Chiari Malformation Type 1 in EPAS1-Associated Syndrome

 , , ,
, , ,
Abstract
:1. Introduction
2. Results
2.1. Summary of Cases
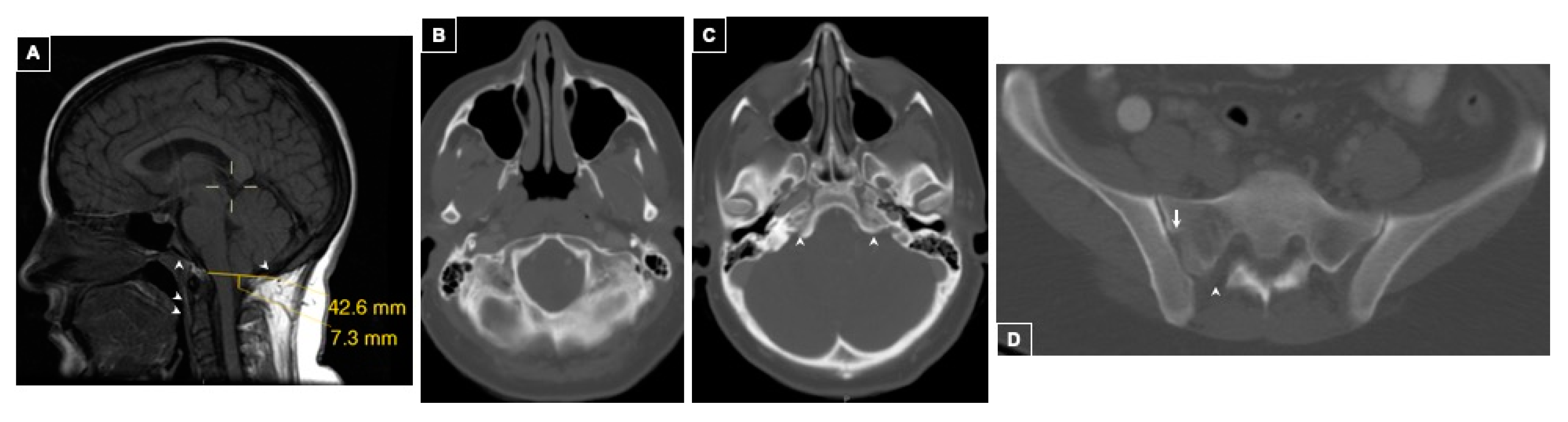
2.1.1. Case 1
History of Present Illness
Imaging Findings
Family History
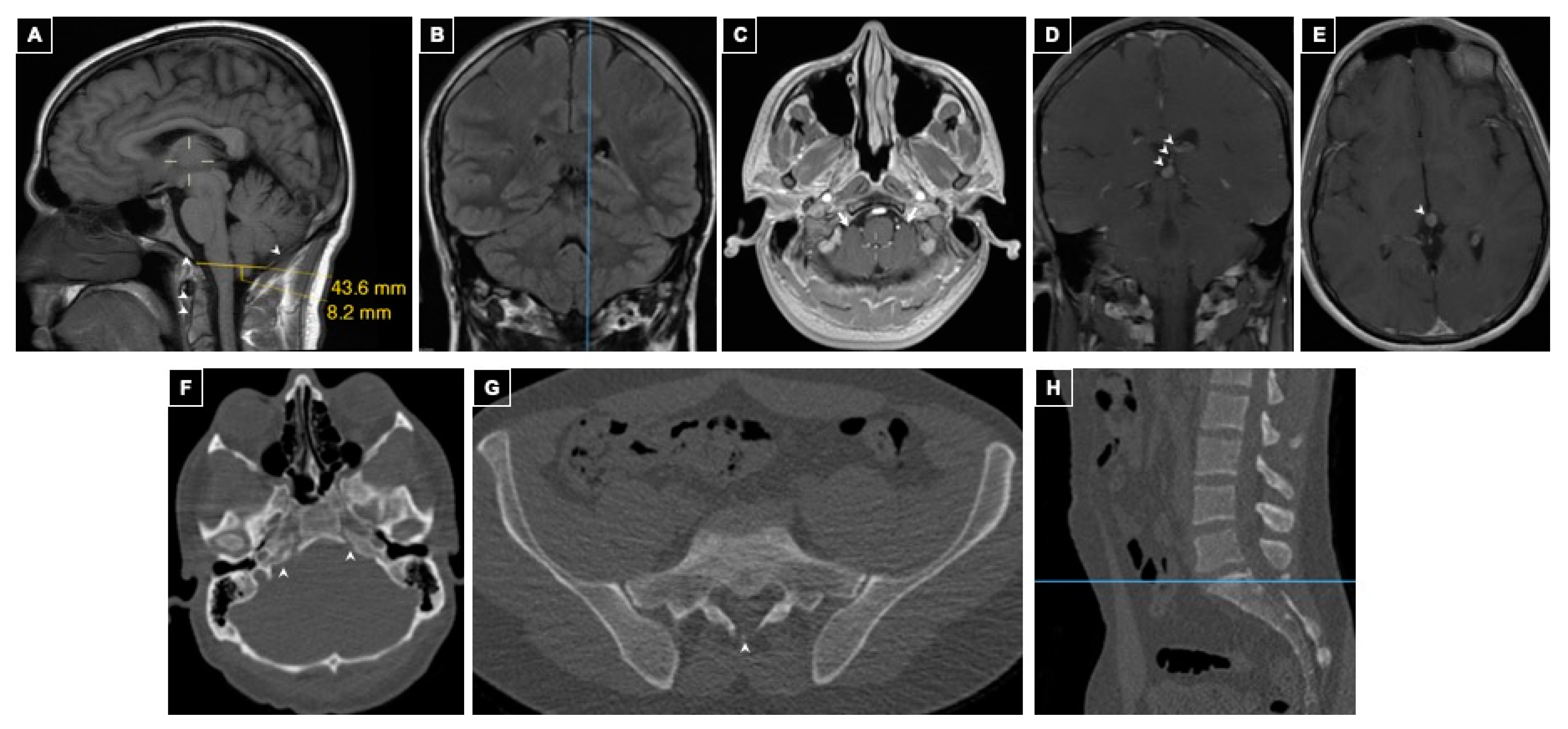
2.1.2. Case 2
History of Present Illness
Imaging Findings
3. Discussion
4. Materials and Methods
4.1. Study Oversight
4.2. Patient Selection and Evaluation
4.3. Patient EPAS1 Mutation Analysis
4.4. Measurements
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| HIF-2α | Hypoxia-inducible factor 2α |
| 18FDG PET/CT | 18F-fluorodeoxyglucose positron emission tomography/computed tomography |
| 123I-MIBG | 123I-metaiodobenzylguanidine |
| EPO | Erythropoietin |
| EPOR | Erythropoietin receptor |
| HIF-1α | Hypoxia-inducible factor 1α |
| JAK2 | Janus kinase 2 |
| PHD | Prolyl hydroxylase |
| PGL/PHEO | Paraganglioma/pheochromocytoma |
| SDHB/C/D | Succinate dehydrogenase B/C/D |
| VHL | von Hippel-Lindau |
| 18F-FDOPA)-PET/CT | l-3,4-dihdroxy-6-[18F]fluorophenylalanine PET/CT scan |
References
- Bano, S.; Chaudhary, S.; Yadav, S. Congenital Malformation of the Brain, Neuroimaging—Clinical Applications; Bright, P., Ed.; InTech: London, UK, 2012; Available online: http://www.intechopen.com/books/neuroimaging-clinical-applications/congenital-malformations-of-the-brain (accessed on 23 January 2019).
- McLone, D.G.; Knepper, P.A. The cause of Chiari II malformation: A unified theory. Pediatr. Neurosci. 1989, 15, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Shoja, M.M.; Johal, J.; Oakes, W.J.; Tubbs, R.S. Embryology and pathophysiology of the Chiari I and II malformations: A comprehensive review. Clin. Anat. 2018, 31, 202–215. [Google Scholar] [CrossRef] [PubMed]
- Kniffin, C.L.; McKusick, V.A. OMIM-Online Mendelian Inheritance in Man: Chiari Malformation Type, I. Published 5/12/1992. Updated 10/07/2016. Available online: https://www.omim.org/entry/118420#title (accessed on 13 January 2019).
- Urbizu, A.; Khan, T.N.; Ashley-Koch, A.E. Genetic dissection of Chiari malformation type I using endophenotypes and stratification. J. Rare Dis. Res. Treat. 2016, 2, 35–42. [Google Scholar]
- Dunwoodie, S.L. The Role of Hypoxia in Development of the Mammalian Embryo. Dev. Cell 2009, 17, 755–773. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.Y.; Park, K.H.; Yu, H.G.; Kook, E.; Song, W.H.; Gyuseok, L.; Koh, J.T.; Shin, H.I.; Choi, J.Y.; Huh, Y.H.; et al. Controlling hypoxia-inducible factor-2α is critical for maintaining bone homeostasis in mice. Bone Res. 2019, 7, 14. [Google Scholar] [CrossRef]
- Bess, S.; Varma, V. Embryology and Anatomy: Spine/Spinal Cord. In The Growing Spine: Management of Spinal Disorders in Young Children; Akbarnia, B.A., Yazici, M., Thompson, G.H., Eds.; Springer: Berlin/Heidelberg, Germany, 2010; pp. 3–12. [Google Scholar]
- Zhuang, Z.; Yang, C.; Lorenzo, F.; Merino, M.; Fojo, T.; Kebebew, E.; Popovic, V.; Stratakis, C.A.; Prchal, J.T.; Pacak, K. Somatic HIF2A Gain-of-Function Mutations in Paraganglioma with Polycythemia. N. Engl. J. Med. 2012, 367, 922–930. [Google Scholar] [CrossRef]
- Bogdanov, E.I.; Heiss, J.D.; Mendelevich, E.G.; Mikhaylov, I.M.; Haass, A. Clinical and neuroimaging features of “idiopathic” syringomyelia. Neurology 2004, 62, 791–794. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention. Data and Statistics on Spina Bifida|CDC. Washington, DC, USA: Center for Disease Control, 13 September 2018. Available online: https://www.cdc.gov/ncbddd/spinabifida/data.html (accessed on 5 February 2019).
- Marin-Padilla, M. Notochordal-basichondrocranium relationships: Abnormalities in experimental axial skeletal (dysraphic) disorders. J. Embryol. Exp. Morphol. 1979, 53, 15–38. [Google Scholar] [PubMed]
- Marina-Padilla, M.; Marina-Padilla, T.M. Morphogenesis of experimentally induced Arnold-Chiari malformation. J. Neurol Sci. 1981, 50, 29–55. [Google Scholar] [CrossRef]
- Nishikawa, M.; Sakamoto, H.; Hakuba, A.; Nakanishi, N.; Inoue, Y. Pathogenesis of Chiari malformation: A morphometric study of the posterior cranial fossa. J. Neurosurg. 1997, 86, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Taylor, D.G.; Mastorakos, P.; Jane, J.A., Jr.; Oldfield, E. Two distinct populations of Chiari I malformation based on presence or absence of posterior fossa crowdedness on magnetic resonance imaging. J. Neurosurg. 2017, 126, 1934–1940. [Google Scholar] [CrossRef] [PubMed]
- Ogul, H.; Kantarci, M. Unusual Association: Cerebral Arteriovenous Malformation and Chiari Type I Malformation. J. Craniofac. Surg. 2017, 28, e376–e377. [Google Scholar] [CrossRef] [PubMed]
- Kao, S.C.; Waziri, M.H.; Smith, W.L.; Sato, Y.; Yuh, W.T.; Franken, E.A., Jr. MR imaging of the craniovertebral junction, cranium, and brain in children with achondroplasia. Am. J. Roentgenol. 1989, 153, 565–569. [Google Scholar] [CrossRef] [PubMed]
- Pinter, N.K.; McVige, J.; Mechtler, L. Basilar invagination, basilar impression, and platybasia: Clinical and imaging aspects. Curr. Pain Headache Rep. 2016, 20, 49. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Patient | ||
|---|---|---|
| Age at Onset of Diagnosed Condition (years) | ||
| Polycythemia (HCT >51.0%) | 7 | 2.3 |
| EPO (mIU/mL) | 165.0 (3.7–31.5) | 101 (2.6–18.5) |
| PGL/PHEO | 36 | 14 |
| Ampullary Somatostatinoma | 36 | − |
| Mutation Analysis—Tumors and Circulating Leukocytes | ||
| EPAS1 Gain-of-Function Mutation | Y532C | D539N |
| EPOR, HIF-1α, JAK2, PHD1/2, SDHB/C/D, VHL | Negative | Negative |
| Clinical Characteristics | ||
| Blood Pressure (mm Hg) | ||
| At Presentation to NIH | 111/69 | 141/85 |
| Heart Rate (bpm) | 71 | 75 |
| Chiari Malformation Features | ||
| Lowest Cerebellar Tonsillar Position (mm) | 7 | 8 |
| Position of Right Cerebellar Tonsil (mm) | 4 | 6 |
| Position of Left Cerebellar Tonsil (mm) | 7 | 8 |
| Shape of Cerebellar Tonsils | Pegged | Round |
| Ventral CSF Space (12 ± 2.3 mm) [8] | 4 | 9 |
| Dorsal CSF Space (19 ± 2.3 mm) [8] | 0 | 2 |
| Pontomedullary Junction to Foramen Magnum (19 ± 3 mm) [10] | 14 | 15 |
| Tectal Beaking | No | No |
| Supraoccipital Bone Length (41 ± 5 mm) [10] | 35 | 46 |
| Clival Length (43.4 ± 4.4 mm) [10] | 41 | 44 |
| Tentorial Angle (27–52°) [10] | 50.40 | 63.07 |
| Klaus Index Height (38.0 ± 5 mm) [10] | 49 | 51 |
| Boogaard Angle (133.8 ± 6.5°) [10] | 130 | 114 |
| McGregor Line (<4.5 mm) | <0 | <0 |
| Posterior Fossa Height (32 ± 3 mm) [10] | 32 | 39 |
| Uncalcified Petroclival Synchondrosis | Yes | Yes |
| Sacral spina bifida occulta | Yes | Yes |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rosenblum, J.S.; Maggio, D.; Pang, Y.; Nazari, M.A.; Gonzales, M.K.; Lechan, R.M.; Smirniotopoulos, J.G.; Zhuang, Z.; Pacak, K.; Heiss, J.D. Chiari Malformation Type 1 in EPAS1-Associated Syndrome. Int. J. Mol. Sci. 2019, 20, 2819. https://doi.org/10.3390/ijms20112819
Rosenblum JS, Maggio D, Pang Y, Nazari MA, Gonzales MK, Lechan RM, Smirniotopoulos JG, Zhuang Z, Pacak K, Heiss JD. Chiari Malformation Type 1 in EPAS1-Associated Syndrome. International Journal of Molecular Sciences. 2019; 20(11):2819. https://doi.org/10.3390/ijms20112819
Chicago/Turabian StyleRosenblum, Jared S., Dominic Maggio, Ying Pang, Matthew A. Nazari, Melissa K. Gonzales, Ronald M. Lechan, James G. Smirniotopoulos, Zhengping Zhuang, Karel Pacak, and John D. Heiss. 2019. "Chiari Malformation Type 1 in EPAS1-Associated Syndrome" International Journal of Molecular Sciences 20, no. 11: 2819. https://doi.org/10.3390/ijms20112819