonary vasculature; (2) the instigator and perpetuator of disease in injured pulmonary vascular; and (3) the protector of the overloaded right ventricle.
Figure 5.
Three-tier effects of estradiol concept in PAH. The effects on vascular pathobiology, progression, and prognosis of PAH are shown. Estradiol could be viewed as: (1) the protector of heathy pulmonary vasculature; (2) the instigator and perpetuator of disease in injured pulmonary vascular; and (3) the protector of the overloaded right ventricle.


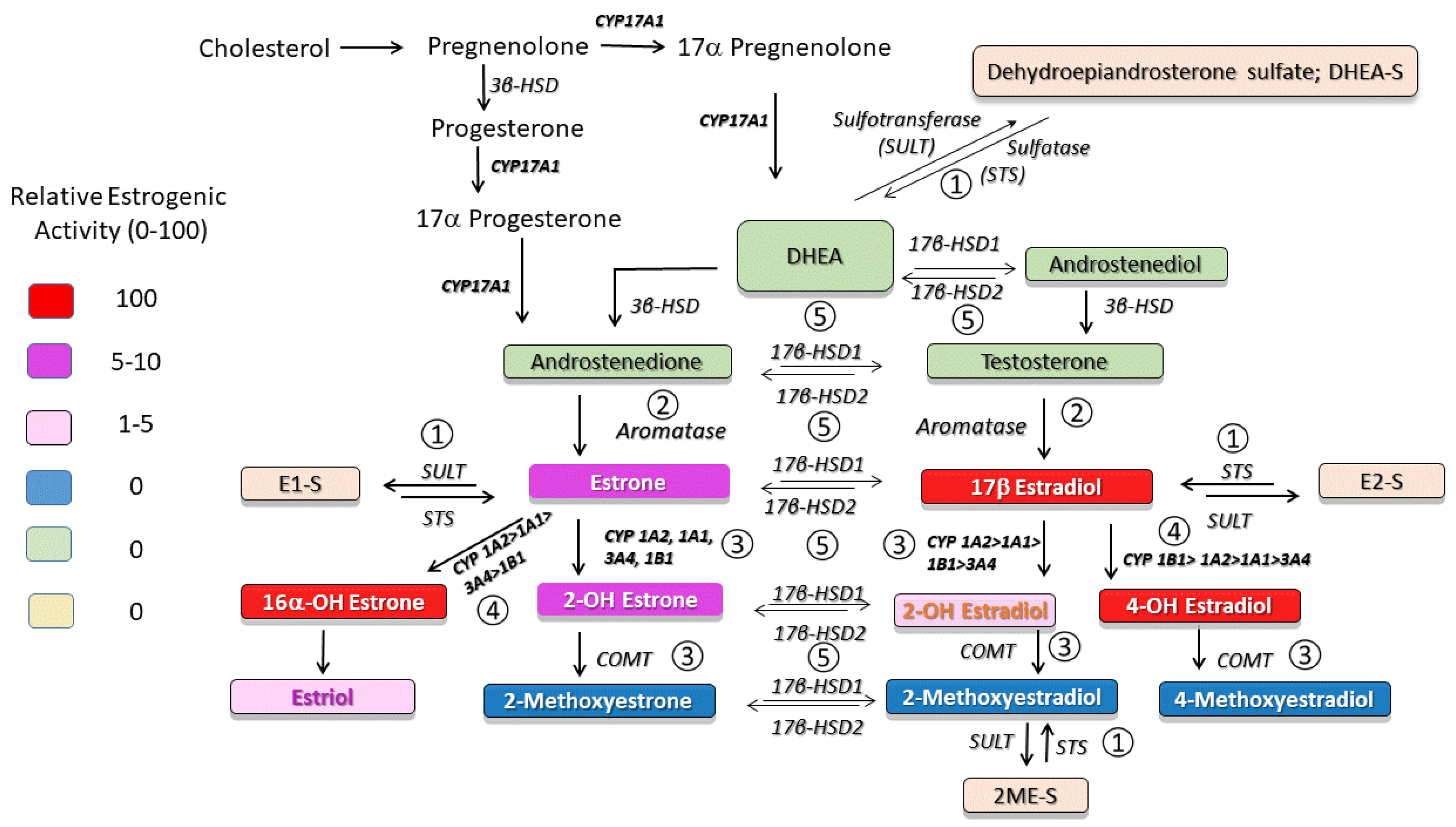{kind=link}
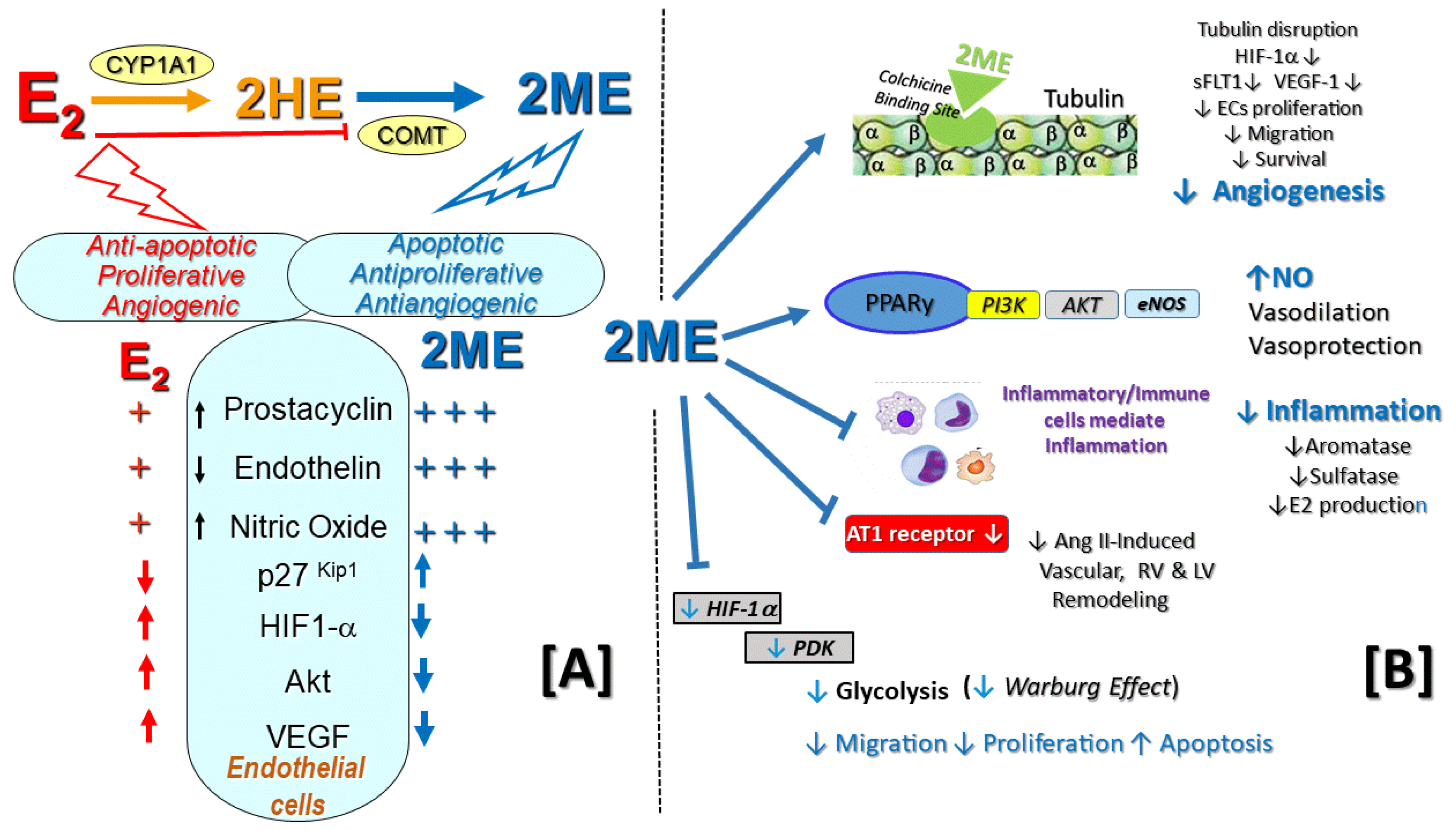{kind=link}
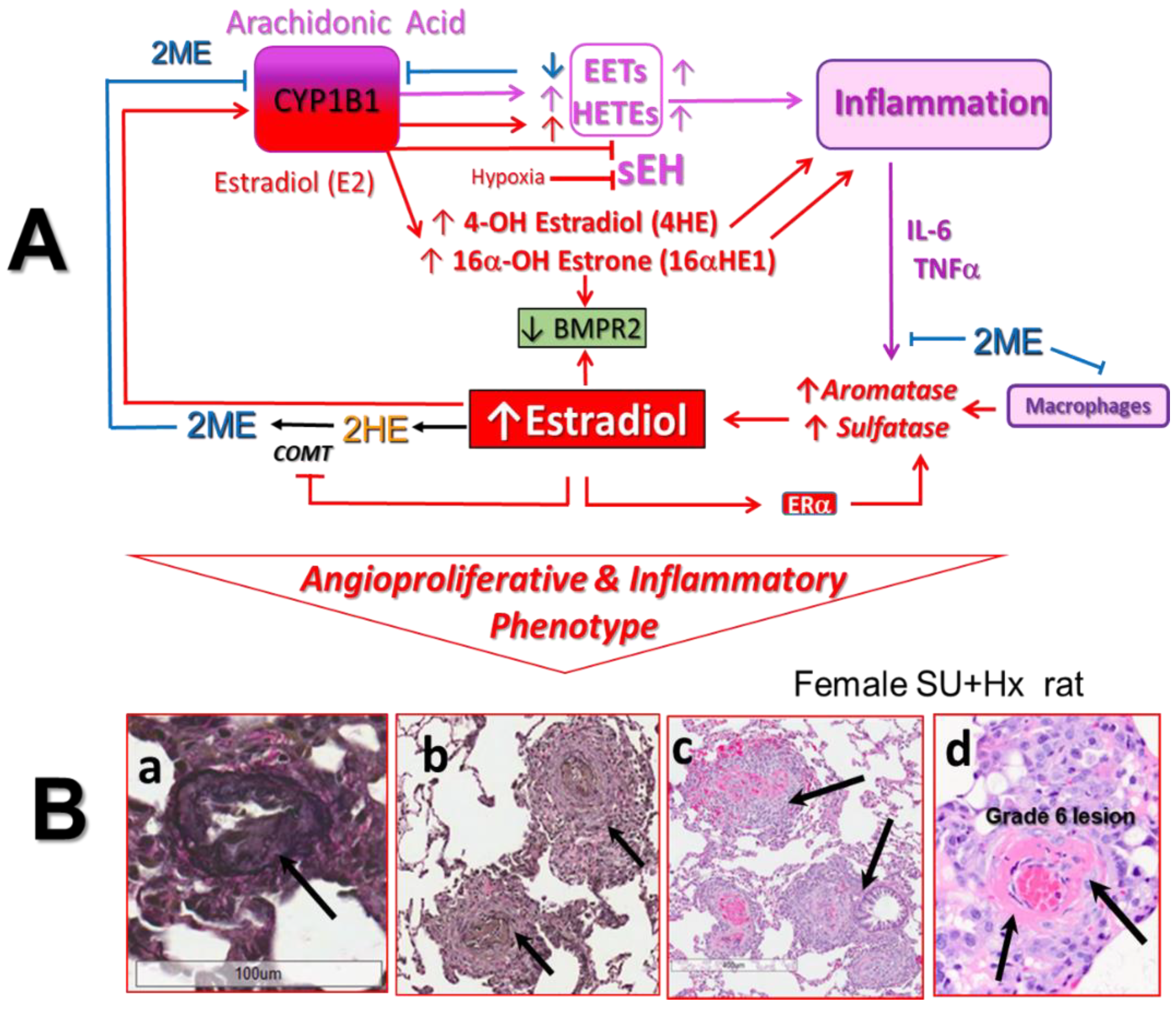{kind=link}
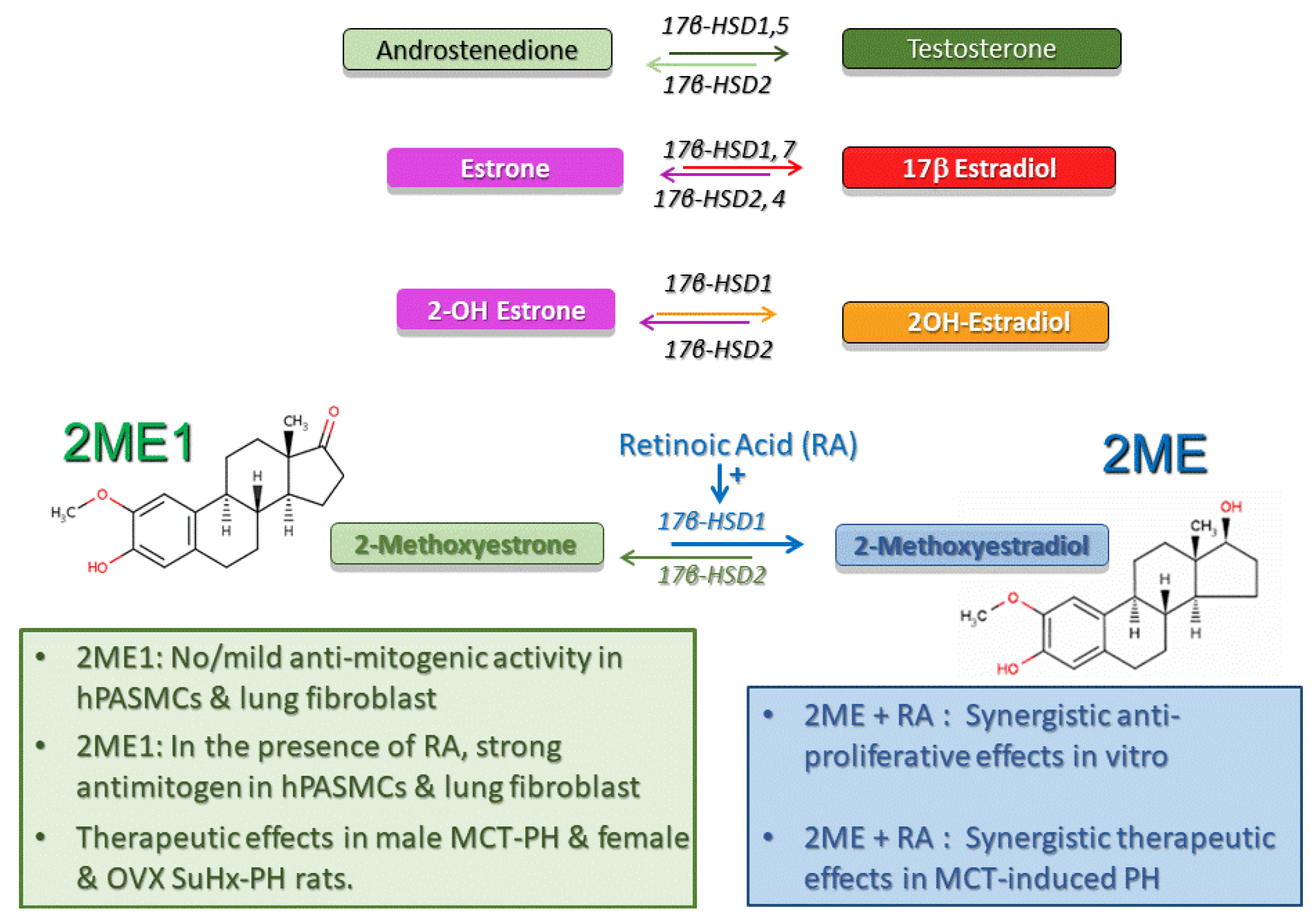{kind=link}
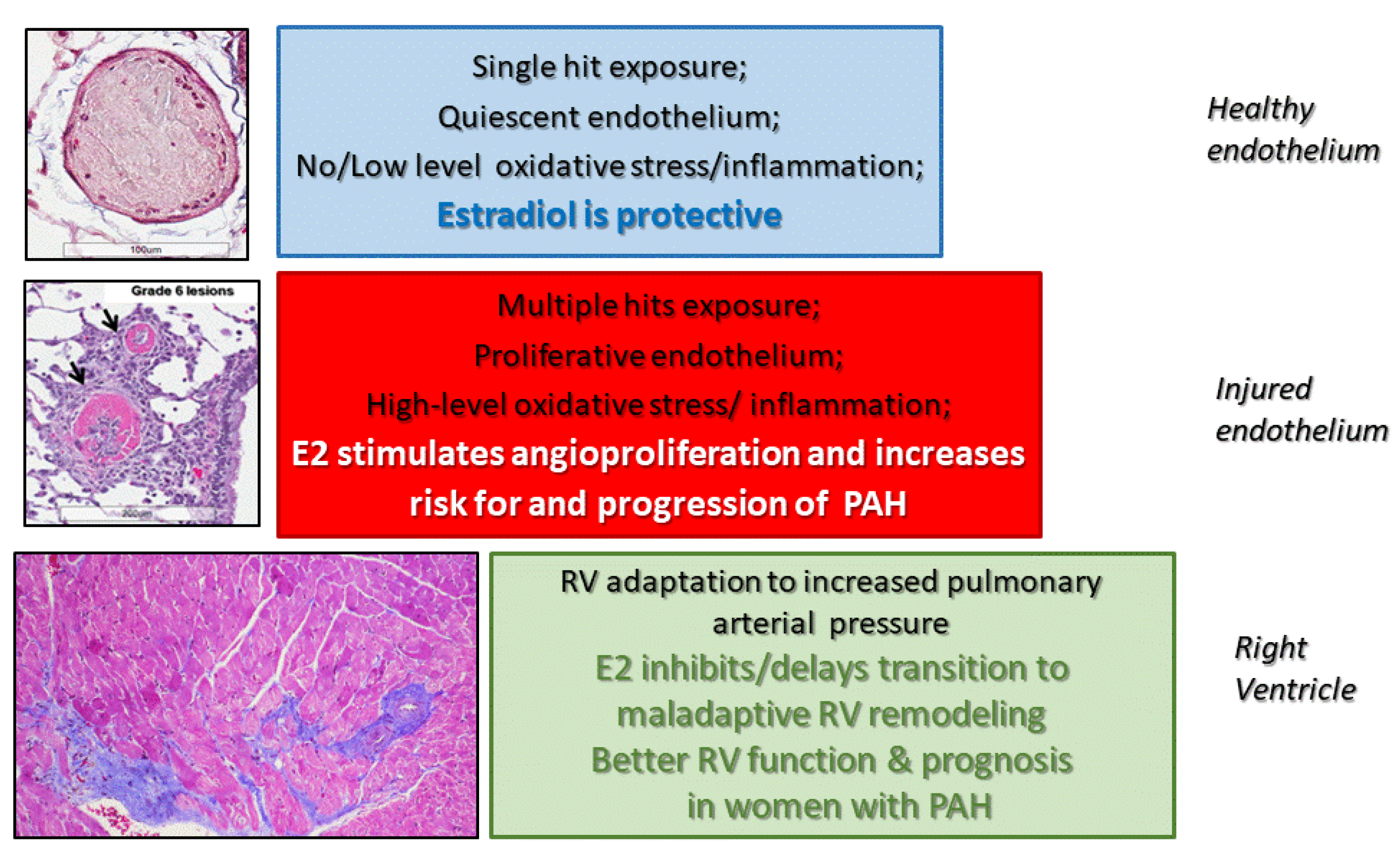{kind=link}
Table 1.
Effects of sex hormones, their precursors, and metabolism inhibitors experimental PH and human PAH.
Table 1.
Effects of sex hormones, their precursors, and metabolism inhibitors experimental PH and human PAH.
| Animal Model PAH Patients | Hormone, Sex, Enzyme, Treatment | Effects/Comments | References |
|---|---|---|---|
| CH- and MCT-PH in rats | Estradiol (E2) | Female sex protective; E2 attenuate PH and RV and vascular remodeling; OVX exacerbates PH | Reviewed previously in Tofovic SP, 2010 [8] |
| MCT female and OVX rats | Estradiol | E2 attenuates whereas OVX exacerbates PH; ↓ E2 levels and aromatase, ↑ Cyp 1A1 and Cyp1B1 | Yuan P. et al., 2013 [48] |
| Su + Hx male and female rats | Female versus Male | Female: greater pulmonary vascular remodeling; Male: more severe RV failure, lower survival rate | Rafikova et al., 2015 [49] |
| Chronic hypoxia (CH)- induced PH in rats | DHEA | Prevents or reverses PH; Inhibits PH and augments vasodilator responsiveness to NO | Bonnet S. et al., 2003; Oka M. et al., 2007 [37,39] |
| MCT + pneumonectomy rat | DHEA | Rescue treatment attenuates PH and vascular remodeling and eliminates late mortality | Homma N. et al., 2008 [40] |
| MCT rat | DHEA | Reduces PH and RV hypertrophy | Paulin R. et al., 2011 [41] |
| Su + Hx rat | DHEA | Attenuates PH; restores RV structure/function | Alzoubi A. et al., 2012 [44] |
| CH-PH rats Rat-pup model of CH-PH | DHEA | Reverses CH/reoxygenation-induced RV dysfunction; Attenuates PH, RV, and vascular remodeling in infant rats | Dumas de La Roque E. et al., 2012; 2013 [43,50] |
| Pregnant Sheep | DHEA | Vasodilator effects on fetal pulmonary circulation | Sharma D. et al., 2018 [42] |
| Rat model of left heart failure (LHF)-induced PH | DHEA | Attenuates LHF-induced PH and RV and vascular remodeling | Zhang YT. et al., 2019 [51] |
| PM women, idiopathic, CTD- or CHD-PAH | Low DHEA-S High E2 | Increased risk and severity of PAH in postmenopausal women | Baird GL et al., 2018 [47] |
| Men with idiopathic, heritable, or CTD-PAH | Low DHEA-S High E2 | Associated with risk of PAH; ↑ E2, shorter 6MWD; ↑ DHEA-S, lower right atrial pressure | Ventetuolo CE et al., 2016 [46] |
| Patients with COPD | DHEA | Improves PH in COPD patients | D. La Roque E. et al., 2012 [45] |
| Men with idiopathic PAH | Estradiol Testosterone (T) Progesterone (P) | ↑ E2 and E2/T ratio and ↓ T and P associated with ↑ risk of PAH; high E2 independently associated with higher mortality | Wu W-H. et al., 2018 [52] |
| Premenopausal women with idiopathic PAH | FSH Progesterone | ↑ FSH and ↓ P tended to be associated with high risk of IPAH and mortality among patients | Zhang Y-X et al., 2019 [53] |
| Su+Hx female rats | Anastrozole | Reduces PH and number of occlusive and PLX lesions, but has no effect on RV remodeling. | Tofovic SP. et al., 2013 [54] * |
| CH mice and Su+Hx rats | Anastrozole | Attenuates PH only in female animals | Mair KM. et al., 2014 [55] |
| Su+Hx rats | Metformin | ↓ circulating estrogens and aromatase levels/activity, and attenuates PH and vascular and RV remodeling | Dean A. et al., 2016 [56] |
| BMPR2 mutant mice | Anastrozole + Fulvestrant | Prevents and Reverses PH and reduces BMPR2 mutation associated with metabolic defects | Chen X. et al., 2017 [57] |
| Portopulmonary hypertension | Estradiol Aromatase | Irrespective of gender, in patients with liver disease, ↑ aromatase and ↑ E2 levels increase risk of portopulmonary PH | Roberts KE. et al., 2009 [58] |
| PAH patients | Anastrozole | Reduces serum E2 and E1 levels, significantly increases 6MWD, but has no effect on RV function | Kawut SM et al., 2017 [59] |
| Su + Hx OVX female rats | Estradiol | Protects RV function and restores RV ventricular–vascular coupling efficiency | Liu A. et al., 2017 [60] |
| Su + Hx male, female and OVX rats | Estradiol | Endogenous and exogenous E2 exerts protective effects on baseline RV function and after an acute exercise challenge | Frump, AL. et al., 2015 [15] Lahm T. et al., 2016 [13] |
| Su + Hx female and OVX mice | Estradiol | E2 has no effect on PH, yet reduces proximal conduit arteries stiffness and improves ventricular–vascular coupling; no data on RV remodeling available | Liu A. et al., 2015 [61] |
| Su + Hx female and OVX mice | Estradiol | In absence of RV remodeling in diseased animals, improves RV function (enhances RV contractility in response to PH and preserves cardiac reserve) | Liu A. et al., 2014 [62] |
| MCT in Apo E deficient female mice | Estradiol | Rescue treatment reduces PH and RV hypertrophy | Umar S. et al., 2017 [63] |
| Female and OVX mice overexpressing SERT | Estradiol | Only female, SERT+ mice develop PAH, OVX abolishes and E2 treatment of OVX + SERT mice reestablishes PH. | White K. et al., 2011 [64] |
| MCT OVX rats | Progesterone | Attenuates PH, and RV and vascular remodeling | Tofovic SP. et al., 2009 [65] |
↑—increased; ↓—reduced; RV—right ventricle; PM—postmenopausal; OVX—ovariectomy; CTD—connective tissue disease; CHD—congenital heart disease; SERT—serotonin transporter; *—published in form of abstract.
Table 2.
Effects of estradiol metabolites in PH.
| Animal Model | Estradiol Metabolite | Effects Comments | Reference |
|---|---|---|---|
| MCT male rats | 2-methoxyestradiol 2-hydroxyestardiol | Attenuate development or progression of PH | Tofovic SP et al., 2005 [12] |
| MCT OVX and female rats | 2-methoxyestradiol | Ovariectomy exacerbates PH, whereas 2ME attenuates PH in OVX rats; no estrogenic effects | Tofovic SP et al., 2006 [26] |
| MCT male rats | 2-ethoxyestradiol | Synthetic metabolite; attenuates PH, lung inflammation, and RV and vascular modeling, and eliminates late mortality | Tofovic SP et al., 2008 [22] |
| Bleomycin-induced PH and lung fibrosis, OVX female rats | 2-methoxyestradiol | Ovariectomy exacerbates whereas 2ME attenuates PH, inflammation, fibrosis and vascular remodeling | Tofovic SP et al., 2009 [21] |
| MCT male rats | 2-methoxyestradiol | Dose-dependent therapeutic effects on PH, lung inflammation and RV and vascular remodeling; no estrogenic effects | Tofovic SP et al., 2010 [113] |
| MCT male rats | 2-methoxyestradiol | Synergistic effects with bosentan or sildenafil on amelioration of PH, lung inflammation, and vascular remodeling | Tofovic SP et al., 2010 [12] |
| CH male and female rats | 2-methoxyestradiol | Attenuates development and retards the progression of PH; decreases HIF-1α expression and reduces elevated hematocrit | Wang L. et al., 2017; Hao S. et al., 2019 Docherty CK. et al., 2019, Tofovic SP. et al., 2005 * [77,78,79,114] |
| MCT male rats | 2-methoxyestradiol +/− retinoic acid | Synergistic effects of 2ME with retinoic acid on amelioration of PH | Tofovic SP et al., 2008 [115] * |
| MCT OVX rats; Female and OVX Su+Hx rats | 2-methoxyestrone | Attenuates MCT-induced PH, inflammation and RV and vascular remodeling; attenuates Su+Hx-induced PH, RV dysfunction, and remodeling and number of occlusive lesions | Tofovic SP. et al., 2008, Hu J. et al., 2016 [116,117] * |
| Su+Hx OVX rats | 4-hydroxyestradiol | Has no effect on PH, but attenuates RV hypertrophy | Tofovic SP. et al., 2013 [54] * |
| Obese ZDSD rats | 2-hydroxyestradiol | Reduces HbA1c and attenuates metabolic syndrome-induced PH in male rats | Tofovic SP. et al., 2014 [118] * |
| Female mice | 16α - hydroxyestrone | Induces mild PH and RV and vascular remodeling | White K. et al., 2012 [119] |
| BMPR2 mutant mice | 16α -hydroxyestrone | Exacerbates BMPR2-associated PH | Chen X. et al., 2016 [120] |
*—Published in form of abstract; MCT—monocrotaline; CH—chronic hypoxia. HbA1c-Glycosilated hemoglobin; BMPR2-bone morphogenetic protein receptor type 2.
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Tofovic, S.P.; Jackson, E.K. Estradiol Metabolism: Crossroads in Pulmonary Arterial Hypertension. Int. J. Mol. Sci. 2020, 21, 116. https://doi.org/10.3390/ijms21010116
AMA Style
Tofovic SP, Jackson EK. Estradiol Metabolism: Crossroads in Pulmonary Arterial Hypertension. International Journal of Molecular Sciences. 2020; 21(1):116. https://doi.org/10.3390/ijms21010116
Chicago/Turabian StyleTofovic, Stevan P., and Edwin K. Jackson. 2020. "Estradiol Metabolism: Crossroads in Pulmonary Arterial Hypertension" International Journal of Molecular Sciences 21, no. 1: 116. https://doi.org/10.3390/ijms21010116
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.