BANK1 Transethnic Finemapping
Using the high quality filtered genome-wide data from the European and African American genome-wide association scans, we performed a univariate genetic logistic regression association analysis of
BANK1 using an additive model. In Europeans, 1133 imputed markers mapped on
BANK1 were tested for association. We selected a set of 307 markers with a
p value lower than 0.05 (
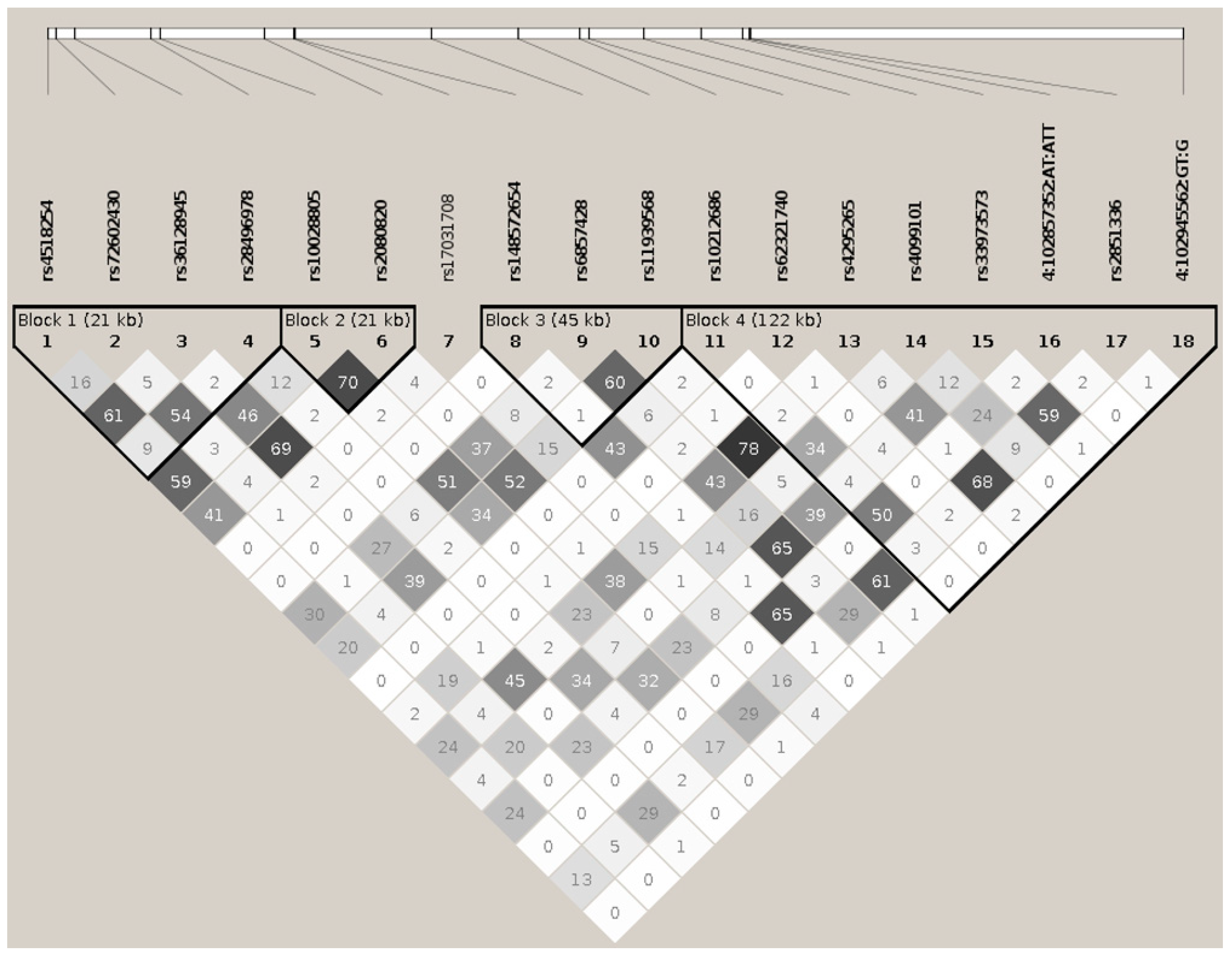
Supplemental Table S1A and Supplemental Figure S1A). Eighteen tagging markers captured all 307 (100%) alleles at
r2 ≥0.8, representing the 18 respective linkage disequilibrium groups organized in four haplotypic blocks. Only one marker remained as an independent signal, rs17031708 (
Figure 1,
Table 1 and
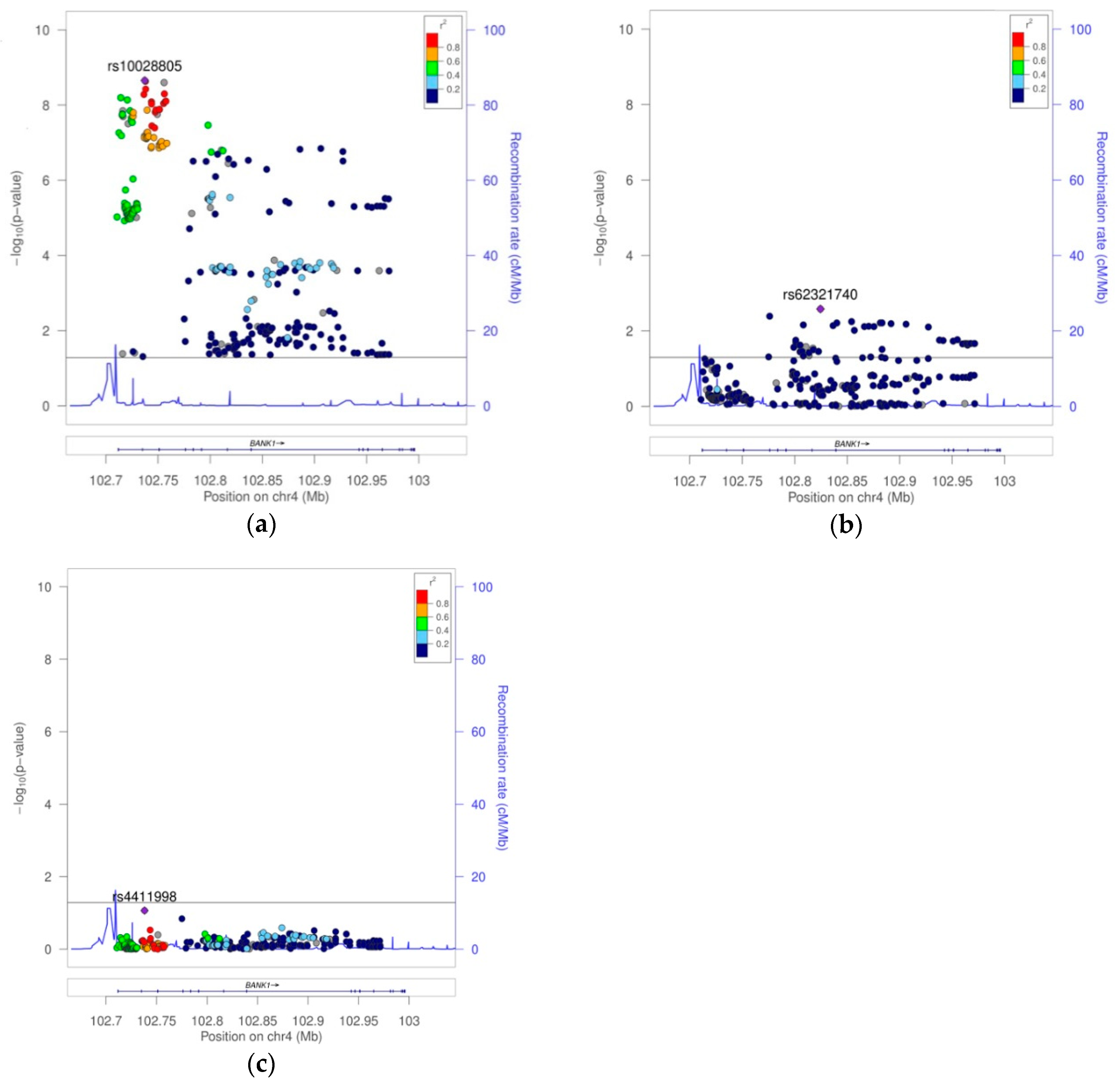
Supplemental Table S1A). The best hit for association was the SNP rs10028805, (haplotypic block two, and linkage disequilibrium group five), with OR = 0.8177, confidence interval (CI) = 95% (0.7656, 0.8735), and a Pcorr = 2.22E−09 (
Figure 2a,
Table 1, and
Supplemental Table S1A). Conditioning on rs10028805, the association disappeared, except that of the five low frequency variants (MAF < 5%), rs148572654, rs10212686, rs62321740, 4:102857352:AT:ATT, and 4:102945562:GT:G (
Table 1 and
Figure 2b), representing linkage groups 8, 11, 12, 16, and 19, respectively. Only by conditioning on rs10028805 and three of these low frequency variants, namely, rs148572654, rs62321740, and 4:102945562:GT:G, did all of the signals of association in
BANK1 disappear (
Table 1,
Supplemental Table S1A, and
Figure 2c). In addition, these four markers constituted the haplotypes significantly associated with the affected phenotype. The best was the allelic combination ‘1111’, corresponding to a risk haplotype with frequencies of 62% in the cases and 56% in the controls, and a value of
p = 7.64E−13 (
Table 2,
Figure 3a).
In African Americans, 1752 imputed SNPs were mapped on
BANK1, of which 260 showed association test
p values lower than 0.05 (
Supplemental Table S1B and Supplemental Figure S1B). Running the tagger tool, a set of 50 markers captured all 260 (100%) alleles at
r2 ≥0.8, representing their 50 respective linkage disequilibrium groups (
Table 3 and
Supplemental Table S1B). These 50 tagging markers were organized in 8 haplotypic blocks and 11 independent signals (
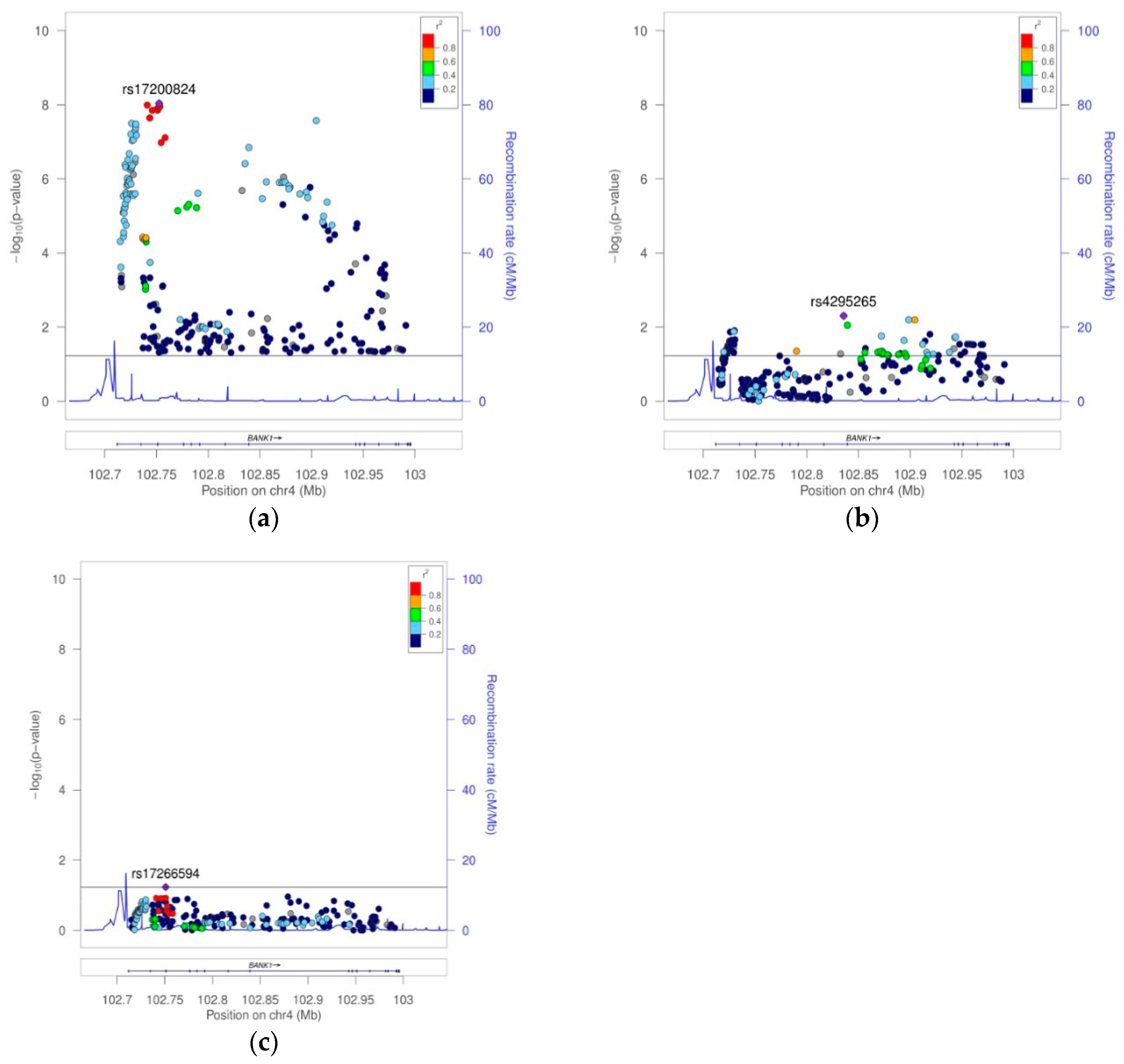
Figure 4). The best hit was the SNP rs17200824 with OR = 0.6787, CI = 95% (0.5947, 0.7747), and a Pcorr = 9.24E−09. This best-hit was in linkage group 13, the same as the branch-point site SNP rs17266594 and rs10516487 (R61H) (
Figure 5a,
Table 3, and
Supplemental Table S1B). After conditioning on rs17200824, the association signals remained in 11 of the tagging markers (
Figure 5b,
Table 3, and
Supplemental Table S1B). Only after the conditioning on rs17200824 on two of these 11 variants, rs4295265 and rs149302668, had all of the signals on the
BANK1 association disappeared (
Table 3,
Supplemental Table S1B and
Figure 5c). In addition, these two markers constituted haplotypes that are significantly associated with the affected phenotype; the best was the allelic combination ‘11’, corresponding to a risk haplotype with frequencies of 90% in the cases and 84.6% in the controls, and a value of Pcorr = 3.17E−08 (
Table 4 and
Figure 3b). It is noted that rs4295265 (Pcorr = 3.89E–07) was located on intron seven, at 2,319 bp upstream to the exonic SNP rs3733197 (A 353 T, in the ankyrin domain) (Pcorr = 1.44E−07), described as being associated to SLE1 and sharing the same haplotypic block eight (pairwise
r2 = 0.75). The SNP rs149302668, located on intron 11, was a low frequency variant (MAF = 0.01348).
In EUR, markers significantly associated with SLE were located to haplotypic blocks one and two, including intron one and intron two–exon two–intron three. In the first haplotypic block, covering intron one, the markers were organized in linkage disequilibrium groups one, two, and three. Group one showed the best signal of association in the block with rs4518254 (Pcorr = 6.36E–09) as a best hit. The second haplotypic block included two linkage disequilibrium groups, five and six, covering intron two–exon two–intron three. Group five contained rs10028805 (intron2) (Pcorr = 2.22E–09) and rs10516486 (L98L, exon2). For group six, rs13106926 (intron2) (Pcorr = 1.36E–08) was the best hit; this group also contained the branch-point site, SNP rs17266594 (intron1), and rs10516487 (R61H, exon two) [
1].
After conditioning on the best-hit, rs10028805, all of the signals of association for the common variation (MAF > 5%) in
BANK1 disappeared; hence, rs10028805 would be the statistically most plausible causal SNP of the
BANK1 association to SLE in the EUR sample, as previously suggested. It has been remarked that, as a result of the LD status among the markers in ‘block two–group five’ (intraBK = 0.96) (
Supplemental Figure S2B), we would expect a similar effect with all of them. We observed that from the conditioning on each of the three SNPs, rs10028805, rs13136297, and rs4411998, the same results were obtained, (=0.99). However, traces of association with a low-frequency variation remained (
Table 1,
Supplemental Table S1A, and
Figure 2b). Only after the conditioning on rs10028805 and three low frequency variants, rs148572654, rs62321740, and 4:102945562:GT:G, did the signals of association in the
BANK1 completely disappear (
Table 1,
Supplemental Table S1A, and
Figure 2c).
In AA, the markers that were significantly associated (10
−8) were located in the same regions as in EUR, intron one, and intron two–exon two–intron three (
Table 2 and
Supplemental Table S1B). Intron one was split in two haplotypic blocks, one and two. In the second block, markers were distributed in four linkage disequilibrium groups (one, two, three, and four) and the best hits were in the third group (equivalent to EUR ‘block one–group three′), SNP rs4699262 (Pcorr = 3.19E–08). Region intron two–exon two–intron three was divided into three haplotypic blocks (three, four, and five) containing 12 linkage disequilibrium groups (
Supplemental Table S1B). The best hits were in the fourth haplotypic block, group 13 (partially equivalent to EUR group six), with the best-hit rs17200824 (Pcorr = 9.24E–09) and the branch-point site SNP rs17266594 (intron one) and rs10516487 (R61H, exon two).
Note that AA group eight, partially equivalent to EUR group six, containing the best-hits, did not contain associated markers at the genome-wide association study (GWAS) level in AA (SNP rs10028805 had a
p value = 4.72E–04) (
Supplemental Table S1B), and was independent of group 13 (AAgroups_8_13 = 0.18 and EURgroups_5_6 = 0.70).
No marker, including the best hit in the AA population, rs17200824, was able to completely eliminate the associations of all of the others, (
Table 3 and
Supplemental Table S1B). Only by conditioning on rs17200824 and two variants, rs4295265 and rs149302668, did all of the signals of the association in
BANK1 disappear (
Table 3,
Supplemental Table S1B, and
Figure 5c). Note that rs4295265 (Pcorr = 3.89E–07) was located on intron seven at 2319 bp upstream to the exonic SNP rs3733197 (Pcorr = 1.44E–07) (exon seven, A353T ankyrin domain), described as SLE associated one. Thus, the elimination of the genetic association in AA required conditioning on the markers located in at least two separate blocks, suggesting two independent but closely related signals, as well as suggestive signals in the intron [
7].
We then performed a trans-racial ‘EUR–AA’ meta-analysis. We selected 165 markers that showed association
p values less than 0.05 in both of the EUR and AA samples (
Supplemental Table S2). The set of meta-analysis best signals for association were located on the region comprising intron two, exon two, and intron three. In EUR, it coincided with haplotypic block two containing linkage disequilibrium groups five and six, while in AA, the same region was divided into three haplotypic blocks containing seven linkage disequilibrium groups (5, 6, 7, 8, 9 10, and 13). Note that the markers in the EUR linkage disequilibrium groups five and six, in the AA sample, were divided into groups of 5, 8, 9, and 10, and 6, 7, and 13 (
Table 5,
Supplemental Table S2, and Supplemental Figure S3B). The meta-analysis of the SNP rs10028805 in groups EUR 5 and AA 8 showed OR = 0.8167, CI = 95% (0.7712, 0.8648), and P
val = 4.33E–12. Interestingly, rs10028805 had a P
val = 4.71 E–04 in the AA sample, clearly under the accepted GWAS threshold. It should be noted that the linkage disequilibrium group AA 13 (and EUR 6) contained the rs71597109, best hit of the meta-analysis, OR = 0.7898, CI = 95% (0.7423, 0.8404), and P
val = 6.86E–14. In addition, SNP rs71597109 shared, with group AA 13, rs17200824 and the markers described as the best causal hits of the
BANK1 association to with SLE, such as the branch-point site SNP rs17266594 on intron two, and the R61H rs10516487 on exon two. We then performed an eQTL analysis using various sources of data available, [
8,
9,
10,
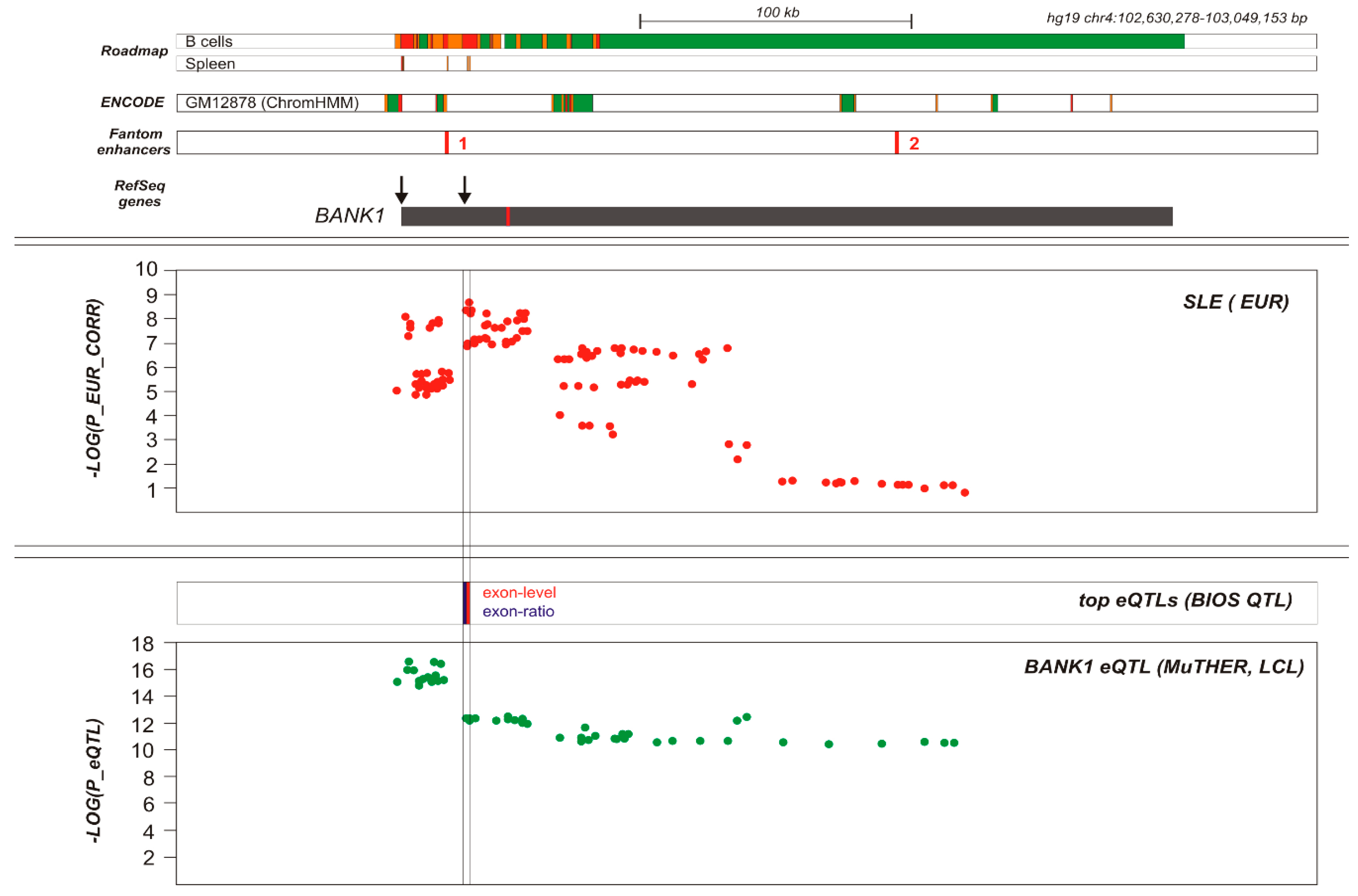
11], as described. The analysis showed that several associated and non-associated SNPs had eQTL effects. In fact, all of the strongly associated SNPs were eQTLs covering a region from the promoter of
BANK1 to UTR-3′/downstream region. However, the strongest eQTLs, according to the BIOS browser10, were localized to intron two, close to exon two, exon two, and intron three, correlating with the most associated SLE signals at the GWAS significance level. These eQTLs were rs13136297 (Cis-eQTL Exon-ratio
p = 5.29E–39) and rs10028805 (Cis-eQTL exon-level
p = 2.74E–52), on the European haploblock two, linkage group five, and the AA haploblock three, linkage group eight. Importantly, risk was associated with an increased expression of
BANK1 (
Supplemental Table S3 and
Figure 6), and importantly, the effects were directly observed in the RNA-Seq data at the exon level of the expression. Using the BIOS data, we also identified a cis-meQTL, specifically rs6833764 containing CpG cg01116491 (
p = 3.64E–35), located in the European haploblock one linkage group one; this SNP is however not associated with SLE in AA (
Supplemental Table S3).
In order to better define the functional effects on the
BANK1 gene expression, we performed an analysis of the ENCODE tracks and investigated the possible causes for the eQTLs. This analysis revealed that the number of annotations in the
BANK1 SNPs increased from the promoter, peaking in the region of association of SLE, centered in intron two and exon two. Using the meta-analysis data, these annotations can be observed in the
Supplemental Table S4 (see the various sheets for transcription factor binding site, histones, and junctions). These annotations showed an enrichment of the splice junctions (
p value = 1.98E–08, Fisher’s exact test), and in various histone marks, particularly histone three lysine 36 27 trimethylation (H3K27me3,
p-value = 1.20E–10) (
Supplemental Table S4), in the lymphoblastoid cell line Gm12878, further supporting a clear regulatory
BANK1 region in this area. The activating histone marks, such as H3K4me1, H3K4me3 in peripheral, and cord blood B cells and CD56+ NK cells, covered the whole associated region. Some spots were also observed in the monocytes and CD34+ hematopoietic stem cells (HSC), but these were clearly less abundant across the region. In addition, the best-hits were enriched for the sites of binding of the transcription factor EZH2 in the Gm12878 cell line (
p value = 2.99E–44). One transcription factor binding site for RUNX3, an activator of the transcription of B cells, was observed in the GM12878 lymphoblastoid cell line, where SNP rs13106926 is located (
Supplemental Table S4) (UCSC genome browser, coordinates chr4: 102,739,791–102,739,791 of GRCh37/Hg19) (
Supplemental Material) 14. This SNP is also found within an active histone mark, histone 3 lysine 27 acetylation mark (H3K27Ac), according to the UCSC browser14. This SNP is an eQTL in lymphoblastoid cell lines, according to the Harvard browser15, but no records were obtained on the BIOS browser. This SNP is located within the European haplotypic block two, group six, and the AA block three, group six, and is strongly associated with SLE (
Supplemental Table S2).
Combining all of the information, our data supports the functional effects limited to the regions of intron two, exon two, and intron three, in both European and African Americans. As expected, the AA population shows a more dispersed haplotypic structure than Europeans, separating the haplotypic block two containing linkage disequilibrium groups five and six, into three haplotypic blocks (three, four, and five) with seven linkage disequilibrium groups (5, 6, 7, 8, 9 10, and 13) (
Supplemental Table S2 and Supplemental Figure S3B).

 ,
, 






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}