Effects of PTEN Loss and Activated KRAS Overexpression on Mechanical Properties of Breast Epithelial Cells

Abstract
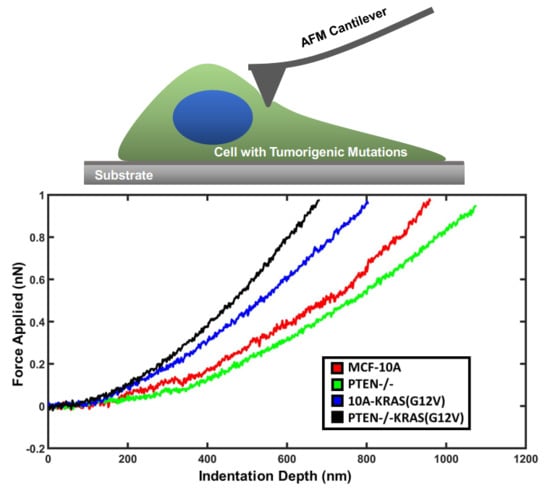
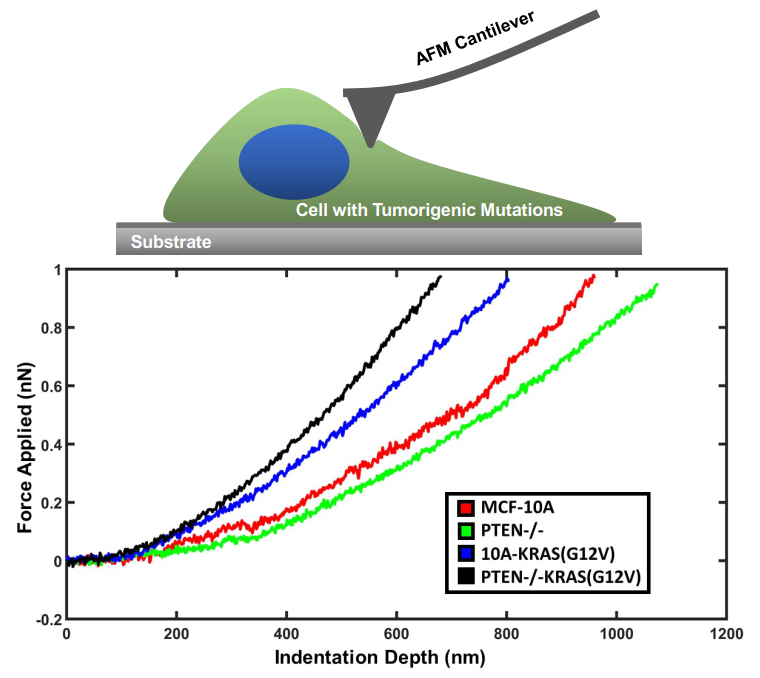
:
{kind=link}
{kind=link}
{kind=link}
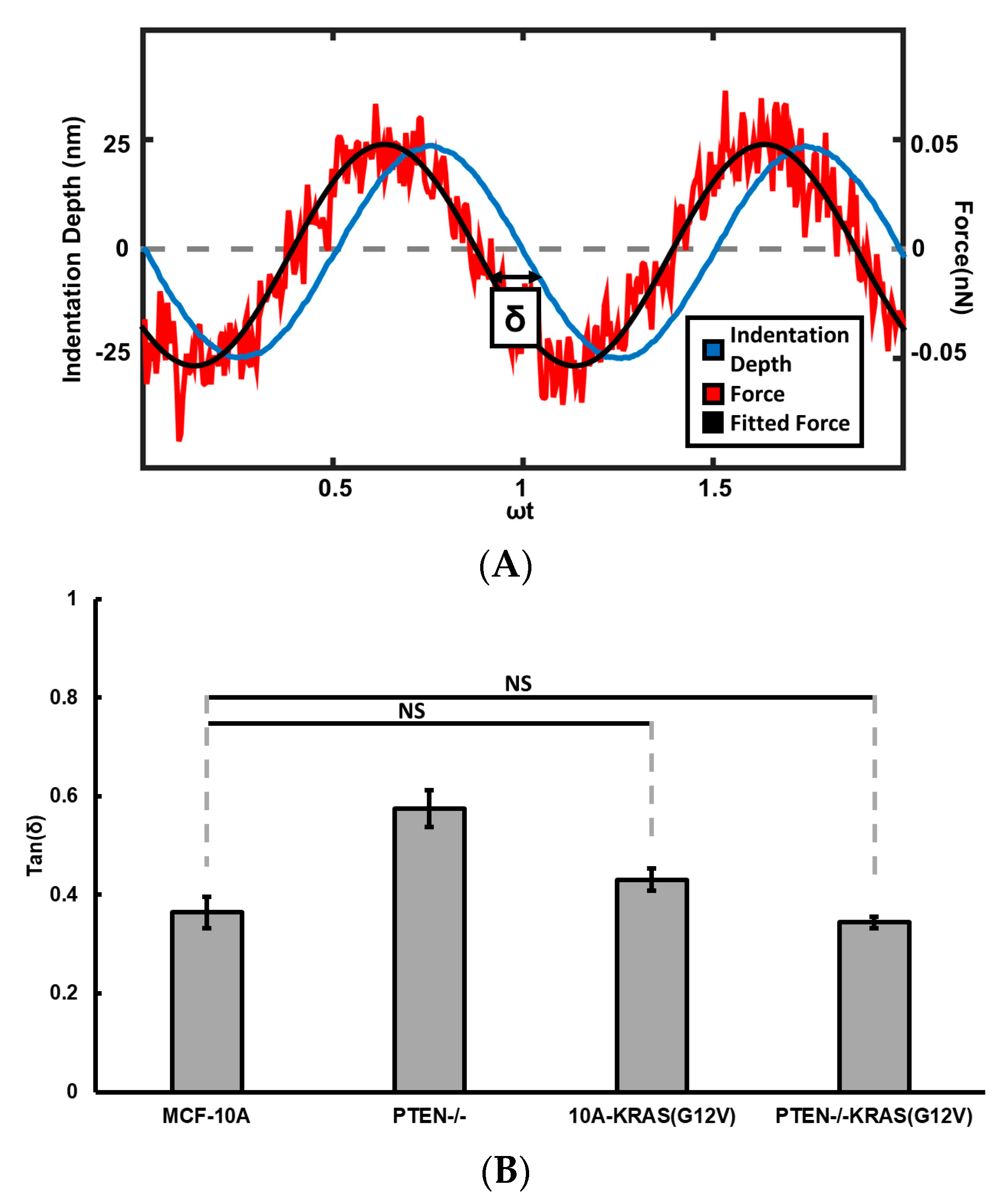{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
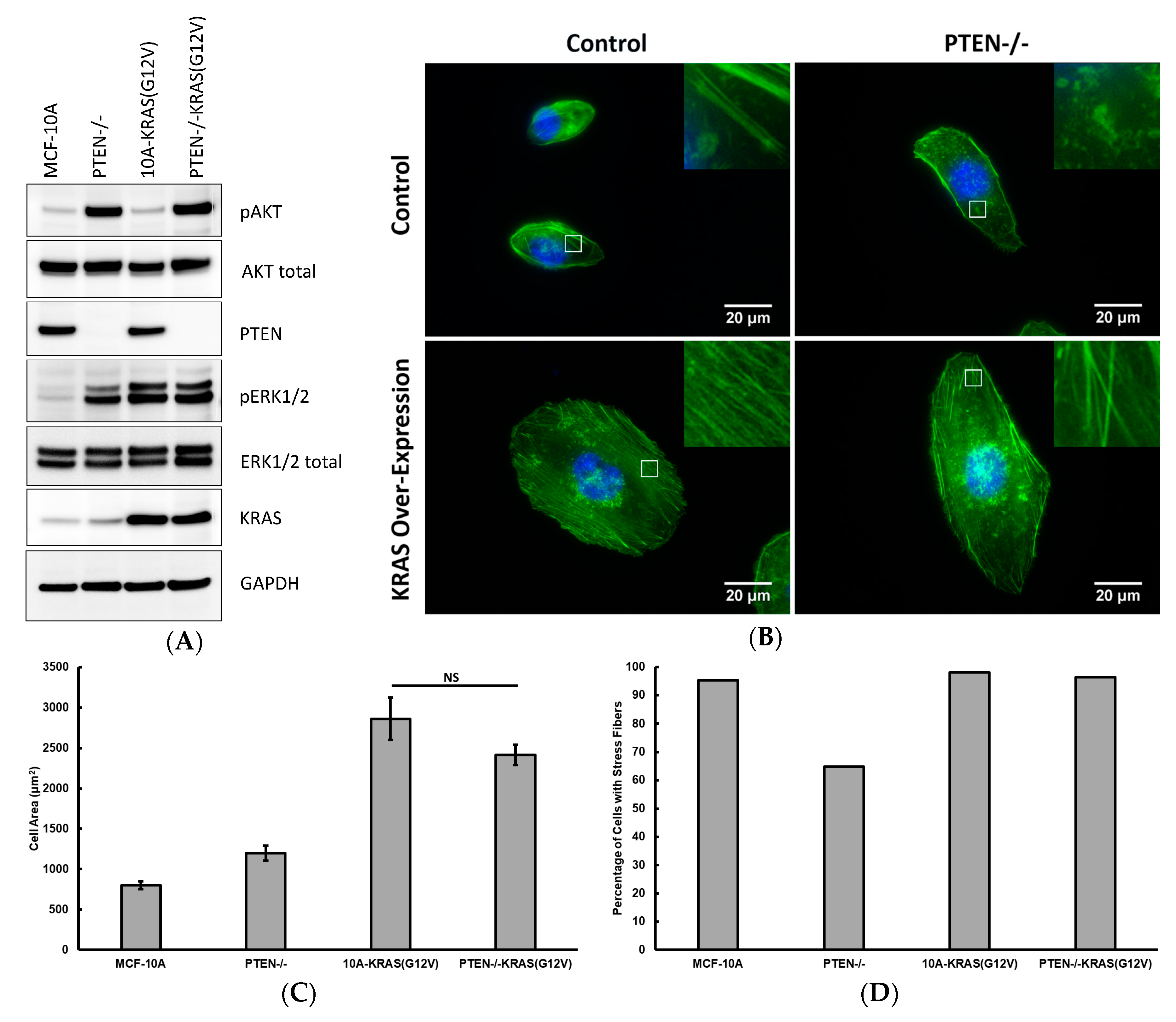
2.1. PTEN Knockout and KRAS Overexpression Modifies Actin Cytoskeleton Structure
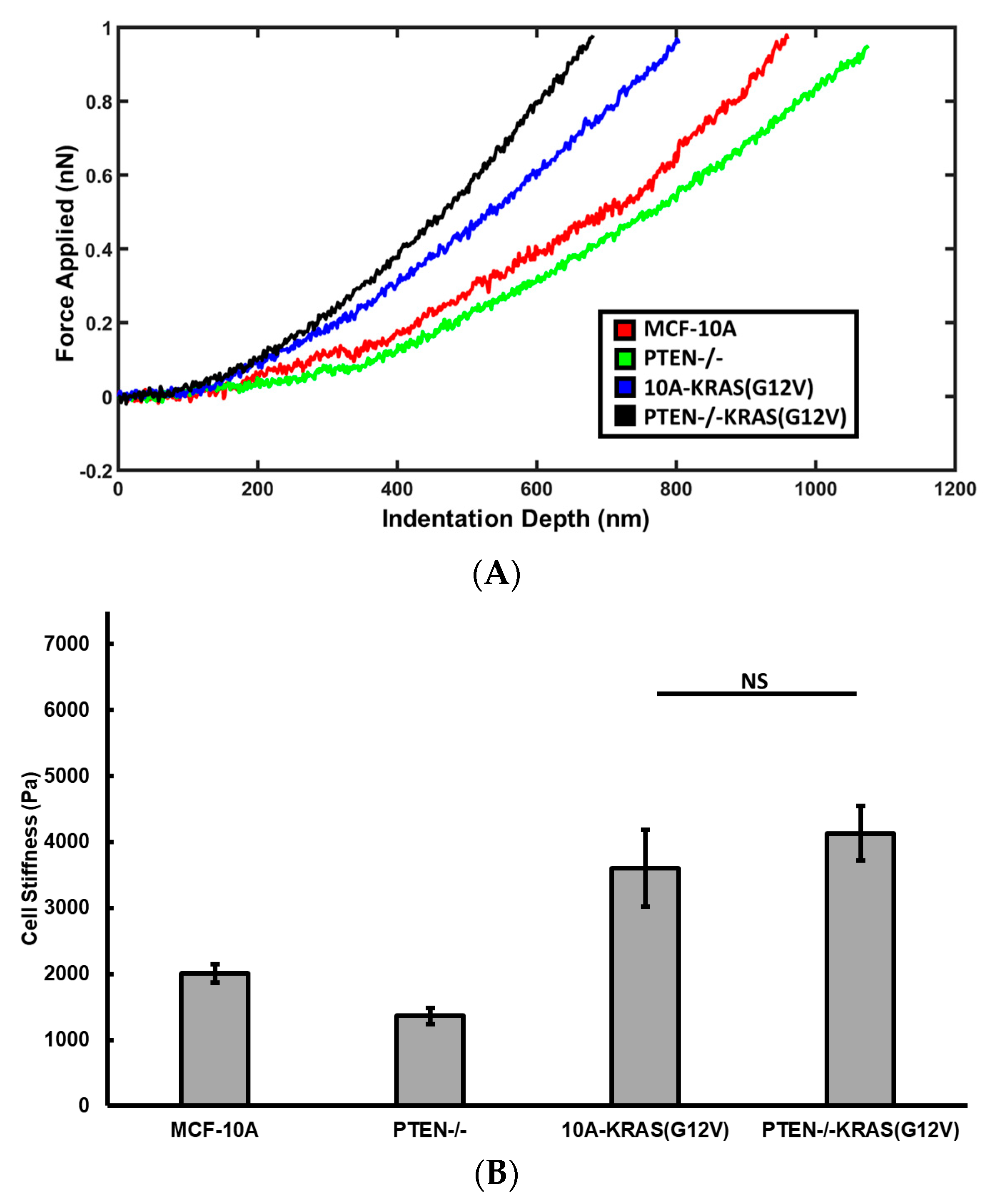
2.2. PTEN Knockout Reduces Cell Stiffness, Activated KRAS Overexpression Increases Cell Stiffness
2.3. PTEN Knockout and Activated KRAS Overexpression Affects Cell Fluidity
2.4. Effects of PTEN Knockout and Activated KRAS Overexpression Depends on Rigidity of Cell Culture Substrate
2.5. Effects of PI3K and ROCK Inhibition on PTEN Loss and Activated KRAS Overexpression Regulated Cell Stiffness
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Cell Culture
4.2. Collagen-Coated Surface Preparation
4.3. Western Blotting
4.4. Immunofluorescence
4.5. AFM Stiffness and Viscoelasticity Assay
4.6. Viscoelasticity Measurements by AFM
Author Contributions
Acknowledgments
Conflicts of Interest
Abbreviations
| PI3K | Phosphatidylinositol 3-kinase |
| MAPK | Mitogen-activated protein kinases |
| PTEN | Phosphatase and tensin homolog |
| AFM | Atomic force microscope |
| ROCK | Rho-associated kinase |
References
- Thompson, K.N.; Whipple, R.A.; Yoon, J.R.; Lipsky, M.; Charpentier, M.S.; Boggs, A.E.; Chakrabarti, K.R.; Bhandary, L.; Hessler, L.K.; Martin, S.S.; et al. The combinatorial activation of the PI3K and Ras/MAPK pathways is sufficient for aggressive tumor formation, while individual pathway activation supports cell persistence. Oncotarget 2015, 6, 35231–35246. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef]
- Kinkade, C.W.; Castillo-Martin, M.; Puzio-Kuter, A.; Yan, J.; Foster, T.H.; Gao, H.; Sun, Y.; Ouyang, X.; Gerald, W.L.; Cordon-Cardo, C.; et al. Targeting AKT/mTOR and ERK MAPK signaling inhibits hormone-refractory prostate cancer in a preclinical mouse model. J. Clin. Investig. 2008, 118, 3051–3064. [Google Scholar] [CrossRef] [PubMed]
- Downward, J. Targeting RAS and PI3K in lung cancer. Nat. Med. 2008, 14, 1315–1316. [Google Scholar] [CrossRef] [PubMed]
- De Roock, W.; Claes, B.; Bernasconi, D.; De Schutter, J.; Biesmans, B.; Fountzilas, G.; Kalogeras, K.T.; Kotoula, V.; Papamichael, D.; Laurent-Puig, P.; et al. Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: A retrospective consortium analysis. Lancet Oncol. 2010, 11, 753–762. [Google Scholar] [CrossRef]
- Ihle, N.T.; Lemos, R., Jr.; Wipf, P.; Yacoub, A.; Mitchell, C.; Siwak, D.; Mills, G.B.; Dent, P.; Kirkpatrick, D.L.; Powis, G. Mutations in the phosphatidylinositol-3-kinase pathway predict for antitumor activity of the inhibitor PX-866 whereas oncogenic Ras is a dominant predictor for resistance. Cancer Res. 2009, 69, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Steelman, L.S.; Chappell, W.H.; Abrams, S.L.; Kempf, R.C.; Long, J.; Laidler, P.; Mijatovic, S.; Maksimovic-Ivanic, D.; Stivala, F.; Mazzarino, M.C.; et al. Roles of the Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR pathways in controlling growth and sensitivity to therapy-implications for cancer and aging. Aging 2011, 3, 192–222. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, T.; Tolcher, A.W.; Papadopoulos, K.P.; Beeram, M.; Rasco, D.W.; Smith, L.S.; Gunn, S.; Smetzer, L.; Mays, T.A.; Kaiser, B.; et al. The clinical effect of the dual-targeting strategy involving PI3K/AKT/mTOR and RAS/MEK/ERK pathways in patients with advanced cancer. Clin. Cancer Res. 2012, 18, 2316–2325. [Google Scholar] [CrossRef] [PubMed]
- Knowles, M.A.; Hurst, C.D. Molecular biology of bladder cancer: New insights into pathogenesis and clinical diversity. Nat. Rev. Cancer 2015, 15, 25–41. [Google Scholar] [CrossRef] [PubMed]
- Aguirre-Ghiso, J.A. The problem of cancer dormancy: Understanding the basic mechanisms and identifying therapeutic opportunities. Cell Cycle 2006, 5, 1740–1743. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, K.; Iwata, M. Stiffness of cancer cells measured with an AFM indentation method. J. Mech. Behav. Biomed. Mater. 2015, 49, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.W.; Mezencev, R.; Kim, B.; Wang, L.J.; McDonald, J.; Sulchek, T. Cell Stiffness Is a Biomarker of the Metastatic Potential of Ovarian Cancer Cells. PLoS ONE 2012, 7, e46609. [Google Scholar] [CrossRef] [PubMed]
- Hou, H.W.; Li, Q.S.; Lee, G.Y.H.; Kumar, A.P.; Ong, C.N.; Lim, C.T. Deformability study of breast cancer cells using microfluidics. Biomed. Microdevices 2009, 11, 557–564. [Google Scholar] [CrossRef] [PubMed]
- Cross, S.E.; Jin, Y.S.; Rao, J.; Gimzewski, J.K. Nanomechanical analysis of cells from cancer patients. Nat. Nanotechnol. 2007, 2, 780–783. [Google Scholar] [CrossRef] [PubMed]
- Tavares, S.; Vieira, A.F.; Taubenberger, A.V.; Araujo, M.; Martins, N.P.; Bras-Pereira, C.; Polonia, A.; Herbig, M.; Barreto, C.; Otto, O.; et al. Actin stress fiber organization promotes cell stiffening and proliferation of pre-invasive breast cancer cells. Nat. Commun. 2017, 8, 15237. [Google Scholar] [CrossRef] [PubMed]
- Pegoraro, A.F.; Janmey, P.; Weitz, D.A. Mechanical Properties of the Cytoskeleton and Cells. Cold Spring Harb. Perspect. Biol. 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Jordan, M.A.; Wilson, L. Microtubules and actin filaments: Dynamic targets for cancer chemotherapy. Curr. Opin. Cell Biol. 1998, 10, 123–130. [Google Scholar] [CrossRef]
- Bhattacharya, K.; Maiti, S.; Mandal, C. PTEN negatively regulates mTORC2 formation and signaling in grade IV glioma via Rictor hyperphosphorylation at Thr1135 and direct the mode of action of an mTORC1/2 inhibitor. Oncogenesis 2016, 5, e227. [Google Scholar] [CrossRef] [PubMed]
- Choi, C.; Helfman, D.M. The Ras-ERK pathway modulates cytoskeleton organization, cell motility and lung metastasis signature genes in MDA-MB-231 LM2. Oncogene 2014, 33, 3668–3676. [Google Scholar] [CrossRef] [PubMed]
- Thomas, G.; Burnham, N.A.; Camesano, T.A.; Wen, Q. Measuring the mechanical properties of living cells using atomic force microscopy. J. Vis. Exp. 2013. [Google Scholar] [CrossRef] [PubMed]
- Hertz, H. Über die Berührung fester elastischer Körper. J. Reine Angew. Math. 1881, 92, 156–171. [Google Scholar]
- Vitolo, M.I.; Weiss, M.B.; Szmacinski, M.; Tahir, K.; Waldman, T.; Park, B.H.; Martin, S.S.; Weber, D.J.; Bachman, K.E. Deletion of PTEN promotes tumorigenic signaling, resistance to anoikis, and altered response to chemotherapeutic agents in human mammary epithelial cells. Cancer Res. 2009, 69, 8275–8283. [Google Scholar] [CrossRef] [PubMed]
- Mendoza, M.C.; Er, E.E.; Blenis, J. The Ras-ERK and PI3K-mTOR pathways: Cross-talk and compensation. Trends Biochem. Sci. 2011, 36, 320–328. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.H.; Lin, H.K.; Lin, I.H.; Chiou, Y.W.; Chen, H.W.; Liu, C.Y.; Harn, H.I.; Chiu, W.T.; Wang, Y.K.; Shen, M.R.; et al. Mechanical phenotype of cancer cells: Cell softening and loss of stiffness sensing. Oncotarget 2015, 6, 20946–20958. [Google Scholar] [CrossRef] [PubMed]
- Park, J.S.; Chu, J.S.; Tsou, A.D.; Diop, R.; Tang, Z.Y.; Wang, A.J.; Li, S. The effect of matrix stiffness on the differentiation of mesenchymal stem cells in response to TGF-beta. Biomaterials 2011, 32, 3921–3930. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Adeyinka, A.; Nui, Y.; Cherlet, T.; Snell, L.; Watson, P.H.; Murphy, L.C. Activated mitogen-activated protein kinase expression during human breast tumorigenesis and breast cancer progression. Clin. Cancer Res. 2002, 8, 1747–1753. [Google Scholar] [PubMed]
- Britten, C.D. PI3K and MEK inhibitor combinations: Examining the evidence in selected tumor types. Cancer Chemother. Pharmacol. 2013, 71, 1395–1409. [Google Scholar] [CrossRef] [PubMed]
- Davis, N.M.; Sokolosky, M.; Stadelman, K.; Abrams, S.L.; Libra, M.; Candido, S.; Nicoletti, F.; Polesel, J.; Maestro, R.; D’Assoro, A.; et al. Deregulation of the EGFR/PI3K/PTEN/Akt/mTORC1 pathway in breast cancer: Possibilities for therapeutic intervention. Oncotarget 2014, 5, 4603–4650. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Hoeflich, K.P.; O’Brien, C.; Boyd, Z.; Cavet, G.; Guerrero, S.; Jung, K.; Januario, T.; Savage, H.; Punnoose, E.; Truong, T.; et al. In vivo antitumor activity of MEK and phosphatidylinositol 3-kinase inhibitors in basal-like breast cancer models. Clin. Cancer Res. 2009, 15, 4649–4664. [Google Scholar] [CrossRef] [PubMed]
- Mirzoeva, O.K.; Das, D.; Heiser, L.M.; Bhattacharya, S.; Siwak, D.; Gendelman, R.; Bayani, N.; Wang, N.J.; Neve, R.M.; Guan, Y.; et al. Basal subtype and MAPK/ERK kinase (MEK)-phosphoinositide 3-kinase feedback signaling determine susceptibility of breast cancer cells to MEK inhibition. Cancer Res. 2009, 69, 565–572. [Google Scholar] [CrossRef] [PubMed]
- Pereira, C.B.; Leal, M.F.; de Souza, C.R.; Montenegro, R.C.; Rey, J.A.; Carvalho, A.A.; Assumpcao, P.P.; Khayat, A.S.; Pinto, G.R.; Demachki, S.; et al. Prognostic and predictive significance of MYC and KRAS alterations in breast cancer from women treated with neoadjuvant chemotherapy. PLoS ONE 2013, 8, e60576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, P.J.; Usary, J.E.; Darr, D.B.; Dillon, P.M.; Pfefferle, A.D.; Whittle, M.C.; Duncan, J.S.; Johnson, S.M.; Combest, A.J.; Jin, J.; et al. Combined PI3K/mTOR and MEK inhibition provides broad antitumor activity in faithful murine cancer models. Clin. Cancer Res. 2012, 18, 5290–5303. [Google Scholar] [CrossRef] [PubMed]
- Saini, K.S.; Loi, S.; de Azambuja, E.; Metzger-Filho, O.; Saini, M.L.; Ignatiadis, M.; Dancey, J.E.; Piccart-Gebhart, M.J. Targeting the PI3K/AKT/mTOR and Raf/MEK/ERK pathways in the treatment of breast cancer. Cancer Treat. Rev. 2013, 39, 935–946. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Munoz, A.; Gallego, E.; de Luque, V.; Perez-Rivas, L.G.; Vicioso, L.; Ribelles, N.; Lozano, J.; Alba, E. Lack of evidence for KRAS oncogenic mutations in triple-negative breast cancer. BMC Cancer 2010, 10, 136. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.P.; Roth, A.; Goya, R.; Oloumi, A.; Ha, G.; Zhao, Y.; Turashvili, G.; Ding, J.; Tse, K.; Haffari, G.; et al. The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature 2012, 486, 395–399. [Google Scholar] [CrossRef] [PubMed]
- Sivaraman, V.S.; Wang, H.; Nuovo, G.J.; Malbon, C.C. Hyperexpression of mitogen-activated protein kinase in human breast cancer. J. Clin. Investig. 1997, 99, 1478–1483. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Knowles, E.; O’Toole, S.A.; McNeil, C.M.; Millar, E.K.A.; Qiu, M.R.; Crea, P.; Daly, R.J.; Musgrove, E.A.; Sutherland, R.L. PI3K pathway activation in breast cancer is associated with the basal-like phenotype and cancer-specific mortality. Int. J. Cancer 2010, 126, 1121–1131. [Google Scholar] [CrossRef] [PubMed]
- Reis-Filho, J.S.; Tutt, A.N. Triple negative tumours: A critical review. Histopathology 2008, 52, 108–118. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.S.; Lee, G.Y.; Ong, C.N.; Lim, C.T. AFM indentation study of breast cancer cells. Biochem. Biophys. Res. Commun. 2008, 374, 609–613. [Google Scholar] [CrossRef] [PubMed]
- Physical Sciences-Oncology Centers Network; Agus, D.B.; Alexander, J.F.; Arap, W.; Ashili, S.; Aslan, J.E.; Austin, R.H.; Backman, V.; Bethel, K.J.; Bonneau, R.; et al. A physical sciences network characterization of non-tumorigenic and metastatic cells. Sci. Rep. 2013, 3, 1449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vitolo, M.I.; Boggs, A.E.; Whipple, R.A.; Yoon, J.R.; Thompson, K.; Matrone, M.A.; Cho, E.H.; Balzer, E.M.; Martin, S.S. Loss of PTEN induces microtentacles through PI3K-independent activation of cofilin. Oncogene 2013, 32, 2200–2210. [Google Scholar] [CrossRef] [PubMed]
- Tong, J.; Li, L.; Ballermann, B.; Wang, Z. Phosphorylation and Activation of RhoA by ERK in Response to Epidermal Growth Factor Stimulation. PLoS ONE 2016, 11, e0147103. [Google Scholar] [CrossRef] [PubMed]
- Menter, P. Acrylamide Polymerization—A Practical Approach; Bio-Rad Laboratories: Hercules, CA, USA, 2000. [Google Scholar]





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Linthicum, W.; Thanh, M.-T.H.; Vitolo, M.I.; Wen, Q. Effects of PTEN Loss and Activated KRAS Overexpression on Mechanical Properties of Breast Epithelial Cells. Int. J. Mol. Sci. 2018, 19, 1613. https://doi.org/10.3390/ijms19061613
Linthicum W, Thanh M-TH, Vitolo MI, Wen Q. Effects of PTEN Loss and Activated KRAS Overexpression on Mechanical Properties of Breast Epithelial Cells. International Journal of Molecular Sciences. 2018; 19(6):1613. https://doi.org/10.3390/ijms19061613
Chicago/Turabian StyleLinthicum, Will, Minh-Tri Ho Thanh, Michele I. Vitolo, and Qi Wen. 2018. "Effects of PTEN Loss and Activated KRAS Overexpression on Mechanical Properties of Breast Epithelial Cells" International Journal of Molecular Sciences 19, no. 6: 1613. https://doi.org/10.3390/ijms19061613