Genetic Epidemiology and Clinical Features of Hereditary Hearing Impairment in the Taiwanese Population

 , , , , ,
, , , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Subject Recruitment and Phenotype Characterization
2.2. Conventional Genetic Examination Using Sanger Sequencing
2.3. Next-Generation Sequence (SGS)-Based Genetic Examination
3. Results
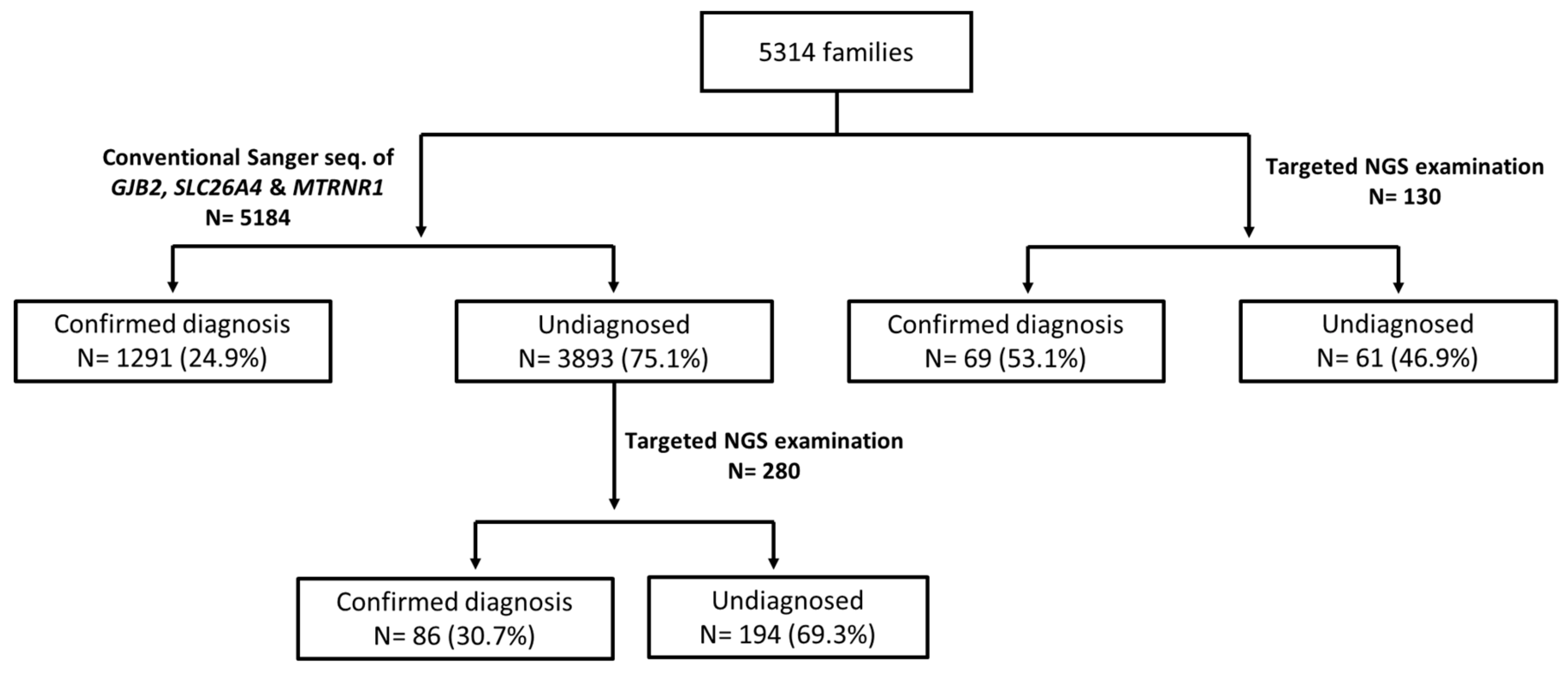
3.1. Diagnostic Yields of the Genetic Examinations
3.2. GJB2 Mutations
3.3. SLC26A4 Mutations
3.4. OTOF Mutations
3.5. MYO15A Mutations
3.6. MTRNR1 Mutations
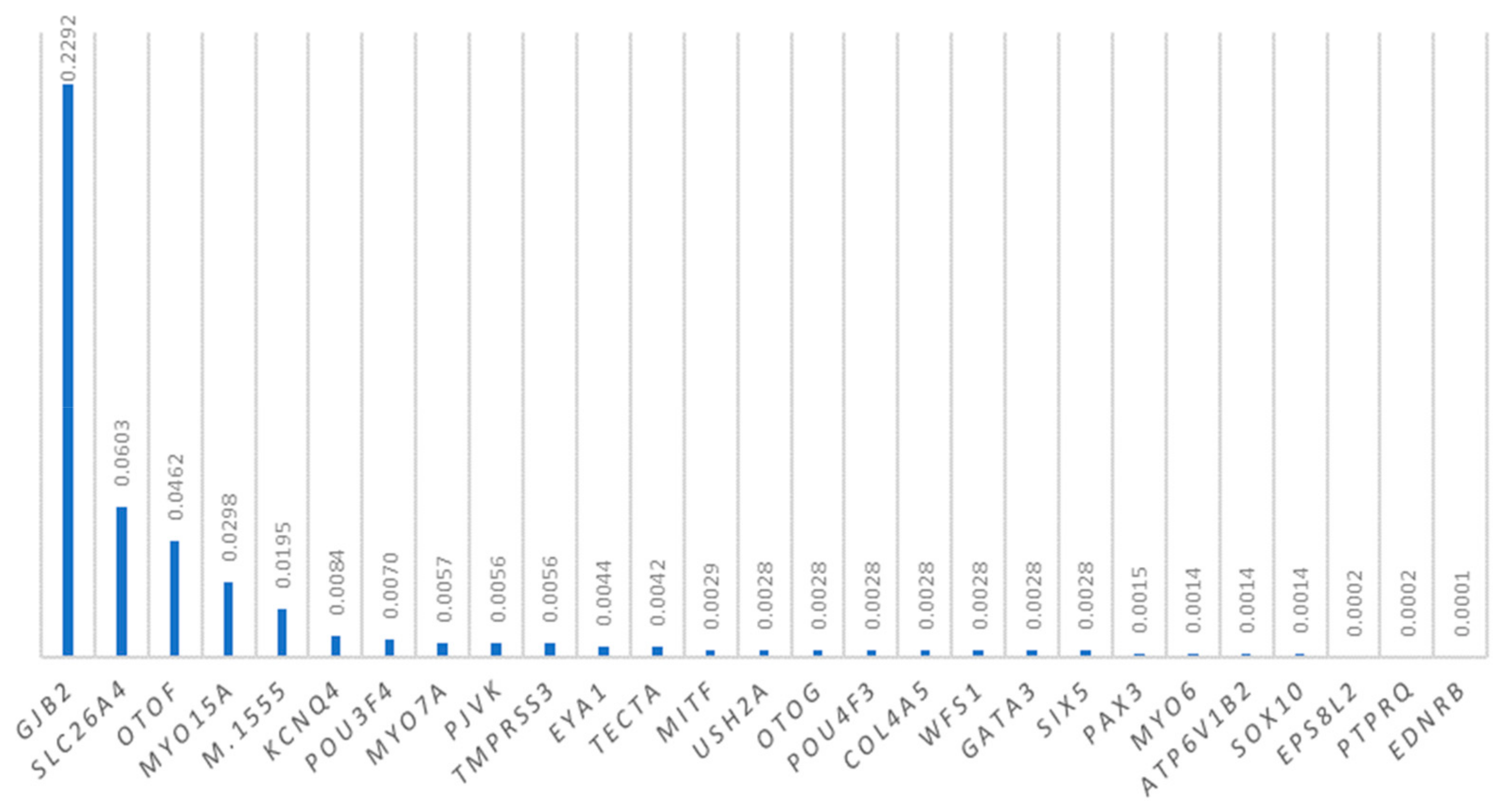
3.7. Mutations in Other Deafness Genes
4. Discussion
4.1. Comparison of Diagnostic Yields among Different Examination Strategies
4.2. GJB2 Mutations
4.3. SLC26A4 Mutations
4.4. OTOF Mutations
4.5. MYO15A Mutations
4.6. MTRNR1 Mutations
4.7. Mutations in Other Deafness Genes
4.8. Strengths and Limitations of the Study
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Morton, C.C.; Nance, W.E. Newborn hearing screening—A silent revolution. N. Engl. J. Med. 2006, 354, 2151–2164. [Google Scholar] [CrossRef] [PubMed]
- Wake, M.; Tobin, S.; Cone-Wesson, B.; Dahl, H.H.; Gillam, L.; McCormick, L.; Poulakis, Z.; Rickards, F.W.; Saunders, K.; Ukoumunne, O.C.; et al. Slight/mild sensorineural hearing loss in children. Pediatrics 2006, 118, 1842–1851. [Google Scholar] [CrossRef] [PubMed]
- Feder, K.P.; Michaud, D.; McNamee, J.; Fitzpatrick, E.; Ramage-Morin, P.; Beauregard, Y. Prevalence of Hearing Loss Among a Representative Sample of Canadian Children and Adolescents, 3 to 19 Years of Age. Ear Hear. 2017, 38, 7–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hilgert, N.; Smith, R.J.; Van Camp, G. Forty-six genes causing nonsyndromic hearing impairment: Which ones should be analyzed in DNA diagnostics? Mutat. Res. 2009, 681, 189–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abe, S.; Usami, S.; Shinkawa, H.; Kelley, P.M.; Kimberling, W.J. Prevalent connexin 26 gene (GJB2) mutations in Japanese. J. Med. Genet. 2000, 37, 41–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwa, H.L.; Ko, T.M.; Hsu, C.J.; Huang, C.H.; Chiang, Y.L.; Oong, J.L.; Chen, C.C.; Hsu, C.K. Mutation spectrum of the connexin 26 (GJB2) gene in Taiwanese patients with prelingual deafness. Genet. Med. 2003, 5, 161–165. [Google Scholar] [CrossRef]
- Park, H.J.; Hahn, S.H.; Chun, Y.M.; Park, K.; Kim, H.N. Connexin26 mutations associated with nonsyndromic hearing loss. Laryngoscope 2000, 110, 1535–1538. [Google Scholar] [CrossRef] [PubMed]
- Gurtler, N.; Kim, Y.; Mhatre, A.; Muller, R.; Probst, R.; Lalwani, A.K. GJB2 mutations in the Swiss hearing impaired. Ear Hear. 2003, 24, 440–447. [Google Scholar] [CrossRef]
- Denoyelle, F.; Weil, D.; Maw, M.A.; Wilcox, S.A.; Lench, N.J.; Allen-Powell, D.R.; Osborn, A.H.; Dahl, H.H.; Middleton, A.; Houseman, M.J.; et al. Prelingual deafness: High prevalence of a 30delG mutation in the connexin 26 gene. Hum. Mol. Genet. 1997, 6, 2173–2177. [Google Scholar] [CrossRef]
- Frei, K.; Szuhai, K.; Lucas, T.; Weipoltshammer, K.; Schofer, C.; Ramsebner, R.; Baumgartner, W.D.; Raap, A.K.; Bittner, R.; Wachtler, F.J.; et al. Connexin 26 mutations in cases of sensorineural deafness in eastern Austria. Eur. J. Hum. Genet. 2002, 10, 427–432. [Google Scholar] [CrossRef] [Green Version]
- Stinckens, C.; Kremer, H.; van Wijk, E.; Hoefsloot, L.H.; Huygen, P.L.; Standaert, L.; Fryns, J.P.; Cremers, C.W. Longitudinal phenotypic analysis in patients with connexin 26 (GJB2) (DFNB1) and connexin 30 (GJB6) mutations. Ann. Otol. Rhinol. Laryngol. 2004, 113, 587–593. [Google Scholar] [CrossRef] [PubMed]
- Morell, R.J.; Kim, H.J.; Hood, L.J.; Goforth, L.; Friderici, K.; Fisher, R.; Van Camp, G.; Berlin, C.I.; Oddoux, C.; Ostrer, H.; et al. Mutations in the connexin 26 gene (GJB2) among Ashkenazi Jews with nonsyndromic recessive deafness. N. Engl. J. Med. 1998, 339, 1500–1505. [Google Scholar] [CrossRef] [PubMed]
- Sirmaci, A.; Akcayoz-Duman, D.; Tekin, M. The c.IVS1+1G>A mutation in the GJB2 gene is prevalent and large deletions involving the GJB6 gene are not present in the Turkish population. J. Genet. 2006, 85, 213–216. [Google Scholar] [CrossRef] [PubMed]
- Mahdieh, N.; Nishimura, C.; Ali-Madadi, K.; Riazalhosseini, Y.; Yazdan, H.; Arzhangi, S.; Jalalvand, K.; Ebrahimi, A.; Kazemi, S.; Smith, R.J.; et al. The frequency of GJB2 mutations and the Delta (GJB6-D13S1830) deletion as a cause of autosomal recessive non-syndromic deafness in the Kurdish population. Clin. Genet. 2004, 65, 506–508. [Google Scholar] [CrossRef] [PubMed]
- Bajaj, Y.; Sirimanna, T.; Albert, D.M.; Qadir, P.; Jenkins, L.; Bitner-Glindzicz, M. Spectrum of GJB2 mutations causing deafness in the British Bangladeshi population. Clin. Otolaryngol. 2008, 33, 313–318. [Google Scholar] [CrossRef] [PubMed]
- Padma, G.; Ramchander, P.V.; Nandur, U.V.; Padma, T. GJB2 and GJB6 gene mutations found in Indian probands with congenital hearing impairment. J. Genet. 2009, 88, 267–272. [Google Scholar] [CrossRef]
- Campbell, C.; Cucci, R.A.; Prasad, S.; Green, G.E.; Edeal, J.B.; Galer, C.E.; Karniski, L.P.; Sheffield, V.C.; Smith, R.J. Pendred syndrome, DFNB4, and PDS/SLC26A4 identification of eight novel mutations and possible genotype-phenotype correlations. Hum. Mutat. 2001, 17, 403–411. [Google Scholar] [CrossRef]
- Coyle, B.; Reardon, W.; Herbrick, J.A.; Tsui, L.C.; Gausden, E.; Lee, J.; Coffey, R.; Grueters, A.; Grossman, A.; Phelps, P.D.; et al. Molecular analysis of the PDS gene in Pendred syndrome. Hum. Mol. Genet. 1998, 7, 1105–1112. [Google Scholar] [CrossRef]
- Tsukamoto, K.; Suzuki, H.; Harada, D.; Namba, A.; Abe, S.; Usami, S. Distribution and frequencies of PDS (SLC26A4) mutations in Pendred syndrome and nonsyndromic hearing loss associated with enlarged vestibular aqueduct: A unique spectrum of mutations in Japanese. Eur. J. Hum. Genet. 2003, 11, 916–922. [Google Scholar] [CrossRef]
- Park, H.J.; Lee, S.J.; Jin, H.S.; Lee, J.O.; Go, S.H.; Jang, H.S.; Moon, S.K.; Lee, S.C.; Chun, Y.M.; Lee, H.K.; et al. Genetic basis of hearing loss associated with enlarged vestibular aqueducts in Koreans. Clin. Genet. 2005, 67, 160–165. [Google Scholar] [CrossRef]
- Dai, P.; Li, Q.; Huang, D.; Yuan, Y.; Kang, D.; Miller, D.T.; Shao, H.; Zhu, Q.; He, J.; Yu, F.; et al. SLC26A4 c.919-2A>G varies among Chinese ethnic groups as a cause of hearing loss. Genet. Med. Off. J. Am. Coll. Med Genet. 2008, 10, 586–592. [Google Scholar] [CrossRef]
- Executive Yuan of R.O.C. Republic of China Yearbook; Executive Yuan of R.O.C.: Taipei, Taiwan, 2016; ISBN 9789860499490. [Google Scholar]
- Chen, C.H.; Yang, J.H.; Chiang, C.W.K.; Hsiung, C.N.; Wu, P.E.; Chang, L.C.; Chu, H.W.; Chang, J.; Song, I.W.; Yang, S.L.; et al. Population structure of Han Chinese in the modern Taiwanese population based on 10,000 participants in the Taiwan Biobank project. Hum. Mol. Genet. 2016, 25, 5321–5331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.C.; Lin, S.Y.; Su, Y.N.; Fang, M.Y.; Chen, S.U.; Hsu, C.J. Preimplantation genetic diagnosis (embryo screening) for enlarged vestibular aqueduct due to SLC26A4 mutation. Audiol. Neuro-Otol. 2010, 15, 311–317. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.C.; Hsu, C.J.; Huang, F.L.; Lin, Y.H.; Lin, Y.H.; Liu, T.C.; Wu, C.M. Timing of cochlear implantation in auditory neuropathy patients with OTOF mutations: Our experience with 10 patients. Clin. Otolaryngol. 2018, 43, 352–357. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.H.; Hsu, C.J.; Lin, Y.H.; Lin, Y.H.; Lee, H.Y.; Wu, C.C.; Liu, T.C. Etiologic and Audiologic Characteristics of Patients With Pediatric-Onset Unilateral and Asymmetric Sensorineural Hearing Loss. JAMA Otolaryngol. Head Neck Surg. 2017, 143, 912–919. [Google Scholar] [CrossRef]
- Sennaroglu, L.; Saatci, I. A new classification for cochleovestibular malformations. Laryngoscope 2002, 112, 2230–2241. [Google Scholar] [CrossRef] [PubMed]
- McClay, J.E.; Tandy, R.; Grundfast, K.; Choi, S.; Vezina, G.; Zalzal, G.; Willner, A. Major and minor temporal bone abnormalities in children with and without congenital sensorineural hearing loss. Arch. Otolaryngol. Head Neck Surg. 2002, 128, 664–671. [Google Scholar] [CrossRef]
- Wu, C.C.; Chen, Y.S.; Chen, P.J.; Hsu, C.J. Common clinical features of children with enlarged vestibular aqueduct and Mondini dysplasia. Laryngoscope 2005, 115, 132–137. [Google Scholar] [CrossRef]
- Wu, C.C.; Chen, P.J.; Chiu, Y.H.; Lu, Y.C.; Wu, M.C.; Hsu, C.J. Prospective mutation screening of three common deafness genes in a large Taiwanese cohort with idiopathic bilateral sensorineural hearing impairment reveals a difference in the results between families from hospitals and those from rehabilitation facilities. Audiol. Neurotol. 2008, 13, 172–181. [Google Scholar] [CrossRef]
- Wu, C.C.; Yeh, T.H.; Chen, P.J.; Hsu, C.J. Prevalent SLC26A4 mutations in patients with enlarged vestibular aqueduct and/or Mondini dysplasia: A unique spectrum of mutations in Taiwan, including a frequent founder mutation. Laryngoscope 2005, 115, 1060–1064. [Google Scholar] [CrossRef]
- Wu, C.C.; Chen, P.J.; Hsu, C.J. Specificity of SLC26A4 mutations in the pathogenesis of inner ear malformations. Audiol. Neurootol. 2005, 10, 234–242. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.C.; Lin, Y.H.; Lu, Y.C.; Chen, P.J.; Yang, W.S.; Hsu, C.J.; Chen, P.L. Application of massively parallel sequencing to genetic diagnosis in multiplex families with idiopathic sensorineural hearing impairment. PLoS ONE 2013, 8, e57369. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.H.; Wu, C.C.; Hsu, T.Y.; Chiu, W.Y.; Hsu, C.J.; Chen, P.L. Identification of a novel GATA3 mutation in a deaf Taiwanese family by massively parallel sequencing. Mutat. Res. 2015, 771, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.H.; Lin, Y.H.; Lu, Y.C.; Liu, T.C.; Chen, C.Y.; Hsu, C.J.; Chen, P.L.; Wu, C.C. A novel missense variant in the nuclear localization signal of POU4F3 causes autosomal dominant non-syndromic hearing loss. Sci. Rep. 2017, 7, 7551. [Google Scholar] [CrossRef]
- Lin, Y.H.; Wu, C.C.; Lin, Y.H.; Lu, Y.C.; Chen, C.S.; Liu, T.C.; Chen, P.L.; Hsu, C.J. Targeted Next-Generation Sequencing Facilitates Genetic Diagnosis and Provides Novel Pathogenetic Insights into Deafness with Enlarged Vestibular Aqueduct. J. Mol. Diagn. 2019, 21, 138–148. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.Y.; Lin, Y.H.; Liu, T.C.; Lin, Y.H.; Tseng, L.H.; Yang, T.H.; Chen, P.L.; Wu, C.C.; Hsu, C.J. Prediction Model for Audiological Outcomes in Patients With GJB2 Mutations. Ear Hear. 2019. [Google Scholar] [CrossRef]
- Wu, C.C.; Lu, Y.C.; Chen, P.J.; Yeh, P.L.; Su, Y.N.; Hwu, W.L.; Hsu, C.J. Phenotypic analyses and mutation screening of the SLC26A4 and FOXI1 genes in 101 Taiwanese families with bilateral nonsyndromic enlarged vestibular aqueduct (DFNB4) or Pendred syndrome. Audiol. Neurootol. 2010, 15, 57–66. [Google Scholar] [CrossRef]
- Wu, C.C.; Chiu, Y.H.; Chen, P.J.; Hsu, C.J. Prevalence and clinical features of the mitochondrial m.1555A>G mutation in Taiwanese patients with idiopathic sensorineural hearing loss and association of haplogroup F with low penetrance in three families. Ear Hear. 2007, 28, 332–342. [Google Scholar] [CrossRef]
- Zhao, H.; Li, R.H.; Wang, Q.J.; Yan, Q.F.; Deng, J.H.; Han, D.Y.; Bai, Y.D.; Young, W.Y.; Guan, M.X. Maternally inherited aminoglycoside-induced and nonsyndromic deafness is associated with the novel C1494T mutation in the mitochondrial 12S rRNA gene in a large chinese family. Am. J. Hum. Genet. 2004, 74, 139–152. [Google Scholar] [CrossRef]
- Moassass, F.; Al-Halabi, B.; Nweder, M.S.; Al-Achkar, W. Investigation of the mtDNA mutations in Syrian families with non-syndromic sensorineural hearing loss. Int. J. Pediatr. Otorhi. 2018, 113, 110–114. [Google Scholar] [CrossRef]
- Su, C.C.; Yang, J.J.; Shieh, J.C.; Su, M.C.; Li, S.Y. Identification of novel mutations in the KCNQ4 gene of patients with nonsyndromic deafness from Taiwan. Audiol. Neuro-Otol. 2007, 12, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Su, M.C.; Yang, J.J.; Su, C.C.; Hsin, C.H.; Li, S.Y. Identification of novel variants in the Myosin VIIA gene of patients with nonsyndromic hearing loss from Taiwan. Int. J. Pediatr. Otorhinolaryngol. 2009, 73, 811–815. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.H.; Wu, C.C.; Lu, Y.C.; Lin, Y.H.; Su, Y.N.; Hwu, W.L.; Yu, I.S.; Hsu, C.J. Mutation screening of the EYA1, SIX1, and SIX5 genes in an east asian cohort with branchio-oto-renal syndrome. Laryngoscope 2012, 122, 1130–1136. [Google Scholar] [CrossRef] [PubMed]
- Shearer, A.E.; DeLuca, A.P.; Hildebrand, M.S.; Taylor, K.R.; Gurrola, J.; Scherer, S.; Scheetz, T.E.; Smith, R.J.H. Comprehensive genetic testing for hereditary hearing loss using massively parallel sequencing. Proc. Natl. Acad. Sci. USA 2010, 107, 21104–21109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brownstein, Z.; Friedman, L.M.; Shahin, H.; Oron-Karni, V.; Kol, N.; Abu Rayyan, A.; Parzefall, T.; Lev, D.; Shalev, S.; Frydman, M.; et al. Targeted genomic capture and massively parallel sequencing to identify genes for hereditary hearing loss in Middle Eastern families. Genome Biol. 2011, 12, R89. [Google Scholar] [CrossRef] [PubMed]
- Miyagawa, M.; Naito, T.; Nishio, S.Y.; Kamatani, N.; Usami, S. Targeted exon sequencing successfully discovers rare causative genes and clarifies the molecular epidemiology of Japanese deafness patients. PLoS ONE 2013, 8, e71381. [Google Scholar] [CrossRef]
- Sommen, M.; Schrauwen, I.; Vandeweyer, G.; Boeckx, N.; Corneveaux, J.J.; van den Ende, J.; Boudewyns, A.; De Leenheer, E.; Janssens, S.; Claes, K.; et al. DNA Diagnostics of Hereditary Hearing Loss: A Targeted Resequencing Approach Combined with a Mutation Classification System. Hum. Mutat. 2016, 37, 812–819. [Google Scholar] [CrossRef]
- Sun, Y.; Yuan, J.; Wu, L.; Li, M.; Cui, X.; Yan, C.; Du, L.; Mao, L.; Man, J.; Li, W.; et al. Panel-based NGS reveals disease-causing mutations in hearing loss patients using BGISEQ-500 platform. Medicine 2019, 98, e14860. [Google Scholar] [CrossRef]
- Lu, C.Y.; Tsao, P.N.; Ke, Y.Y.; Lin, Y.H.; Lin, Y.H.; Hung, C.C.; Su, Y.N.; Hsu, W.C.; Hsieh, W.S.; Huang, L.M.; et al. Concurrent Hearing, Genetic, and Cytomegalovirus Screening in Newborns, Taiwan. J. Pediatr. 2018, 199, 144–150. [Google Scholar] [CrossRef]
- Wu, C.C.; Tsai, C.H.; Hung, C.C.; Lin, Y.H.; Lin, Y.H.; Huang, F.L.; Tsao, P.N.; Su, Y.N.; Lee, Y.L.; Hsieh, W.S.; et al. Newborn genetic screening for hearing impairment: A population-based longitudinal study. Genet. Med. 2017, 19, 6–12. [Google Scholar] [CrossRef]
- Li, L.; Lu, J.; Tao, Z.; Huang, Q.; Chai, Y.; Li, X.; Huang, Z.; Li, Y.; Xiang, M.; Yang, J.; et al. The p.V37I exclusive genotype of GJB2: A genetic risk-indicator of postnatal permanent childhood hearing impairment. PLoS ONE 2012, 7, e36621. [Google Scholar] [CrossRef] [PubMed]
- Wattanasirichaigoon, D.; Limwongse, C.; Jariengprasert, C.; Yenchitsomanus, P.T.; Tocharoenthanaphol, C.; Thongnoppakhun, W.; Thawil, C.; Charoenpipop, D.; Pho-iam, T.; Thongpradit, S.; et al. High prevalence of V37I genetic variant in the connexin-26 (GJB2) gene among non-syndromic hearing-impaired and control Thai individuals. Clin. Genet. 2004, 66, 452–460. [Google Scholar] [CrossRef] [PubMed]
- Erdenechuluun, J.; Lin, Y.H.; Ganbat, K.; Bataakhuu, D.; Makhbal, Z.; Tsai, C.Y.; Lin, Y.H.; Chan, Y.H.; Hsu, C.J.; Hsu, W.C.; et al. Unique spectra of deafness-associated mutations in Mongolians provide insights into the genetic relationships among Eurasian populations. PLoS ONE 2018, 13, e0209797. [Google Scholar] [CrossRef] [PubMed]
- Snoeckx, R.L.; Huygen, P.L.; Feldmann, D.; Marlin, S.; Denoyelle, F.; Waligora, J.; Mueller-Malesinska, M.; Pollak, A.; Ploski, R.; Murgia, A.; et al. GJB2 mutations and degree of hearing loss: A multicenter study. Am. J. Hum. Genet. 2005, 77, 945–957. [Google Scholar] [CrossRef] [PubMed]
- Oguchi, T.; Ohtsuka, A.; Hashimoto, S.; Oshima, A.; Abe, S.; Kobayashi, Y.; Nagai, K.; Matsunaga, T.; Iwasaki, S.; Nakagawa, T.; et al. Clinical features of patients with GJB2 (connexin 26) mutations: Severity of hearing loss is correlated with genotypes and protein expression patterns. J. Hum. Genet. 2005, 50, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.K.; Schrijver, I.; Chang, K.W. Connexin-26-associated deafness: Phenotypic variability and progression of hearing loss. Genet. Med. 2010, 12, 174–181. [Google Scholar] [CrossRef]
- Kenna, M.A.; Feldman, H.A.; Neault, M.W.; Frangulov, A.; Wu, B.L.; Fligor, B.; Rehm, H.L. Audiologic phenotype and progression in GJB2 (Connexin 26) hearing loss. Arch. Otolaryngol. Head Neck Surg. 2010, 136, 81–87. [Google Scholar] [CrossRef]
- Tsukada, K.; Nishio, S.; Usami, S. A large cohort study of GJB2 mutations in Japanese hearing loss patients. Clin. Genet. 2010, 78, 464–470. [Google Scholar] [CrossRef] [PubMed]
- Azaiez, H.; Booth, K.T.; Ephraim, S.S.; Crone, B.; Black-Ziegelbein, E.A.; Marini, R.J.; Shearer, A.E.; Sloan-Heggen, C.M.; Kolbe, D.; Casavant, T.; et al. Genomic Landscape and Mutational Signatures of Deafness-Associated Genes. Am. J. Hum. Genet. 2018, 103, 484–497. [Google Scholar] [CrossRef] [Green Version]
- Tsukada, K.; Nishio, S.; Hattori, M.; Usami, S. Ethnic-Specific Spectrum of GJB2 and SLC26A4 Mutations: Their Origin and a Literature Review. Ann. Oto. Rhinol. Laryngol. 2015, 124, 61s–76s. [Google Scholar] [CrossRef]
- Song, M.J.; Lee, S.T.; Lee, M.K.; Ji, Y.; Kim, J.W.; Ki, C.S. Estimation of carrier frequencies of six autosomal-recessive Mendelian disorders in the Korean population. J. Hum. Genet. 2012, 57, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Nishio, S.Y.; Usami, S. Deafness gene variations in a 1120 nonsyndromic hearing loss cohort: Molecular epidemiology and deafness mutation spectrum of patients in Japan. Ann. Otol. Rhinol. Laryngol. 2015, 124, 49S–60S. [Google Scholar] [CrossRef] [PubMed]
- Shearer, A.E.; Kolbe, D.L.; Azaiez, H.; Sloan, C.M.; Frees, K.L.; Weaver, A.E.; Clark, E.T.; Nishimura, C.J.; Black-Ziegelbein, E.A.; Smith, R.J.H. Copy number variants are a common cause of non-syndromic hearing loss. Genome Med. 2014, 6. [Google Scholar] [CrossRef] [PubMed]
- Pique, L.M.; Brennan, M.L.; Davidson, C.J.; Schaefer, F.; Greinwald, J.; Schrijver, I. Mutation analysis of the SLC26A4, FOXI1 and KCNJ10 genes in individuals with congenital hearing loss. Peerj 2014, 2. [Google Scholar] [CrossRef] [PubMed]
- Rendtorff, N.D.; Schrijver, I.; Lodahl, M.; Rodriguez-Paris, J.; Johnsen, T.; Hansen, E.C.; Nickelsen, L.A.A.; Tumer, Z.; Fagerheim, T.; Wetke, R.; et al. SLC26A4 mutation frequency and spectrum in 109 Danish Pendred syndrome/DFNB4 probands and a report of nine novel mutations. Clin. Genet. 2013, 84, 388–391. [Google Scholar] [CrossRef] [PubMed]
- Miyagawa, M.; Nishio, S.Y.; Usami, S.; Deafness Gene Study, C. Mutation spectrum and genotype-phenotype correlation of hearing loss patients caused by SLC26A4 mutations in the Japanese: A large cohort study. J. Hum. Genet. 2014, 59, 262–268. [Google Scholar] [CrossRef]
- Wang, Q.J.; Zhao, Y.L.; Rao, S.Q.; Guo, Y.F.; Yuan, H.; Zong, L.; Guan, J.; Xu, B.C.; Wang, D.Y.; Han, M.K.; et al. A distinct spectrum of SLC26A4 mutations in patients with enlarged vestibular aqueduct in China. Clin. Genet. 2007, 72, 245–254. [Google Scholar] [CrossRef]
- Pang, X.H.; Chai, Y.C.; Chen, P.H.; He, L.X.; Wang, X.W.; Wu, H.; Yang, T. Mono-allelic mutations of SLC26A4 is over-presented in deaf patients with non-syndromic enlarged vestibular aqueduct. Int. J. Pediatr. Otorhinolaryngol. 2015, 79, 1351–1353. [Google Scholar] [CrossRef]
- Albert, S.; Blons, H.; Jonard, L.; Feldmann, D.; Chauvin, P.; Loundon, N.; Sergent-Allaoui, A.; Houang, M.; Joannard, A.; Schmerber, S.; et al. SLC26A4 gene is frequently involved in nonsyndromic hearing impairment with enlarged vestibular aqueduct in Caucasian populations. Eur. J. Hum. Genet. 2006, 14, 773–779. [Google Scholar] [CrossRef]
- Azaiez, H.; Yang, T.; Prasad, S.; Sorensen, J.L.; Nishimura, C.J.; Kimberling, W.J.; Smith, R.J. Genotype-phenotype correlations for SLC26A4-related deafness. Hum. Genet. 2007. [Google Scholar] [CrossRef]
- Pryor, S.P.; Madeo, A.C.; Reynolds, J.C.; Sarlis, N.J.; Arnos, K.S.; Nance, W.E.; Yang, Y.; Zalewski, C.K.; Brewer, C.C.; Butman, J.A.; et al. SLC26A4/PDS genotype-phenotype correlation in hearing loss with enlargement of the vestibular aqueduct (EVA): Evidence that Pendred syndrome and non-syndromic EVA are distinct clinical and genetic entities. J. Med. Genet. 2005, 42, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Manchaiah, V.K.; Zhao, F.; Danesh, A.A.; Duprey, R. The genetic basis of auditory neuropathy spectrum disorder (ANSD). Int. J. Pediatr. Otorhinolaryngol. 2011, 75, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Moser, T.; Starr, A. Auditory neuropathy—Neural and synaptic mechanisms. Nat. Rev. Neurol. 2016, 12, 135–149. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Liu, M.; Wu, X.; Zong, L.; Jiang, H. Targeted next generation sequencing reveals OTOF mutations in auditory neuropathy spectrum disorder. Int. J. Pediatr. Otorhinolaryngol. 2018, 115, 19–23. [Google Scholar] [CrossRef]
- Chiu, Y.H.; Wu, C.C.; Lu, Y.C.; Chen, P.J.; Lee, W.Y.; Liu, A.Y.; Hsu, C.J. Mutations in the OTOF gene in Taiwanese patients with auditory neuropathy. Audiol. Neurootol. 2010, 15, 364–374. [Google Scholar] [CrossRef] [PubMed]
- Rehman, A.U.; Bird, J.E.; Faridi, R.; Shahzad, M.; Shah, S.; Lee, K.; Khan, S.N.; Imtiaz, A.; Ahmed, Z.M.; Riazuddin, S.; et al. Mutational Spectrum of MYO15A and the Molecular Mechanisms of DFNB3 Human Deafness. Hum. Mutat. 2016, 37, 991–1003. [Google Scholar] [CrossRef]
- Motavaf, M.; Soveizi, M.; Maleki, M.; Mahdieh, N. MYO15A splicing mutations in hearing loss: A review literature and report of a novel mutation. Int. J. Pediatr. Otorhinolaryngol. 2017, 96, 35–38. [Google Scholar] [CrossRef]
- Miyagawa, M.; Nishio, S.Y.; Hattori, M.; Moteki, H.; Kobayashi, Y.; Sato, H.; Watanabe, T.; Naito, Y.; Oshikawa, C.; Usami, S. Mutations in the MYO15A gene are a significant cause of nonsyndromic hearing loss: Massively parallel DNA sequencing-based analysis. Ann. Otol. Rhinol. Laryngol. 2015, 124, 158s–168s. [Google Scholar] [CrossRef]
- Usami, S.; Nishio, S.Y.; Nagano, M.; Abe, S.; Yamaguchi, T. Simultaneous screening of multiple mutations by invader assay improves molecular diagnosis of hereditary hearing loss: A multicenter study. PLoS ONE 2012, 7, e31276. [Google Scholar] [CrossRef]
- Bashir, R.; Fatima, A.; Naz, S. Prioritized sequencing of the second exon of MYO15A reveals a new mutation segregating in a Pakistani family with moderate to severe hearing loss. Eur. J. Med Genet. 2012, 55, 99–102. [Google Scholar] [CrossRef] [Green Version]
- Bai, X.; Nian, S.; Feng, L.; Ruan, Q.; Luo, X.; Wu, M.; Yan, Z. Identification of novel variants in MYO15A, OTOF, and RDX with hearing loss by next-generation sequencing. Mol. Genet. Genom. Med. 2019. [Google Scholar] [CrossRef] [PubMed]
- Fattahi, Z.; Shearer, A.E.; Babanejad, M.; Bazazzadegan, N.; Almadani, S.N.; Nikzat, N.; Jalalvand, K.; Arzhangi, S.; Esteghamat, F.; Abtahi, R.; et al. Screening for MYO15A gene mutations in autosomal recessive nonsyndromic, GJB2 negative Iranian deaf population. Am. J. Med Genet. Part A 2012, 158, 1857–1864. [Google Scholar] [CrossRef] [PubMed]
- Woo, H.M.; Park, H.J.; Baek, J.I.; Park, M.H.; Kim, U.K.; Sagong, B.; Koo, S.K. Whole-exome sequencing identifies MYO15A mutations as a cause of autosomal recessive nonsyndromic hearing loss in Korean families. BMC Med Genet. 2013, 14, 72. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.Y.; Lee, C.; Han, J.H.; Kim, M.Y.; Park, H.R.; Kim, N.; Park, W.Y.; Oh, D.Y.; Choi, B.Y. Expansion of phenotypic spectrum of MYO15A pathogenic variants to include postlingual onset of progressive partial deafness. BMC Med Genet. 2018, 19, 29. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.Y.; Kim, A.R.; Kim, N.K.; Lee, C.; Lee, K.Y.; Jeon, W.S.; Koo, J.W.; Oh, S.H.; Park, W.Y.; Kim, D.; et al. Identification and Clinical Implications of Novel MYO15A Mutations in a Non-consanguineous Korean Family by Targeted Exome Sequencing. Mol. Cells 2015, 38, 781–788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, F.; Xu, L.; Xiao, Y.; Li, J.; Bai, X.; Wang, H. Three MYO15A Mutations Identified in One Chinese Family with Autosomal Recessive Nonsyndromic Hearing Loss. Neural Plast. 2018, 2018, 5898025. [Google Scholar] [CrossRef] [PubMed]
- Cengiz, F.B.; Duman, D.; Sirmaci, A.; Tokgoz-Yilmaz, S.; Erbek, S.; Ozturkmen-Akay, H.; Incesulu, A.; Edwards, Y.J.; Ozdag, H.; Liu, X.Z.; et al. Recurrent and private MYO15A mutations are associated with deafness in the Turkish population. Genet. Test. Mol. Biomark. 2010, 14, 543–550. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.C.; Hung, C.C.; Lin, S.Y.; Hsieh, W.S.; Tsao, P.N.; Lee, C.N.; Su, Y.N.; Hsu, C.J. Newborn Genetic Screening for Hearing Impairment: A Preliminary Study at a Tertiary Center. PLoS ONE 2011, 6. [Google Scholar] [CrossRef]
- Bitner-Glindzicz, M.; Pembrey, M.; Duncan, A.; Heron, J.; Ring, S.M.; Hall, A.; Rahman, S. Prevalence of mitochondrial 1555A—>G mutation in European children. N. Engl. J. Med. 2009, 360, 640–642. [Google Scholar] [CrossRef] [PubMed]
- Igumnova, V.; Veidemane, L.; Viksna, A.; Capligina, V.; Zole, E.; Ranka, R. The prevalence of mitochondrial mutations associated with aminoglycoside-induced deafness in ethnic Latvian population: The appraisal of the evidence. J. Hum. Genet. 2019, 64, 199–206. [Google Scholar] [CrossRef]
- Yao, G.D.; Li, S.X.; Chen, D.L.; Feng, H.Q.; Zhao, S.B.; Liu, Y.J.; Guo, L.L.; Yang, Z.M.; Zhang, X.F.; Sun, C.X.; et al. Combination of hearing screening and genetic screening for deafness-susceptibility genes in newborns. Exp. Ther. Med. 2014, 7, 218–222. [Google Scholar] [CrossRef] [PubMed]
- Soini, H.K.; Karjalainen, M.K.; Hinttala, R.; Rautio, A.; Hallman, M.; Uusimaa, J. Mitochondrial hearing loss mutations among Finnish preterm and term-born infants. Audiol. Res. 2017, 7, 189. [Google Scholar] [CrossRef] [PubMed]
- Gopel, W.; Berkowski, S.; Preuss, M.; Ziegler, A.; Kuster, H.; Felderhoff-Muser, U.; Gortner, L.; Mogel, M.; Hartel, C.; Herting, E.; et al. Mitochondrial mutation m.1555A > G as a risk factor for failed newborn hearing screening in a large cohort of preterm infants. BMC Pediatr. 2014, 14. [Google Scholar] [CrossRef] [PubMed]
- Bindu, L.H.; Reddy, P.P. Genetics of aminoglycoside-induced and prelingual non-syndromic mitochondrial hearing impairment: A review. Int. J. Audiol. 2008, 47, 702–707. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Gene | No. of Patients | Inheritance Pattern | Associated Syndrome in Ref | Associated Syndrome in our Patients |
|---|---|---|---|---|
| 86 probands with confirmed diagnosis in the second-phase targeted NGS screening | ||||
| OTOF | 22 | AR | - | - |
| MYO15A | 18 | AR | - | - |
| KCNQ4 | 5 | AD | - | - |
| POU3F4 | 4 | X-linked | - | - |
| MYO7A | 5 | AD/AR | Usher syndrome 1B | Non-syndromic |
| EYA1 | 7 | AD | Branchio-oto-renal syndrome | Branchio-oto-renal syndrome |
| TECTA | 3 | AD/AR | - | - |
| MITF | 2 | AD | Waardenburg syndrome | Waardenburg syndromic |
| POU4F3 | 2 | AD | - | - |
| PJVK | 1 | AR | - | - |
| COL4A5 | 2 | X-linked | Alport syndrome | Alport syndrome |
| WFS1 | 2 | AD/AR | Wolfram syndrome | Non-syndromic |
| GATA3 | 2 | AD | HDR syndrome | Non-syndromic |
| SIX5 | 1 | AD | Branchio-oto-renal syndrome | Branchio-oto-renal syndrome |
| TMPRSS3 | 2 | AR | - | - |
| PAX3 | 1 | AD | Waardenburg syndrome | Waardenburg syndrome |
| USH2A | 2 | AR | Usher syndrome 2A | - |
| MYO6 | 2 | AD/AR | - | - |
| OTOG | 1 | AR | - | - |
| ATP6V1B2 | 1 | AD | Congenital deafness with onychodystrophy | Congenital deafness with onychodystrophy |
| SOX10 | 1 | AD | Waardenburg syndrome | Waardenburg syndrome |
| 69 probands with confirmed diagnosis in directly targeted NGS screening | ||||
| GJB2 | 42 | AR | - | - |
| MYO15A | 8 | AR | - | - |
| SLC26A4 | 5 | AR | Pendred syndrome | Non-syndromic |
| OTOF | 4 | AR | - | |
| EYA1 | 2 | AD | Branchio-oto-renal syndrome | Branchio-oto-renal syndrome |
| KCNQ4 | 1 | AD | - | - |
| POU3F4 | 1 | X-linked | - | - |
| MYO7A | 1 | AD/AR | Usher syndrome, type 1B | Non syndromic |
| MITF | 1 | AD | Waardenburg syndrome | Waardenburg syndrome |
| PAX3 | 1 | AD | Waardenburg syndrome | Waardenburg syndrome |
| EDNRB | 1 | AD | - | - |
| EPS8L2 | 1 | AR | - | - |
| PTPRQ | 1 | AD/AR | - | - |
| Nucleotide Change | Amino Acid Change | Allele Count (ratio%) | Popmax AF (population) from gnomAD |
|---|---|---|---|
| Recessive mutations | |||
| c.109G>A | p.V37I | 1608 (85.9) | 0.08345 (EAS) |
| c.235delC | p.L79Cfs *3 | 215 (11.5) | 0.006515 (EAS) |
| c.299_300delAT | p.H100Rfs *14 | 31 (1.7) | 0.0009023 (EAS) |
| c.427C>T | p.R143W | 6 (0.3) | 0.0007227 (AFR) |
| c.508_511dupAACG | p.A171Qfs *40 | 5 (0.3) | 0.0003008 (EAS) |
| c.176_191del | p.G59Afs *18 | 2 (0.1) | 0.0001631 (EAS) |
| c.95G>A | p.R32H | 1 (0.1) | 0.000008859 (EUP-NF) |
| c.107T>C | p.L36P | 1 (0.1) | 0.0001002 (EAS) |
| c.571T>C | p.F191L | 1 (0.1) | 0.001854 (EAS) |
| Dominant mutations | |||
| c.263C>T | p.A88V | 1 (0.1) | no data |
| c.428G>A | p.R143Q | 1 (0.1) | no data |
| Total | 1872 (100.0) |
| Nucleotide Change | Amino Acid Change | Allele Count (ratio%) | Popmax AF (population) from gnomAD |
|---|---|---|---|
| Bi-allelic SLC26A4 mutations | |||
| c.919-2A >G | NA | 473 (73.2) | 0.005064 (EAS) |
| c.2168A >G | p.H723R | 39 (6.0) | 0.001604 (EAS) |
| c.1229C >T | p.T410M | 17 (2.6) | 0.0005879 (SA) |
| c.1160C >T | p.A387V | 6 (0.9) | 0.00005438 (EAS) |
| c.754T >C | p.S252P | 5 (0.8) | 0.00005437 (EAS) |
| c.1115C >T | p.A372V | 5 (0.8) | No data |
| c.916dupG | p.V306Gfs *24 | 5 (0.8) | 0.0001631 (EAS) |
| c.164+1G >C | NA | 4 (0.6) | No data |
| c.706C >G | p.L236V | 3 (0.5) | 0.0002602 (LAT) |
| c.1343C >T | p.S448L | 3 (0.5) | 0.0001088 (EAS) |
| c.439A >G | p.M147V | 2 (0.3) | 0.0001087 (EAS) |
| c.1079C >T | p.A360V | 2 (0.3) | 0.000641 (EAS) |
| c.1173C >A | p.S391R | 2 (0.3) | No data |
| c.1318A >T | p.K440 * | 2 (0.3) | No data |
| c.1489G >C | p.G497R | 2 (0.3) | 0.00005013 (EAS) |
| c.2086C >T | p.Q696 * | 2 (0.3) | 0.0001695 (EAS) |
| c.2T >G | p.M1R | 1 (0.2) | 0.00007383 (EAS) |
| c.230A >T | p.K77I | 1 (0.2) | No data |
| c.235C >T | p.R79 * | 1 (0.2) | No data |
| c.241A >G | p.K81E | 1 (0.2) | No data |
| c.387delC | p.F130Lfs *15 | 1 (0.2) | No data |
| c.416-1G >A | NA | 1 (0.2) | 0.00005437 (EAS) |
| c.697G >C | p.V233L | 1 (0.2) | 0.001353 (EAS) |
| c.918+2T >C | NA | 1 (0.2) | 0.0001387 (OTH) |
| c.1001+1G >A | NA | 1 (0.2) | 0.0003977 (EUP-NF) |
| c.1105A >T | p.K369 * | 1 (0.2) | No data |
| c.1174A >T | p.N392Y | 1 (0.2) | 0.00005438 (EAS) |
| c.1226G >A | p.R409H | 1 (0.2) | 0.0001977 (LAT) |
| c.1489G >A | p.G497S | 1 (0.2) | 0.00005013 (EAS) |
| c.1520delT | p.L507 * | 1 (0.2) | No data |
| c.1544+6T >C | NA | 1 (0.2) | No data |
| c.1614+1G >A | NA | 1 (0.2) | 0.00003525 (EUP-NF) |
| c.1615-2A >G | NA | 1 (0.2) | 0.000008827 (EUP-NF) |
| c.1658C >A | p.P553H | 1 (0.2) | 0.00005438 (EAS) |
| c.1693_1694insA | p.C565 * | 1 (0.2) | No data |
| c.1707+1G >A | NA | 1 (0.2) | No data |
| c.1786C >T | p.Q596 * | 1 (0.2) | No data |
| c.1829C >A | p.S610 * | 1 (0.2) | No data |
| c.1975G >C | p.V659L | 1 (0.2) | 0.0002006 (EAS) |
| c.2086C >T | p.Q696T * | 1 (0.2) | 0.0001695 (EAS) |
| c.2089G >C | p.D697H | 1 (0.2) | No data |
| c.2107C >G | p.L703V | 1 (0.2) | No data |
| c.2162C >T | p.T721M | 1 (0.2) | 0.0002895 (LAT) |
| g.-1066C_6602Adel | NA | 1 (0.2) | No data |
| Mono-allelic SLC26A4 mutations | |||
| c.919-2A >G | NA | 44 (6.8) | 0.005064 (EAS) |
| c.916_917insG | p.V306Gfs *24 | 1 (0.2) | 0.0001631 (EAS) |
| c.974_977 delCTGGinsTTAAATTA | p.A325Vfs *6 | 1 (0.2) | No data |
| Total | 646 (100.0) | ||
| Nucleotide Change | Amino Acid Change | Allele Count (ratio%) | Popmax AF (population) from gnomAD |
|---|---|---|---|
| Bi-allelic OTOF mutations | |||
| c.5098G>C | p.E1700Q | 30 (57.7) | 0.006774 (EAS) |
| c.2521G>A | p.E841K | 3 (5.8) | 0.0002202 (EAS) |
| c.1498C>T | p.R500 * | 1 (1.9) | 0.0001387 (OTH) |
| c.2279T>C | p.L760P | 1 (1.9) | no data |
| c.3704_3719del | p.D1235Afs *30 | 1 (1.9) | no data |
| c.3894+5G>C | NA | 1 (1.9) | no data |
| c.4023+1G>A | NA | 1 (1.9) | 0.01178 (EAS) |
| c.4030C>T | p.R1344 * | 1 (1.9) | no data |
| c.4961-1G>A | NA | 1 (1.9) | no data |
| c.5197G>A | p.E1733K | 1 (1.9) | 0.00005439 (EAS) |
| c.5203C>T | p.R1735W | 1 (1.9) | 0.000008792 (EUP-NF) |
| c.5335C>T | p.H1779Y | 1 (1.9) | no data |
| c.5566C>T | p.R1856W | 1 (1.9) | 0.0001003 (EAS) |
| Mono-allelic OTOF mutations | |||
| c.5098G>C | p.E1700Q | 6 (11.5) | 0.006774 (EAS) |
| c.4023+1G>A | NA | 1 (1.9) | 0.01178 (EAS) |
| c.4227+5G>C | NA | 1 (1.9) | 0.001414 (EAS) |
| Total | 52 (100.0) | ||
| Nucleotide Change | Amino Acid Change | Allele Count (ratio%) | Popmax AF (population) from gnomAD |
|---|---|---|---|
| c.3524dupA | p.Ser1176ValfsTer14 | 9 (20.5) | 0.002152 (EAS) |
| c.10250_10252delCCT | p.Ser3417del | 8 (18.2) | 0.0004095 (EAS) |
| c.8182C>G | p.Arg2728Gly | 4 (9.1) | 0.0002226 (EAS) |
| c.3757-32_3757-1del | NA | 3 (6.8) | 0.0003583 (EAS) |
| c.5964+3G>A | NA | 2 (4.5) | 0.0003908 (EAS) |
| c.9408G>C | p.Trp3136Cys | 2 (4.5) | 0.0003583 (EAS) |
| c.10111C>T | p.Gln3371 * | 1 (2.3) | 0.00006473 (AFR) |
| c.10258_10260delTTC | p.Phe3420del | 1 (2.3) | 0.0001112 (EAS) |
| c.3844C>T | p.Arg1282Trp | 1 (2.3) | 0.0001403 (OTH) |
| c.4101C>A | p.Asn1367Lys | 1 (2.3) | no data |
| c.4457G>T | p.Gly1486Val | 1 (2.3) | no data |
| c.4642G>A | p.Ala1548Thr | 1 (2.3) | 0.0001239 (AFR) |
| c.4760T>C | p.Leu1587Pro | 1 (2.3) | no data |
| c.4761_4762insGTTTCTAT | p.Asp1588Valfs *11 | 1 (2.3) | no data |
| c.5421_5437del | p.Glu1808Glyfs *41 | 1 (2.3) | no data |
| c.5443C>A | p.Gln1815Lys | 1 (2.3) | no data |
| c.5977C>T | p.Arg1993Trp | 1 (2.3) | 0.0001712 (EAS) |
| c.6281G>A | p.Arg2094His | 1 (2.3) | 0.000008965 (EUP-NF) |
| c.6956+1G>A | NA | 1 (2.3) | 0.00006504 (EAS) |
| c.7708_7709insCA | p.Gln2571Hisfs * 35 | 1 (2.3) | 0.000641 (EAS) |
| c.7986dupG | p.Glu2663Glyfs * 4 | 1 (2.3) | no data |
| c.8602-1G>C | NA | 1 (2.3) | no data |
| Total | 52 (100.0) |
| Nucleotide Change | Amino Acid Change | Allele Count | Popmax AF (population) from gnomAD | References |
| KCNQ4 | ||||
| c.546C>G | p.F182L | 3 | 0.004576 (EAS) | Our cohort; Ref [42] |
| c.2014G>A | p.V672M | 2 | No data | Our cohort |
| c.2039C>T | p.S680F | 1 | 0.0002006 (EAS) | Our cohort; Ref [33] |
| MYO7A | ||||
| c.689C>T | p.A230V | 1 | No data | Our cohort |
| c.1142C>T | p.T381M | 1 | 0.0007619 (EAS) | Our cohort; Ref [33,43] |
| c.2557C>T | p.R853C | 1 | No data | Our cohort |
| c.6335C>G | p.S2112 * | 1 | No data | Our cohort |
| c.6470T>C | p.I2157T | 1 | 0.00005563 (EAS) | Our cohort |
| c.592+1G>A | NA | 1 | 0.00005563 (EAS) | Our cohort |
| POU3F4 | ||||
| c.346dupG | p.A116Gfs *77 | 2 | No data | Our cohort |
| c.919_921delGAG | p.E307del | 1 | No data | Our cohort |
| c.950T>A | p.L317 * | 1 | No data | Our cohort |
| c.1084T>C | p.*362Rext *113 | 1 | No data | Our cohort |
| EYA1 | ||||
| c.1081C>T | p.R361 * | 2 | No data | Our cohort; Ref [36] |
| c.1540_1542delCTG | p.L514del | 2 | No data | Our cohort |
| c.385C>T | p.Q129 * | 1 | No data | Our cohort |
| c.403G>A | p.G135S | 1 | 0.0007082 (EAS) | Our cohort |
| c.466C>T | p.Q156 * | 1 | No data | Our cohort; Ref [44] |
| c.1360+5G>A | NA | 1 | No data | Our cohort; Ref [36] |
| c.1735delG | p.D579Tfs *60 | 1 | No data | Our cohort; Ref [44] |
| TECTA | ||||
| c.5372C>G | p.P1791R | 1 | 0.001035 (EAS) | Our cohort |
| c.5471G>A | p.G1824D | 1 | 0.0005513 (EAS) | Our cohort |
| c.6062G>A | p.R2021H | 1 | 0.000008791 (EUP-NF) | Our cohort |
| POU4F3 | ||||
| c.491C>G | p.P164R | 1 | 0.0004023 (EAS) | Our cohort |
| c.982A>G | p.K328E | 1 | No data | Our cohort; Ref [35] |
| MITF | ||||
| c.862dupA | p.I288Nfs *9 | 1 | No data | Our cohort |
| c.1078C>T | p.R360 * | 1 | No data | Our cohort |
| c.938-1G>A | NA | 1 | No data | Our cohort |
| PJVK | ||||
| c.406C>T | p.R136 * | 1 | 0.00008316 (AFR) | Our cohort |
| c.593C>A | p.A198D | 1 | 0.0005119 (EAS) | Our cohort |
| COL4A5 | ||||
| c.367delG | p.G123Dfs *32 | 1 | No data | Our cohort |
| c.796C>T | p.R266 * | 1 | No data | Our cohort |
| WFS1 | ||||
| c.2051C>T | p.A684V | 2 | No data | Our cohort |
| SIX5 | ||||
| c.1872dupC | p.A625Rfs *15 | 1 | No data | Our cohort |
| GATA3 | ||||
| c.149delT | p.F51Lfs *144 | 1 | No data | Our cohort; Ref [34] |
| c.477delG | p.D160Tfs *35 | 1 | No data | Our cohort |
| TMPRSS3 | ||||
| c.916G>A | p.A306T | 2 | 0.0006014 (EAS) | Our cohort |
| c.432delA | p.Q144Hfs *8 | 1 | 0.0005012 (EAS) | Our cohort |
| c.743C>T | p.T248M | 1 | 0.0004894 (EAS) | Our cohort |
| PAX3 | ||||
| c.52C>T | p.Q18 * | 1 | No data | Our cohort |
| c.587-2A>G | NA | 1 | 0.000109 (EAS) | Our cohort |
| USH2A | ||||
| c.1614C>A | p.C538* | 1 | No data | Our cohort |
| c.4576G>A | p.G1526R | 1 | 0.0002521 (EAS) | Our cohort |
| c.7045dupT | p.W2349Lfs *8 | 1 | No data | Our cohort |
| c.8559-2A>G | NA | 1 | 0.0004353 (EAS) | Our cohort |
| MYO6 | ||||
| c.3736delC | p.P1246Qfs *39 | 1 | No data | Our cohort |
| c.1675-2A>C | NA | 1 | No data | Our cohort |
| OTOG | ||||
| c.3582C>A | p.Y1194 * | 1 | 0.0009720 (EAS) | Our cohort |
| c.4023_4045del | p.Q1342Pfs *104 | 1 | No data | Our cohort |
| ATP6V1B2 | ||||
| c.1516C>T | p.R506 * | 1 | No data | Our cohort |
| SOX10 | ||||
| c.314_315del | p.K105Tfs *28 | 1 | No data | Our cohort |
| PTPRQ | ||||
| c.6087-3T>G | NA | 3 | 0.002970 (EAS) | Our cohort |
| c.3181delC | p.L1061Ffs *11 | 1 | No data | Our cohort |
| EPS8L2 | ||||
| c.1304G>A | p.W435 * | 2 | 0.0001784 (EAS) | Our cohort |
| EDNRB | ||||
| c.754-2A>G | NA | 1 | No data | Our cohort |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, C.-C.; Tsai, C.-Y.; Lin, Y.-H.; Chen, P.-Y.; Lin, P.-H.; Cheng, Y.-F.; Wu, C.-M.; Lin, Y.-H.; Lee, C.-Y.; Erdenechuluun, J.; et al. Genetic Epidemiology and Clinical Features of Hereditary Hearing Impairment in the Taiwanese Population. Genes 2019, 10, 772. https://doi.org/10.3390/genes10100772
Wu C-C, Tsai C-Y, Lin Y-H, Chen P-Y, Lin P-H, Cheng Y-F, Wu C-M, Lin Y-H, Lee C-Y, Erdenechuluun J, et al. Genetic Epidemiology and Clinical Features of Hereditary Hearing Impairment in the Taiwanese Population. Genes. 2019; 10(10):772. https://doi.org/10.3390/genes10100772
Chicago/Turabian StyleWu, Chen-Chi, Cheng-Yu Tsai, Yi-Hsin Lin, Pey-Yu Chen, Pei-Hsuan Lin, Yen-Fu Cheng, Che-Ming Wu, Yin-Hung Lin, Chee-Yee Lee, Jargalkhuu Erdenechuluun, and et al. 2019. "Genetic Epidemiology and Clinical Features of Hereditary Hearing Impairment in the Taiwanese Population" Genes 10, no. 10: 772. https://doi.org/10.3390/genes10100772