Genetic Analysis and Evolutionary Changes of the Torque teno sus Virus

Abstract
:1. Introduction
2. Results
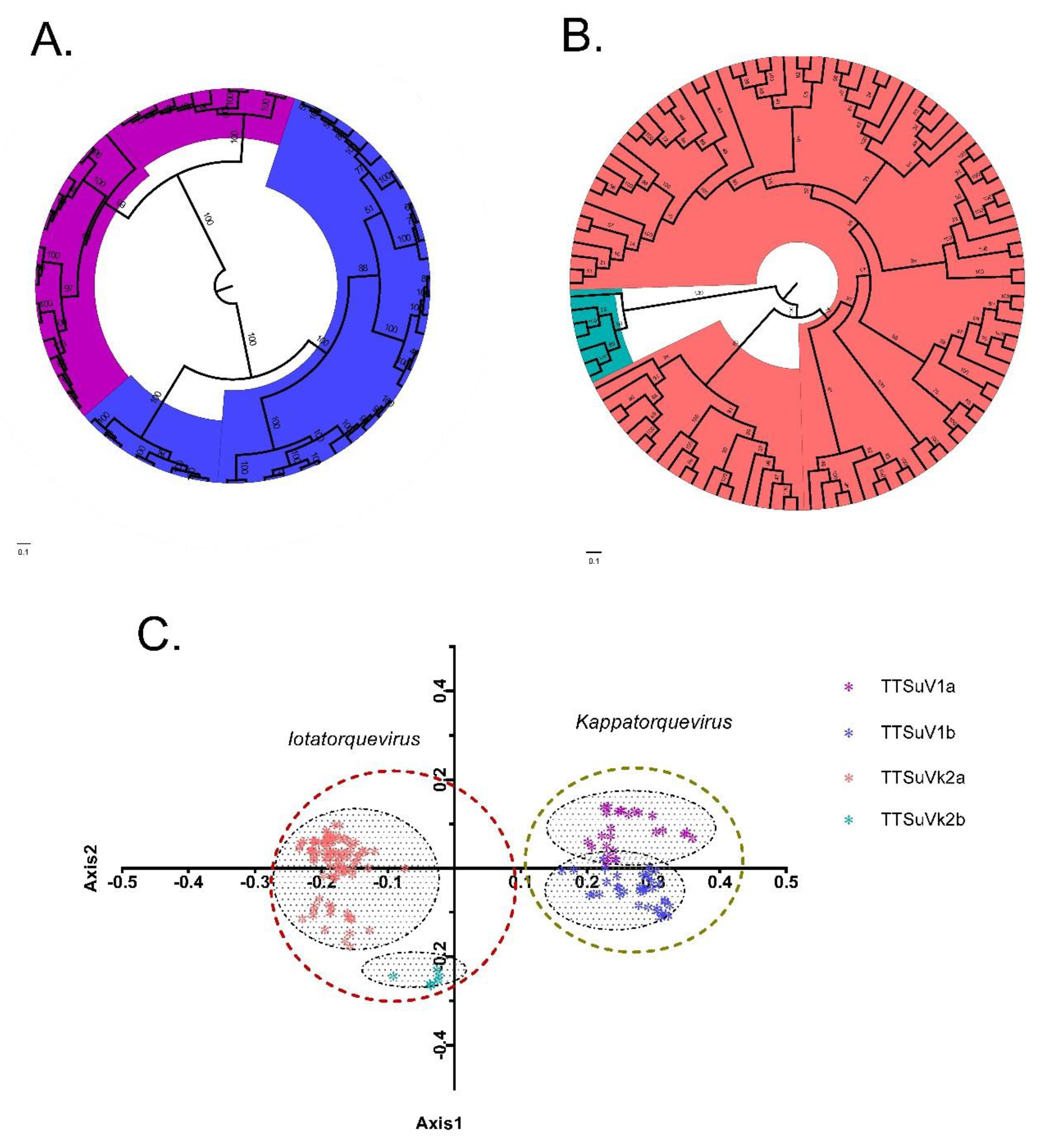
2.1. Recombination and Phylogeny
2.2. Principal Component Analysis
2.3. Codon Usage Analysis
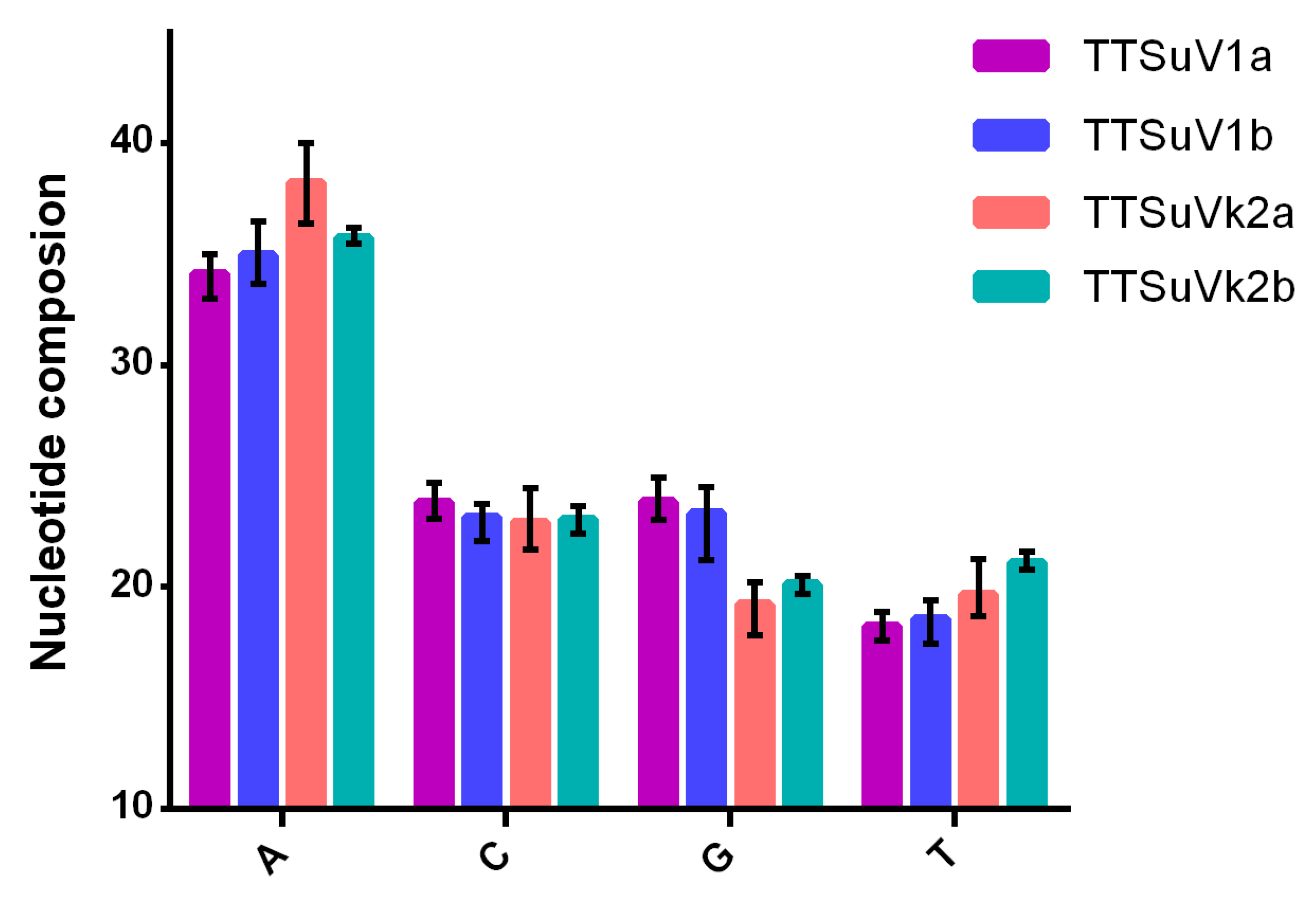
2.3.1. Nucleotide and Codon Composition
2.3.2. RSCU in Different Species
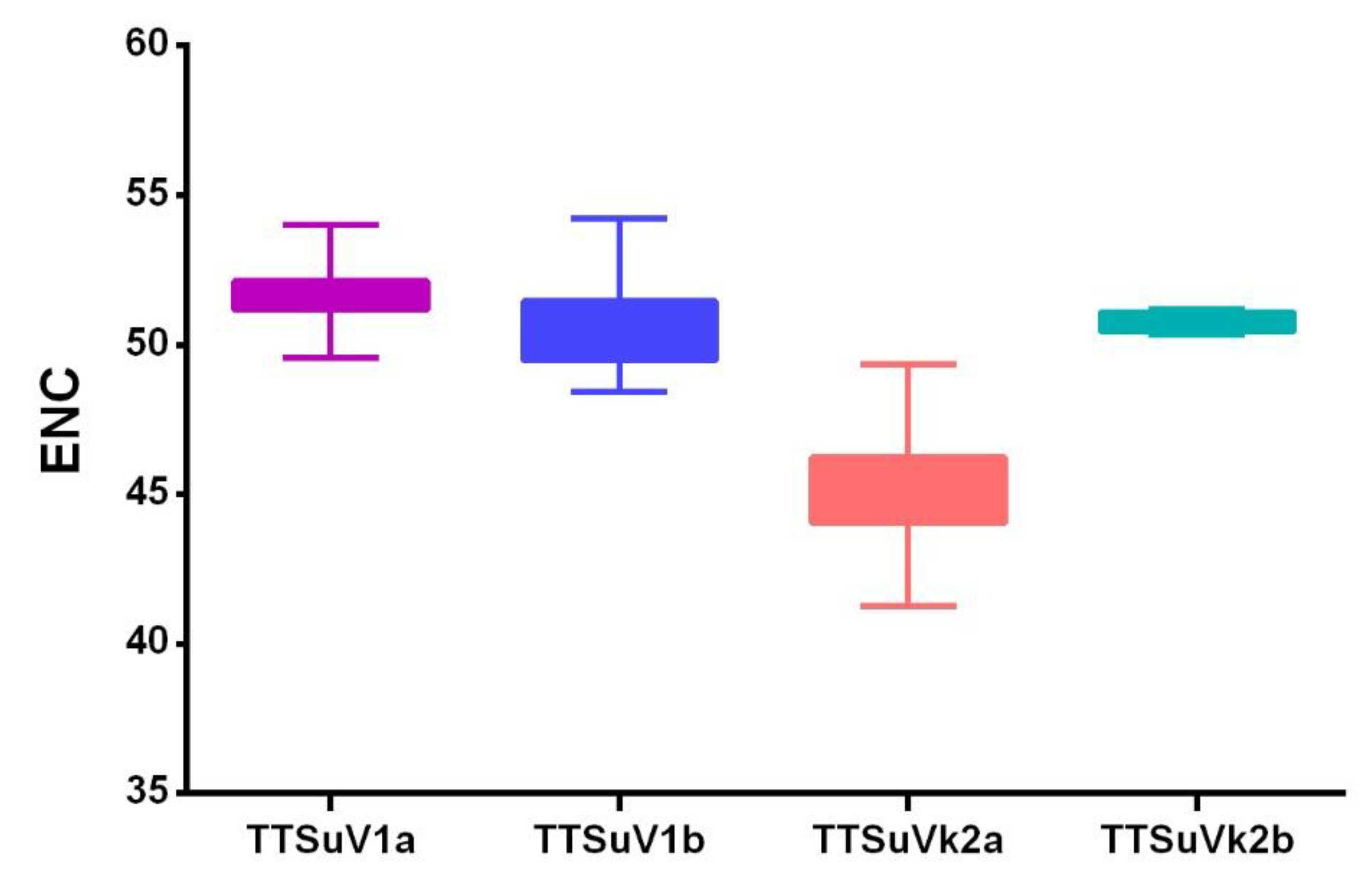
2.3.3. TTSuV Codon Usage Bias
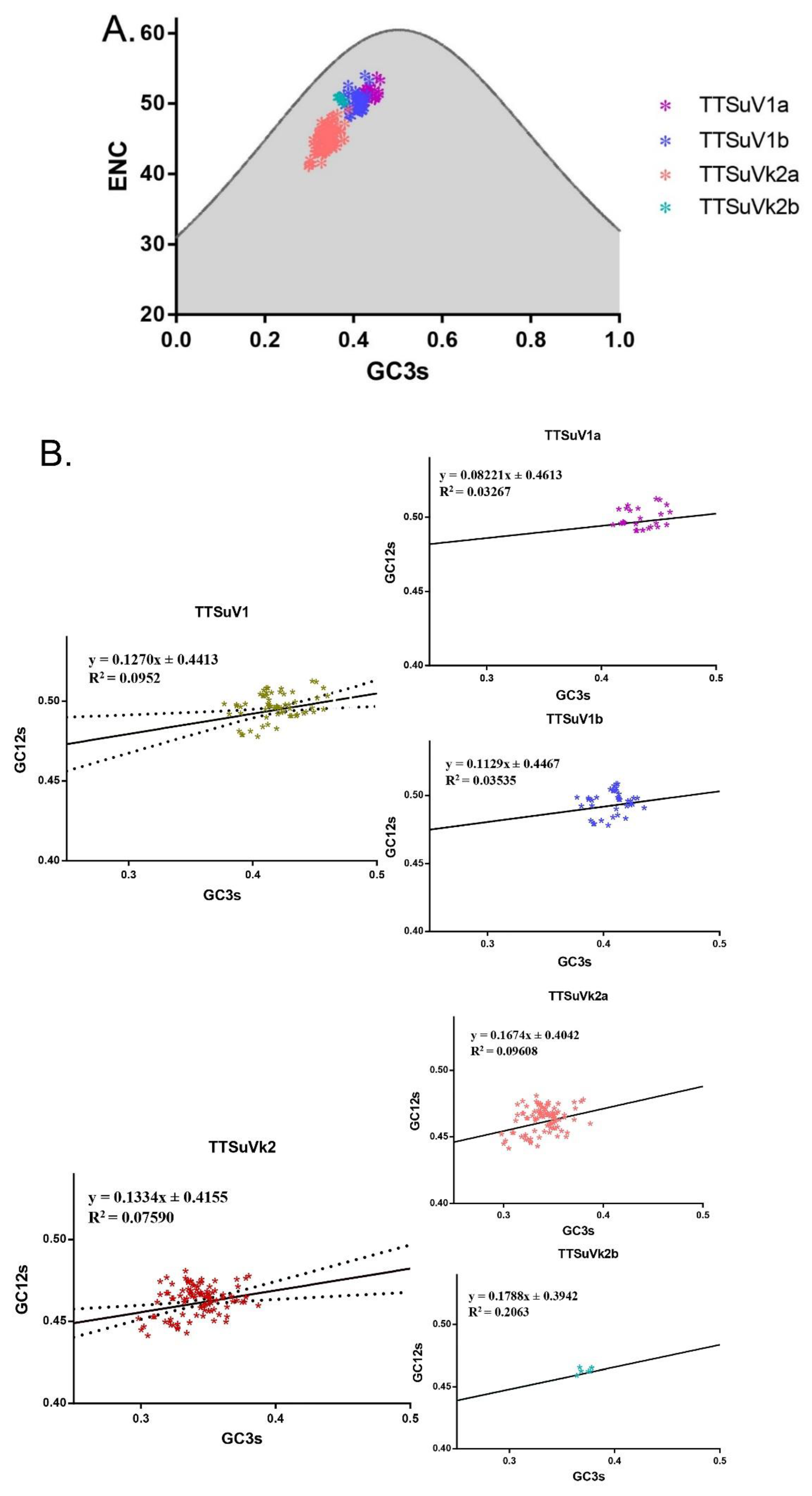
2.3.4. Factors Shaping the Codon Usage of TTSuV
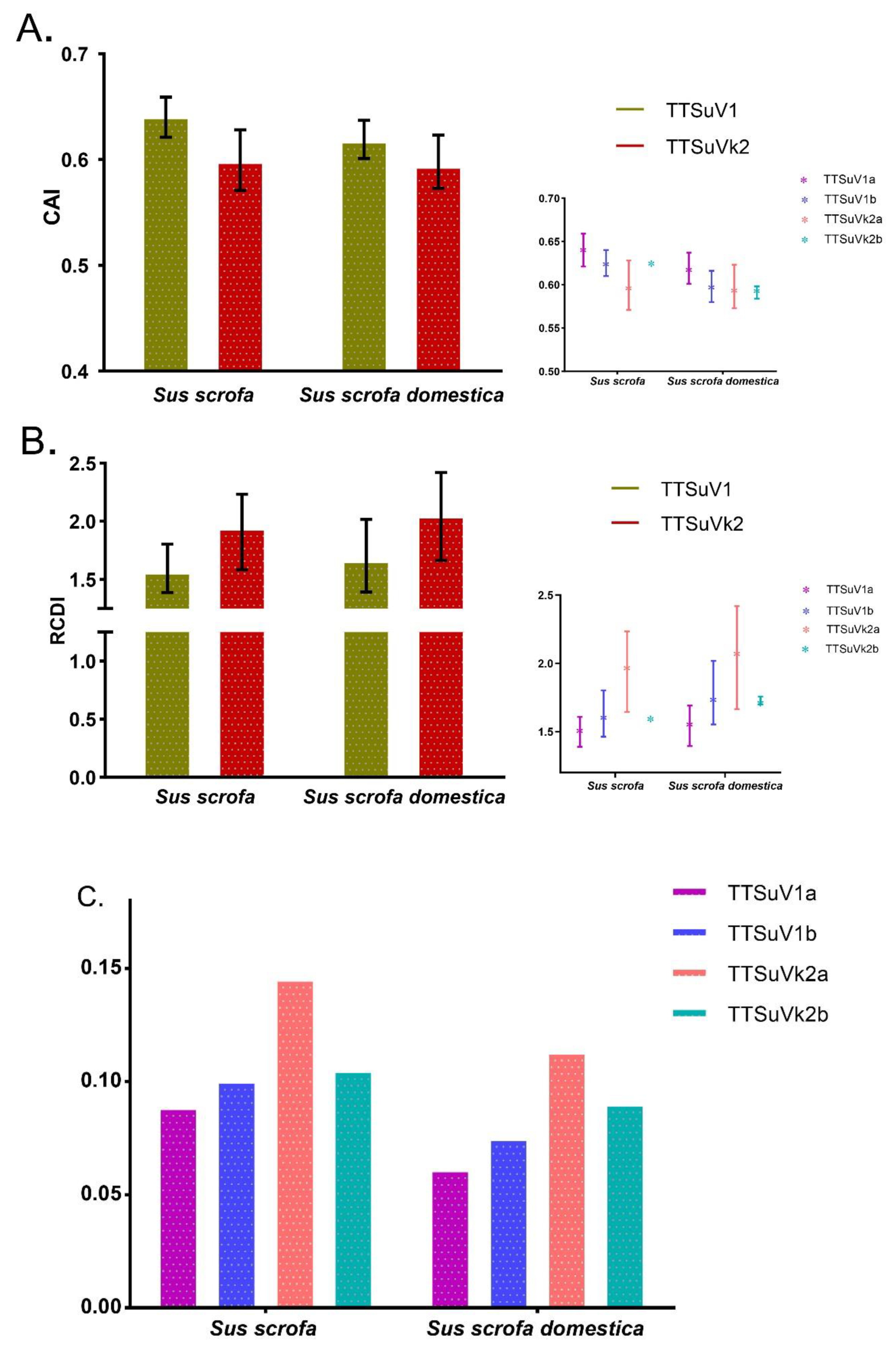
2.3.5. Species-Specific Codon Adaptation and Deoptimization Pattern of TTSuV
2.3.6. Selection Pressure on TTSuV is Species-Specific
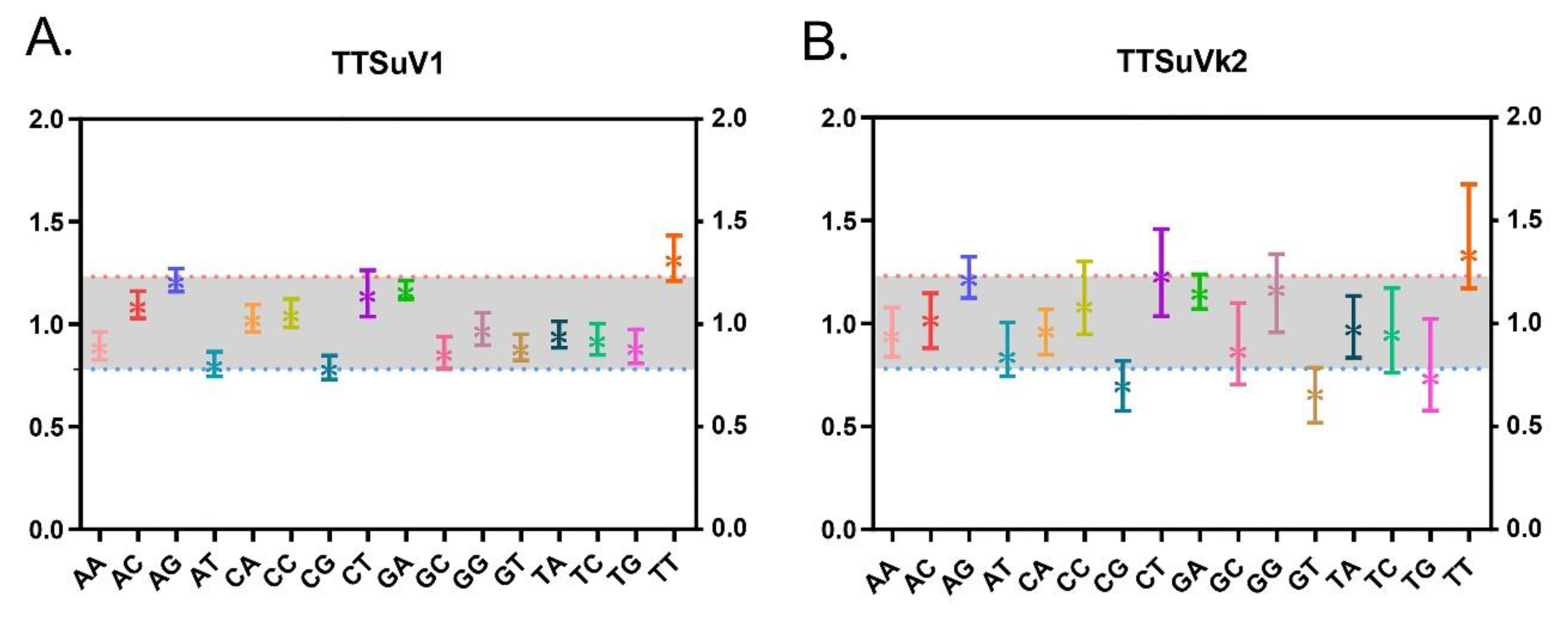
2.4. TTSuV Dinucleotide Abundance
3. Discussion
4. Materials and Methods
4.1. Sequence Data
4.2. Recombination and Phylogenetic Analysis
4.3. Codon Usage Analysis
4.3.1. Sequence Composition
4.3.2. Relative Synonymous Codon Usage
4.4. Principal Component Analysis
4.5. Effective Number of Codons Analysis
4.6. Neutrality Analysis
Codon Adaptation Index and Relative Codon Deoptimization Index
4.7. Similarity Index
4.8. Dinucleotide Abundance Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Ng, T.F.F.; Suedmeyer, W.K.; Wheeler, E.; Gulland, F.; Breitbart, M. Novel anellovirus discovered from a mortality event of captive California sea lions. J. Gen. Virol. 2009, 90, 1256–1261. [Google Scholar] [CrossRef] [PubMed]
- Tian, L.; Shen, X.; Murphy, R.W.; Shen, Y. The adaptation of codon usage of +ssRNA viruses to their hosts. Infect. Genet. Evol. 2018, 63, 175–179. [Google Scholar] [CrossRef] [PubMed]
- Sibila, M.; Martínez-Guinó, L.; Huerta, E.; Mora, M.; Grau-Roma, L.; Kekarainen, T.; Segalés, J. Torque teno virus (TTV) infection in sows and suckling piglets. Vet. Microbiol. 2009, 137, 354–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okamoto, H.; Takahashi, M.; Nishizawa, T.; Tawara, A.; Fukai, K.; Muramatsu, U.; Naito, Y.; Yoshikawa, A. Genomic characterization of TT viruses (TTVs) in pigs, cats and dogs and their relatedness with species-specific TTVs in primates and tupaias. J. Gen. Virol. 2002, 83, 1291–1297. [Google Scholar] [CrossRef] [PubMed]
- Cornelissen-Keijsers, V.; Jiménez-Melsió, A.; Sonnemans, D.; Cortey, M.; Segalés, J.; van den Born, E.; Kekarainen, T. Discovery of a novel Torque teno sus virus species: Genetic characterization, epidemiological assessment and disease association. J. Gen. Virol. 2012, 93, 2682–2691. [Google Scholar] [CrossRef] [PubMed]
- Biagini, P. Classification of TTV and Related Viruses (Anelloviruses). In TT Viruses: The Still Elusive Human Pathogens; de Villiers, E.-M., Hausen, H.Z., Eds.; Springer: Berlin/Heidelberg, Germany, 2009; pp. 21–33. [Google Scholar]
- Zheng, H.; Ye, L.; Fang, X.; Li, B.; Wang, Y.; Xiang, X.; Kong, L.; Wang, W.; Zeng, Y.; Ye, L.; et al. Torque Teno Virus (SANBAN Isolate) ORF2 Protein Suppresses NF-κB Pathways via Interaction with IκB Kinases. J. Virol. 2007, 81, 11917–11924. [Google Scholar] [CrossRef] [PubMed]
- Cadar, D.; Kiss, T.; Ádám, D.; Cságola, A.; Novosel, D.; Tuboly, T. Phylogeny, spatio-temporal phylodynamics and evolutionary scenario of Torque teno sus virus 1 (TTSuV1) and 2 (TTSuV2) in wild boars: Fast dispersal and high genetic diversity. Vet. Microbiol. 2013, 166, 200–213. [Google Scholar] [CrossRef]
- Cortey, M.; Pileri, E.; Segalés, J.; Kekarainen, T. Globalisation and global trade influence molecular viral population genetics of Torque Teno Sus Viruses 1 and 2 in pigs. Vet. Microbiol. 2012, 156, 81–87. [Google Scholar] [CrossRef]
- Li, G.; He, W.; Zhu, H.; Bi, Y.; Wang, R.; Xing, G.; Zhang, C.; Zhou, J.; Yuen, K.-Y.; Gao, G.F.; et al. Origin, Genetic Diversity, and Evolutionary Dynamics of Novel Porcine Circovirus 3. Adv. Sci. 2018, 5, 1800275. [Google Scholar] [CrossRef]
- Cortey, M.; Macera, L.; Segalés, J.; Kekarainen, T. Genetic variability and phylogeny of Torque teno sus virus 1 (TTSuV1) and 2 (TTSuV2) based on complete genomes. Vet. Microbiol. 2011, 148, 125–131. [Google Scholar] [CrossRef]
- He, W.; Auclert, L.Z.; Zhai, X.; Wong, G.; Zhang, C.; Zhu, H.; Xing, G.; Wang, S.; He, W.; Li, K.; et al. Interspecies Transmission, Genetic Diversity, and Evolutionary Dynamics of Pseudorabies Virus. J. Infect. Dis. 2018, 219, 1705–1715. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.; Bera, B.C.; Greenbaum, B.D.; Bhatia, S.; Sood, R.; Selvaraj, P.; Anand, T.; Tripathi, B.N.; Virmani, N. Revelation of Influencing Factors in Overall Codon Usage Bias of Equine Influenza Viruses. PLoS ONE 2016, 11, e0154376. [Google Scholar] [CrossRef] [PubMed]
- Grantham, R.; Gautier, C.; Gouy, M.; Mercier, R.; Pavé, A. Codon catalog usage and the genome hypothesis. Nucleic Acids Res. 1980, 8, r49–r62. [Google Scholar] [CrossRef] [PubMed]
- Moratorio, G.; Iriarte, A.; Moreno, P.; Musto, H.; Cristina, J. A detailed comparative analysis on the overall codon usage patterns in West Nile virus. Infect. Genet. Evol. 2013, 14, 396–400. [Google Scholar] [CrossRef] [PubMed]
- Ramos, N.; Mirazo, S.; Botto, G.; Teixeira, T.F.; Cibulski, S.P.; Castro, G.; Cabrera, K.; Roehe, P.M.; Arbiza, J. High frequency and extensive genetic heterogeneity of TTSuV1 and TTSuVk2a in PCV2- infected and non-infected domestic pigs and wild boars from Uruguay. Vet. Microbiol. 2018, 224, 78–87. [Google Scholar] [CrossRef]
- Martínez, L.; Kekarainen, T.; Sibila, M.; Ruiz-Fons, F.; Vidal, D.; Gortázar, C.; Segalés, J. Torque teno virus (TTV) is highly prevalent in the European wild boar (Sus scrofa). Vet. Microbiol. 2006, 118, 223–229. [Google Scholar] [CrossRef]
- Kekarainen, T.; Segalés, J. Torque Teno Sus Virus in Pigs: An Emerging Pathogen? Transbound. Emerg. Dis. 2012, 59, 103–108. [Google Scholar] [CrossRef]
- Gallei, A.; Pesch, S.; Esking, W.S.; Keller, C.; Ohlinger, V.F. Porcine Torque teno virus: Determination of viral genomic loads by genogroup-specific multiplex rt-PCR, detection of frequent multiple infections with genogroups 1 or 2, and establishment of viral full-length sequences. Vet. Microbiol. 2010, 143, 202–212. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Dai, W.; Dai, D. Synonymous codon usage in TTSuV2, analysis and comparison with TTSuV1. PLoS ONE 2013, 8, e81469. [Google Scholar] [CrossRef]
- Zhang, Z.; Dai, W.; Wang, Y.; Lu, C.; Fan, H. Analysis of synonymous codon usage patterns in torque teno sus virus 1 (TTSuV1). Arch. Virol. 2013, 158, 145–154. [Google Scholar] [CrossRef]
- Butt, A.M.; Nasrullah, I.; Qamar, R.; Tong, Y. Evolution of codon usage in Zika virus genomes is host and vector specific. Emerg. Microbes Infect. 2016, 5, e107. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Baranda, S.; Greenbaum, B.; Manches, O.; Handler, J.; Rabadán, R.; Levine, A.; Bhardwaj, N. Oligonucleotide Motifs That Disappear during the Evolution of Influenza Virus in Humans Increase Alpha Interferon Secretion by Plasmacytoid Dendritic Cells. J. Virol. 2011, 85, 3893–3904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, G.; Ji, S.; Zhai, X.; Zhang, Y.; Liu, J.; Zhu, M.; Zhou, J.; Su, S. Evolutionary and genetic analysis of the VP2 gene of canine parvovirus. BMC Genom. 2017, 18, 534. [Google Scholar] [CrossRef] [PubMed]
- Nasrullah, I.; Butt, A.M.; Tahir, S.; Idrees, M.; Tong, Y. Genomic analysis of codon usage shows influence of mutation pressure, natural selection, and host features on Marburg virus evolution. BMC Evol. Biol. 2015, 15, 174. [Google Scholar] [CrossRef] [PubMed]
- Schat, K. TT Viruses: The Still Elusive Human Pathogens; Springer: Berlin/Heidelberg, Germany, 2009; pp. 151–183. [Google Scholar]
- Dave, U.; Srivathsan, A.; Kumar, S. Analysis of codon usage pattern in the viral proteins of chicken anaemia virus and its possible biological relevance. Infect. Genet. Evol. 2019, 69, 93–106. [Google Scholar] [CrossRef]
- Chen, Y.; Sun, J.; Tong, X.; Xu, J.; Deng, H.; Jiang, Z.; Jiang, C.; Duan, J.; Li, J.; Zhou, P.; et al. First analysis of synonymous codon usage in porcine circovirus. Arch. Virol. 2014, 159, 2145–2151. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Wang, H.; Wang, S.; Xing, G.; Zhang, C.; Zhang, W.; Liu, J.; Zhang, J.; Su, S.; Zhou, J. Insights into the Genetic and Host Adaptability of Emerging Porcine Circovirus 3. Virulence 2018, 9, 1301–1313. [Google Scholar] [CrossRef]
- Hu, J.-S.; Wang, Q.-Q.; Zhang, J.; Chen, H.-T.; Xu, Z.-W.; Zhu, L.; Ding, Y.-Z.; Ma, L.-N.; Xu, K.; Gu, Y.-X.; et al. The characteristic of codon usage pattern and its evolution of hepatitis C virus. Infect. Genet. Evol. 2011, 11, 2098–2102. [Google Scholar] [CrossRef]
- Mueller, S.; Papamichail, D.; Coleman, J.R.; Skiena, S.; Wimmer, E. Reduction of the Rate of Poliovirus Protein Synthesis through Large-Scale Codon Deoptimization Causes Attenuation of Viral Virulence by Lowering Specific Infectivity. J. Virol. 2006, 80, 9687–9696. [Google Scholar] [CrossRef] [Green Version]
- Carbone, A.; Zinovyev, A.; Kepes, F. Codon adaptation index as a measure of dominating codon bias. Bioinformatics 2003, 19, 2005–2015. [Google Scholar] [CrossRef] [Green Version]
- Kimura, M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 1980, 16, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015, 1, vev003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lole, K.S.; Bollinger, R.C.; Paranjape, R.S.; Gadkari, D.; Kulkarni, S.S.; Novak, N.G.; Ingersoll, R.; Sheppard, H.W.; Ray, S.C. Full-Length Human Immunodeficiency Virus Type 1 Genomes from Subtype C-Infected Seroconverters in India, with Evidence of Intersubtype Recombination. J. Virol. 1999, 73, 152. [Google Scholar] [PubMed]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Keane, T.M.; Creevey, C.J.; Pentony, M.M.; Naughton, T.J.; Mclnerney, J.O. Assessment of methods for amino acid matrix selection and their use on empirical data shows that ad hoc assumptions for choice of matrix are not justified. BMC Evol. Biol. 2006, 6, 29. [Google Scholar] [CrossRef] [PubMed]
- Sharp, P.M.; Li, W.H. Codon usage in regulatory genes in Escherichia coli does not reflect selection for ‘rare’ codons. Nucleic Acids Res. 1986, 14, 7737–7749. [Google Scholar] [CrossRef] [PubMed]
- Wong, E.H.M.; Smith, D.K.; Rabadan, R.; Peiris, M.; Poon, L.L.M. Codon usage bias and the evolution of influenza A viruses. Codon Usage Biases of Influenza Virus. BMC Evol. Biol. 2010, 10, 253. [Google Scholar] [CrossRef] [PubMed]
- Wright, F. The Effective Number of Codons Used in A Gene. Gene 1990, 87, 23–29. [Google Scholar] [CrossRef]
- Xu, C.; Dong, J.; Tong, C.; Gong, X.; Wen, Q.; Zhuge, Q. Analysis of synonymous codon usage patterns in seven different citrus species. Evol. Bioinform. Online 2013, 9, 215–228. [Google Scholar] [CrossRef]
- Zhao, Y.; Zheng, H.; Xu, A.; Yan, D.; Jiang, Z.; Qi, Q.; Sun, J. Analysis of codon usage bias of envelope glycoprotein genes in nuclear polyhedrosis virus (NPV) and its relation to evolution. BMC Genom. 2016, 17, 677. [Google Scholar] [CrossRef]
- Sueoka, N. Directional mutation pressure and neutral molecular evolution. Proc. Natl. Acad. Sci. USA 1988, 85, 2653. [Google Scholar] [CrossRef] [PubMed]
- Sharp, P.M.; Li, W.H. The codon Adaptation Index--a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res. 1987, 15, 1281–1295. [Google Scholar] [CrossRef] [PubMed]
- Puigbò, P.; Aragonès, L.; Garcia-Vallvé, S. RCDI/eRCDI: A web-server to estimate codon usage deoptimization. BMC Res. Notes 2010, 3, 87. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.-H.; Zhang, J.; Sun, D.-J.; Ma, Q.; Chen, H.-T.; Ma, L.-N.; Ding, Y.-Z.; Liu, Y.-S. The distribution of synonymous codon choice in the translation initiation region of dengue virus. PLoS ONE 2013, 8, e77239. [Google Scholar] [CrossRef] [PubMed]
- Xia, X. DAMBE7: New and Improved Tools for Data Analysis in Molecular Biology and Evolution. Mol. Biol. Evol. 2018, 35, 1550–1552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenbaum, B.D.; Cocco, S.; Levine, A.J.; Monasson, R. Quantitative theory of entropic forces acting on constrained nucleotide sequences applied to viruses. Proc. Natl. Acad. Sci. USA 2014, 111, 5054–5059. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| AA | Codon | TTSuV1 | TTSuV2 | Sus scrofa | Sus scrofa domestica |
|---|---|---|---|---|---|
| F | UUU | 1.1 | 1.2 | 0.79 | 0.59 |
| UUC | 0.9 | 0.8 | 1.21 | 1.41 | |
| L | UUA | 0.93 | 1.73 | 0.32 | 0.3 |
| UUG | 0.32 | 0.21 | 0.67 | 0.49 | |
| CUU | 0.79 | 0.57 | 1.35 | 1.41 | |
| CUC | 0.99 | 1.51 | 1.35 | 1.41 | |
| CUA | 1.74 | 1.68 | 0.33 | 0.28 | |
| CUG | 1.22 | 0.29 | 2.68 | 2.92 | |
| I | AUU | 0.84 | 0.54 | 0.91 | 1.22 |
| AUC | 0.6 | 0.49 | 1.67 | 1.45 | |
| AUA | 1.56 | 1.97 | 0.42 | 0.34 | |
| V | GUU | 0.55 | 0.65 | 0.57 | 0.34 |
| GUC | 0.6 | 0.6 | 1.07 | 1.45 | |
| GUA | 1.81 | 2.24 | 0.34 | 0.17 | |
| GUG | 1.04 | 0.51 | 2.03 | 2.04 | |
| S | UCU | 0.96 | 0.71 | 0.99 | 1.1 |
| UCC | 0.83 | 0.6 | 1.5 | 1 | |
| UCA | 1.28 | 1.51 | 0.73 | 2.45 | |
| UCG | 0.59 | 0.15 | 0.39 | 0.55 | |
| AGU | 1.61 | 1.02 | 0.77 | 0.25 | |
| AGC | 0.73 | 2 | 1.62 | 0.66 | |
| P | CCU | 0.6 | 1.57 | 1.05 | 0.84 |
| CCC | 0.86 | 0.42 | 1.46 | 1.59 | |
| CCA | 1.9 | 0.64 | 0.94 | 0.91 | |
| CCG | 0.65 | 0.71 | 0.56 | 0.66 | |
| T | ACU | 0.69 | 2.33 | 0.83 | 0.77 |
| ACC | 0.65 | 0.31 | 1.68 | 1.2 | |
| ACA | 2.08 | 1.14 | 0.92 | 1.62 | |
| ACG | 0.58 | 0.48 | 0.57 | 0.41 | |
| A | GCU | 0.76 | 2.16 | 0.96 | 0.7 |
| GCC | 1.07 | 0.22 | 1.8 | 1.03 | |
| GCA | 1.6 | 1.03 | 0.74 | 1.92 | |
| GCG | 0.57 | 0.98 | 0.5 | 0.36 | |
| Y | UAU | 0.57 | 1.14 | 0.73 | 0.69 |
| UAC | 1.43 | 0.86 | 1.27 | 1.31 | |
| H | CAU | 0.56 | 0.72 | 0.7 | 0.6 |
| CAC | 1.44 | 1.28 | 1.3 | 1.4 | |
| Q | CAA | 0.81 | 1.5 | 0.44 | 0.29 |
| CAG | 1.19 | 0.5 | 1.56 | 1.71 | |
| N | AAU | 0.77 | 0.93 | 0.79 | 0.82 |
| AAC | 1.23 | 1.07 | 1.21 | 1.18 | |
| K | AAA | 1.38 | 1.6 | 0.76 | 0.91 |
| AAG | 0.62 | 0.4 | 1.24 | 1.09 | |
| D | GAU | 0.6 | 0.75 | 0.8 | 0.81 |
| GAC | 1.4 | 1.25 | 1.2 | 1.19 | |
| E | GAA | 1.22 | 1.39 | 0.72 | 1.24 |
| GAG | 0.78 | 0.61 | 1.28 | 0.76 | |
| C | UGU | 0.88 | 1.3 | 0.79 | 0.81 |
| UGC | 1.12 | 0.7 | 1.21 | 1.19 | |
| R | CGU | 0.48 | 0.15 | 0.44 | 0.4 |
| CGC | 0.88 | 0.95 | 1.31 | 1.09 | |
| CGA | 0.82 | 0.64 | 0.6 | 0.27 | |
| CGG | 0.3 | 0.52 | 1.29 | 0.77 | |
| AGA | 2.8 | 2.95 | 1.12 | 2.75 | |
| AGG | 0.71 | 0.79 | 1.23 | 0.71 | |
| G | GGU | 0.62 | 0.32 | 0.57 | 0.31 |
| GGC | 0.55 | 0.5 | 1.46 | 0.84 | |
| GGA | 2.21 | 2.74 | 0.91 | 1.78 | |
| GGG | 0.61 | 0.44 | 1.05 | 1.07 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, G.; Zhang, W.; Wang, R.; Xing, G.; Wang, S.; Ji, X.; Wang, N.; Su, S.; Zhou, J. Genetic Analysis and Evolutionary Changes of the Torque teno sus Virus. Int. J. Mol. Sci. 2019, 20, 2881. https://doi.org/10.3390/ijms20122881
Li G, Zhang W, Wang R, Xing G, Wang S, Ji X, Wang N, Su S, Zhou J. Genetic Analysis and Evolutionary Changes of the Torque teno sus Virus. International Journal of Molecular Sciences. 2019; 20(12):2881. https://doi.org/10.3390/ijms20122881
Chicago/Turabian StyleLi, Gairu, Wenyan Zhang, Ruyi Wang, Gang Xing, Shilei Wang, Xiang Ji, Ningning Wang, Shuo Su, and Jiyong Zhou. 2019. "Genetic Analysis and Evolutionary Changes of the Torque teno sus Virus" International Journal of Molecular Sciences 20, no. 12: 2881. https://doi.org/10.3390/ijms20122881