Electrophoretic Extraction and Proteomic Characterization of Proteins Buried in Marine Sediments

Abstract
:1. Introduction
2. Experimental Section
2.1. Sediment Collection
2.2. Gel Electrophoresis Protein Extraction
2.3. In Gel Protein Digestion
2.4. Direct Digestion of Sediment
2.5. Mass Spectrometry and Database Searching
2.6. Total Hydrolysable Amino Acid Analysis
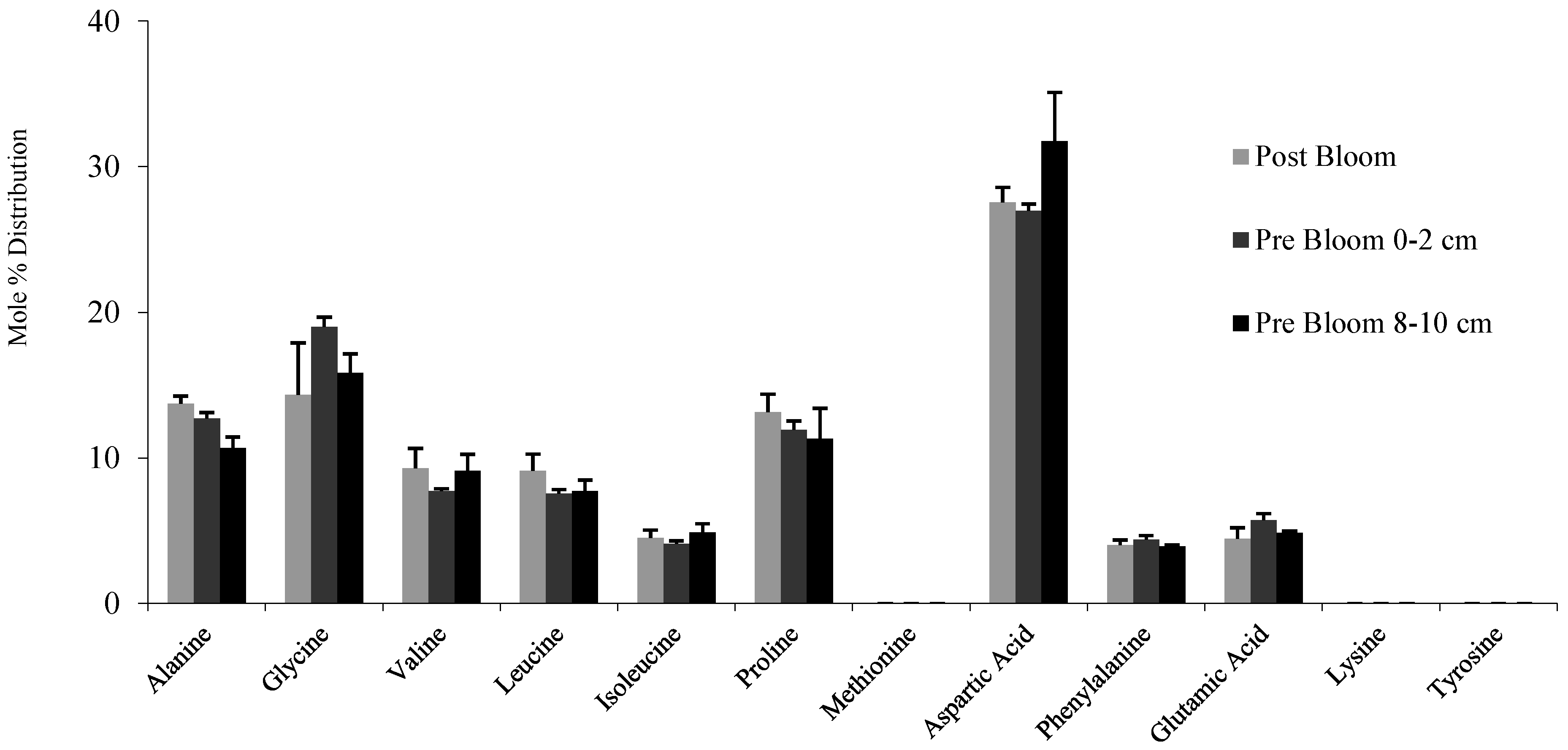
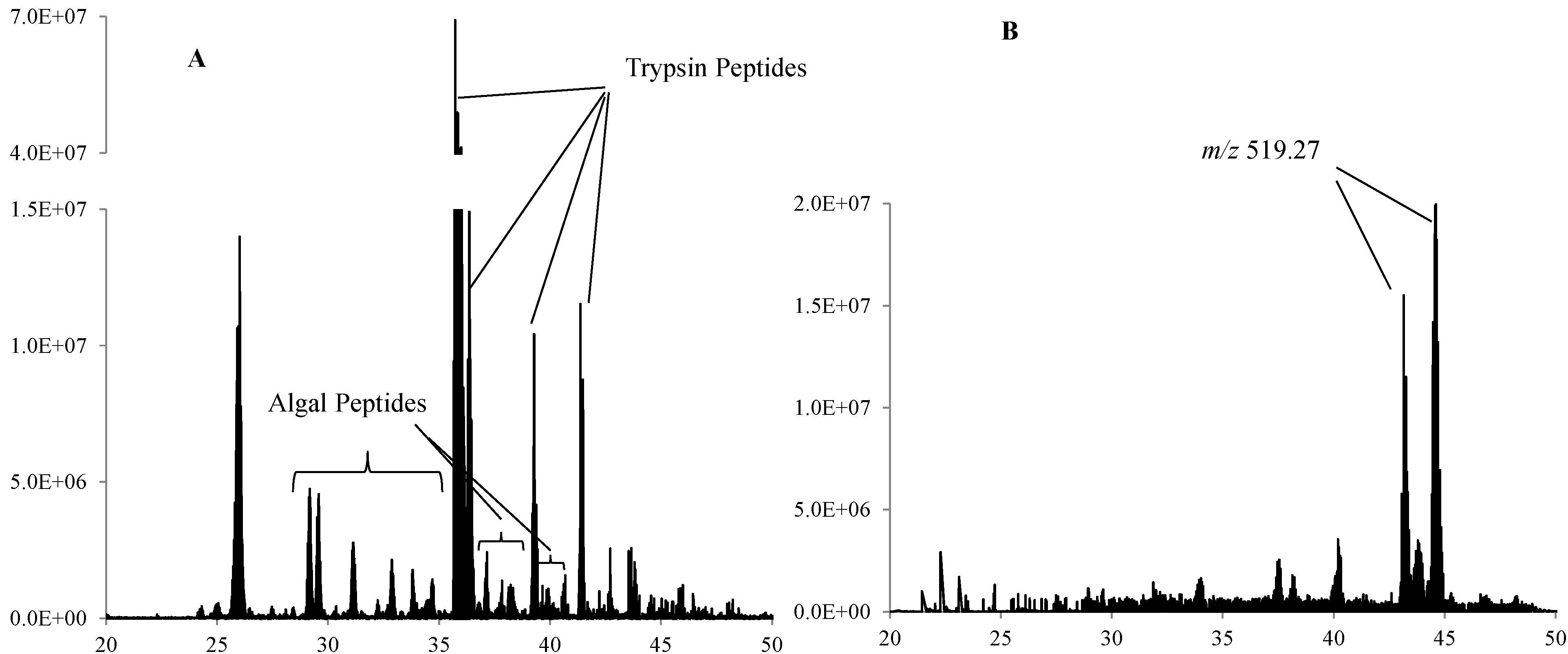
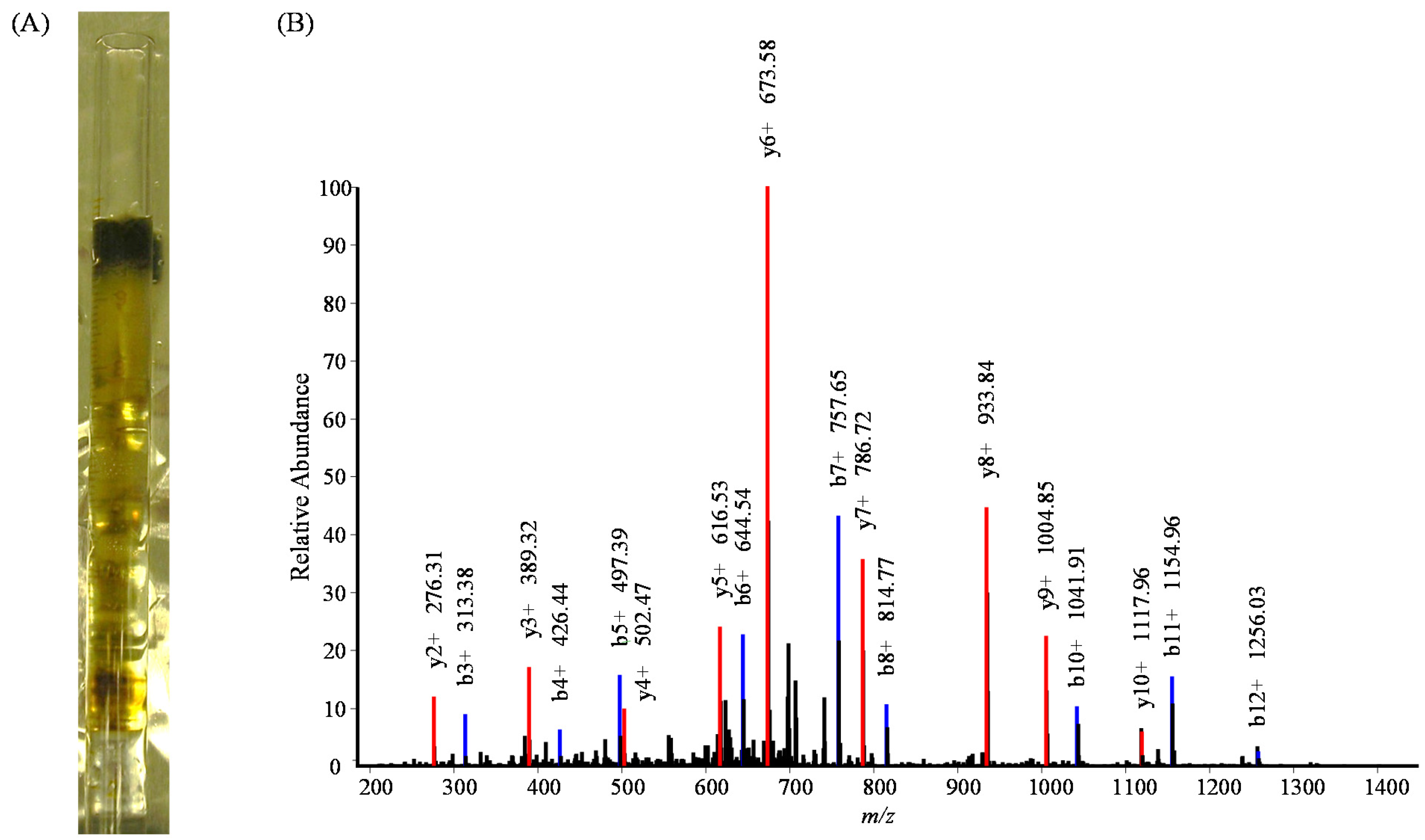
3. Results and Discussion

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | Protein (Annotation)/Peptides | Species | MW | Location | % |
|---|---|---|---|---|---|
| 7 cm Tube Gel | RuBisCO large subunit | T. pseudonana | 54,324.7 | C | 13.3 |
| (gi|118411104|ref|YP_874498.1|) | |||||
| DTDVLALFR | |||||
| FLNCLEGINR | |||||
| MGYWDAAYTVK | |||||
| MSGVDHIHAGTVVGK | |||||
| VALEAMVLAR | |||||
| YESGVIPYAK | |||||
| Histone H4 | T. pseudonana | 11,383.6 | N | 43.7 | |
| (jgi|Thaps3|3184|fgenesh1_pg.C_ chr_3000118) | |||||
| DNIQGITKPAIR | |||||
| ISGLIYEETR | |||||
| VFLENVIR | |||||
| VLRDNIQGITKPAIR | |||||
| ATP-CF1-Beta | T. pseudonana | 51,143.1 | C | 5.3 | |
| (gi|118411134|ref|YP_874528.1|) | |||||
| FTQAGSEVSALLGR | |||||
| IGLFGGAGVGK | |||||
| P2A | T. pseudonana | 56,408.1 | C | 4.7 | |
| (gi|118411113|ref|YP_874507.1|) | |||||
| YQWDSGYFQQEIER | |||||
| TSLESDGVFR | |||||
| FCP-1 | T. pseudonana | 22,628.0 | C | 6.2 | |
| (jgi|Thaps3|268127|estExt_thaps1_ ua_kg.C_chr_10394) | |||||
| IAQLAFLGQVVTR | |||||
| FCP-2 | T. pseudonana | 21,807.2 | C | 6.8 | |
| (jgi|Thaps3|38667|e_gw1.22.164.1) | |||||
| IAQLAFLGNIITR | |||||
| FCP-3 | T. pseudonana | 20,354.4 | C | 6.8 | |
| (jgi|Thaps3|38494|e_gw1.20.149.1) | |||||
| ISQLAFLGQIVTR | |||||
| P2-10P | T. pseudonana | 7,388.0 | C | 21.2 | |
| (gi|118411116|ref|YP_874510.1|) | |||||
| LGEILRPLNAEYGK | |||||
| Putative cysteine synthase | T. pseudonana | 37,867.3 | M | 4.6 | |
| (jgi|Thaps3|38294|e_gw1.19a.45.1) | |||||
| MENLNPGGTGKDRAAR | |||||
| DNA-RNA-P-Gamma | P. marinus | 72,277.0 | S | 1.7 | |
| (gi|33862040|ref|NP_893601.1|) | |||||
| FATSDLNDLYR | |||||
| ATP-PBS | P. marinus | 49,496.0 | S | 3.1 | |
| (gi|33862213|ref|NP_893774.1|) | |||||
| DVSGEGVQQALLK | |||||
| 2 cm Tube Gel | FCP-2 | T. pseudonana | 21,807.2 | C | 7.3 |
| (jgi|Thaps3|38667|e_gw1.22.164.1) | |||||
| DIEGTGNEFVGDFR | |||||
| P2RC-D2 | T. pseudonana | 39,064.0 | C | 3.7 | |
| (gi|118411148|ref|YP_874542.1|) | |||||
| AAEDPEFETFYTK | |||||
| H-DNA-BP | P. marinus | 9,768.0 | S | 24.2 | |
| (gi|33861877|ref|NP_893438.1|) | |||||
| TDVSLVVDAAIETIVDSVVEGK | |||||
| 1D Gel | H-DNA-BP | P. marinus | 9,768.0 | S | 24.2 |
| (gi|33861877|ref|NP_893438.1|) | |||||
| TDVSLVVDAAIETIVDSVVEGK | |||||
| DD | H-DNA-BP | P. marinus | 9,768.0 | S | 24.2 |
| (gi|33861877|ref|NP_893438.1|) | |||||
| TDVSLVVDAAIETIVDSVVEGK |
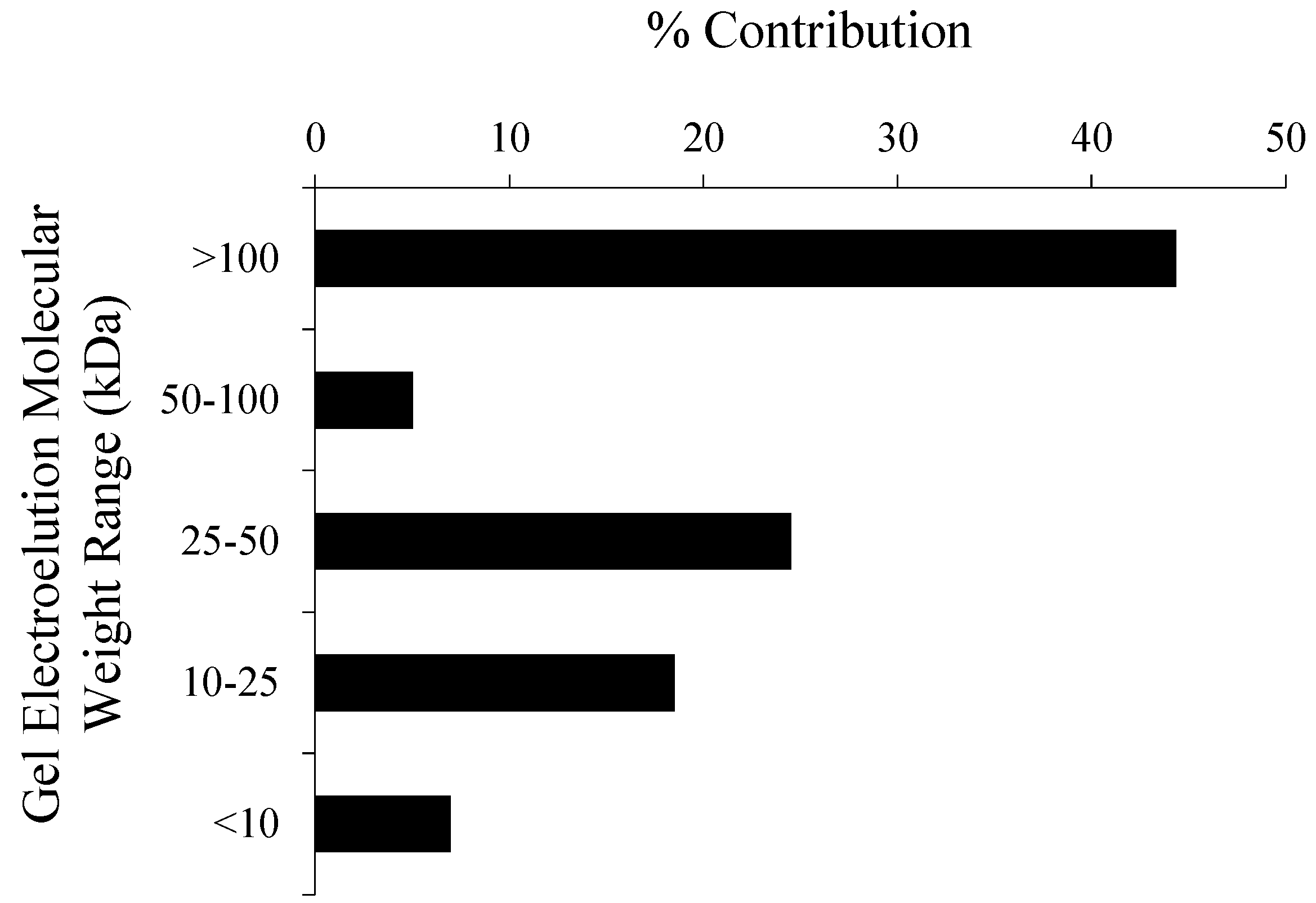
| Gel Section (kDa) | Protein/Peptides | Species | MW | CL | % |
|---|---|---|---|---|---|
| >100 | RuBisCO large subunit | T. pseudonana | 54,324.7 | C | 7.1 |
| MSGVDHIHAGTVVGK | |||||
| VALEAMVLAR | |||||
| FLNCLEGINR | |||||
| Histone H4 | T. pseudonana | 11,383.6 | N | 22.3 | |
| DNIQGITKPAIR | |||||
| VFLENVIR | |||||
| VLRDNIQGITKPAIR | |||||
| FCP-2 | T. pseudonana | 21,807.2 | C | 6.8 | |
| IAQLAFLGNIITR | |||||
| ATP-CF1-Beta | T. pseudonana | 51,143.1 | C | 3.0 | |
| FTQAGSEVSALLGR | |||||
| Putative cysteine synthase | T. pseudonana | 37,867.3 | M | 4.6 | |
| MENLNPGGTGKDRAAR | |||||
| DNA-RNA-P-Gamma | P. marinus | 72,277.0 | S | 1.7 | |
| FATSDLNDLYR | |||||
| ATP-PBS | P. marinus | 49,496.0 | S | 3.1 | |
| DVSGEGVQQALLK | |||||
| 50 to 100 | RuBisCO large subunit | T. pseudonana | 54,324.7 | C | 9.2 |
| DTDVLALFR | |||||
| MSGVDHIHAGTVVGK | |||||
| YESGVIPYAK | |||||
| MGYWDAAYTVK | |||||
| Histone H4 | T. pseudonana | 11,383.6 | N | 21.4 | |
| ISGLIYEETR | |||||
| DNIQGITKPAIR | |||||
| ATP-CF1-Beta | T. pseudonana | 51,143.1 | C | 3.0 | |
| FTQAGSEVSALLGR | |||||
| FCP-2 | T. pseudonana | 21,807.2 | C | 6.8 | |
| IAQLAFLGNIITR | |||||
| 25 to 50 | Histone H4 | T. pseudonana | 11,383.6 | N | 17.5 |
| ISGLIYEETR | |||||
| VFLENVIR | |||||
| P2A | T. pseudonana | 56,408.1 | C | 4.7 | |
| YQWDSGYFQQEIER | |||||
| TSLESDGVFR | |||||
| ATP-CF1-Beta | T. pseudonana | 51,143.1 | C | 3.0 | |
| FTQAGSEVSALLGR | |||||
| FCP-2 | T. pseudonana | 21,807.2 | C | 6.8 | |
| IAQLAFLGNIITR | |||||
| P2-10P | T. pseudonana | 7,388.0 | C | 21.2 | |
| LGEILRPLNAEYGK | |||||
| 10 to 25 | P2-10P | T. pseudonana | 7,388.0 | C | 21.2 |
| LGEILRPLNAEYGK | |||||
| Histone H4 | T. pseudonana | 11,383.6 | N | 29.1 | |
| ISGLIYEETR | |||||
| VFLENVIR | |||||
| DNIQGITKPAIR | |||||
| ATP-CF1-Beta | T. pseudonana | 51,143.1 | C | 5.3 | |
| IGLFGGAGVGK | |||||
| FTQAGSEVSALLGR | |||||
| FCP-1 | T. pseudonana | 22,628.0 | C | 6.2 | |
| IAQLAFLGQVVTR | |||||
| FCP-2 | T. pseudonana | 21,807.2 | C | 6.8 | |
| IAQLAFLGNIITR | |||||
| FCP-3 | T. pseudonana | 20,354.4 | C | 6.8 | |
| ISQLAFLGQIVTR | |||||
| <10 | no IDs |




4. Conclusions
Supplemental Information
Acknowledgments
Author Contributions
Conflicts of Interest
References and Notes
- Falkowski, P.G. Evolution of the nitrogen cycle and its influence of the biological sequestration of CO2 in the ocean. Nature 1997, 387, 272–275. [Google Scholar]
- McCarthy, M.; Pratum, T.; Hedges, J.; Benner, R. Chemical composition of dissolved organic nitrogen in the ocean. Nature 1997, 390, 150–154. [Google Scholar]
- Knicker, H. Solid-State 2-D double cross polarization magic angle spinning N-15 C-13 NMR spectroscopy on degraded algal residues. Org. Geochem. 2000, 31, 337–340. [Google Scholar]
- Zang, X.; Nguyen, R.T.; Harvey, H.R.; Knicker, H.; Hatcher, P.G. Preservation of proteinaceous material during the degradation of the green alga Botryococcus braunii: A solidstate 2D 15N 13C NMR Spectroscopy Study. Geochim. Cosmochim. Acta 2001, 65, 3299–3305. [Google Scholar]
- Cowie, G.L.; Hedges, J.I. Sources and reactivities of amino acids in a coastal marine environment. Limnol. Oceanogr. 1992, 37, 703–724. [Google Scholar]
- Grutters, M.; van Raaphorst, W.; Helder, W. Total hydrolysable amino acid mineralisation in sediments across the northeastern Atlantic continental slope (Goban Spur). Deep-Sea Res. I 2001, 48, 811–832. [Google Scholar]
- Vandewiele, S.; Cowie, G.; Soetaert, K.; Middelburg, J.J. Amino acid biogeochemistry and organic matter degradation state across the Pakistan margin oxygen minimum zone. Deep-Sea Res. II 2009, 56, 318–334. [Google Scholar]
- Fernandes, L.; Garg, A.; Borole, D.V. Amino acid biogeochemistry and bacterial contribution to sediment organic matter along the western margin of the Bay of Bengal. Deep-Sea Res. I 2014, 83, 81–92. [Google Scholar]
- Nunn, B.L.; Timperman, A.T. Marine proteomics. MEPS 2007, 332, 281–289. [Google Scholar]
- Morris, M.M.; Nunn, B.L.; Frazar, C.; Goodlett, D.R.; Ting, Y.S.; Rocap, G. Comparative metaproteomics reveals ocean-scale shifts in microbial nutrient utilization and energy transduction. ISME 2010, 4, 673–685. [Google Scholar]
- Zang, X.; van Heemst, J.D.H.; Dria, K.J.; Hatcher, P.G. Encapsulation of protein in humic acid from a histosol as an explanation for the occurrence of organic nitrogen in soil and sediment. Org. Geochem. 2000, 31, 679–695. [Google Scholar]
- Nguyen, R.T.; Harvey, H.R. Preservation of protein in marine systems: Hydrophobic and other noncovalent associations as major stabilizing forces. Geochim. Cosmochim. Acta 2001, 65, 1467–1480. [Google Scholar]
- Nguyen, R.T.; Harvey, H.R. Preservation via macromolecular associations during Botryococcus braunii decay: Proteins in the Pula Kerogen. Org. Geochem. 2003, 34, 1391–1403. [Google Scholar]
- Ogunseitan, O.A. Direct extraction of proteins from environmental samples. J. Microbiol. Meth. 1993, 17, 273–281. [Google Scholar]
- Nunn, B.L.; Keil, R.G. A comparison of non- hydrolytic methods for extracting amino acids and proteins from coastal marine sediments. Mar. Chem. 2006, 98, 31–42. [Google Scholar]
- Moore, E.K.; Nunn, B.L.; Faux, J.F.; Goodlett, D.R.; Harvey, H.R. Evaluation of electrophoretic protein extraction and database-driven protein identification from marine sediments. Limnol. Oceanogr.Meth. 2012, 10, 353–366. [Google Scholar]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar]
- Reisfeld, R.A.; Lewis, U.J.; Williams, D.E. Disc electrophoresis of basic proteins and peptides on polyacrylamide gels. Nature 1962, 195, 281–283. [Google Scholar]
- Laver, W.G. Structural studies on the protein subunits from three strains of influenza virus. J. Mol. Biol. 1964, 9, 109–124. [Google Scholar]
- Shapiro, A.L.; Vinuela, E.; Maizel, J.V., Jr. Molecular weight estimation of polypeptide chains by electrophoresis in SDS-polyacrylamide gels. Biochem. Biophys. Res. Commun. 1967, 28, 815–820. [Google Scholar]
- Fairbanks, G.; Steck, T.L.; Wallach, D.F.H. Electrophoretic analysis of the major proteins of the human erythrocyte membrane. Biochemistry 1971, 10, 2606–2617. [Google Scholar]
- Maizel, J.V. SDS polyacrylamide gel electrophoresis. Trends Biochem. Sci. 2000, 25, 590–592. [Google Scholar]
- Pederson, T. Turning a PAGE: The overnight sensation of SDS-polycrylamide gel electrophoresis. FASEB J. 2008, 22, 949–953. [Google Scholar]
- Wang, D.Z.; Xie, Z.X.; Zhang, S.F. Marine metaproteomics: Current status and future directions. J. Proteom. 2014, 97, 27–35. [Google Scholar]
- Tran, J.C.; Doucette, A.A. Multiplexed size separation of intact proteins in solution phase for mass spectrometry. Anal. Chem. 2009, 81, 6201–6209. [Google Scholar]
- Botelho, D.; Wall, M.J.; Vieira, D.B.; Fitzsimmons, S.; Liu, F.; Doucette, A. Top-Down and bottom-up proteomics of SDS-containing solutions following mass-based separation. J. Proteome Res. 2010, 9, 2863–2870. [Google Scholar]
- Chourey, K.; Jansson, J.; VerBerkmoes, N.; Shah, M.; Chavarria, K.L.; Tom, L.M.; Brodie, E.L.; Hettich, R.L. Direct cellular lysis/protein extraction protocol for soil metaproteomics. J. Proteome Res. 2010, 9, 6615–6622. [Google Scholar]
- Bastida, F.; Hernández, T.; Garcia, C. Metaproteomics of soils from semiarid environment: Functional and phylogenetic information obtained with different protein extraction methods. J. Proteomics 2014, 101, 31–42. [Google Scholar]
- Moore, E.K.; Brook, B.L.; Goodlett, D.R.; Harvey, H.R. Identifying and tracking proteins through the marine water column: Insights into the inputs and preservation mechanisms of protein in sediments. Geochim. Cosmochim. Acta 2012, 83, 324–359. [Google Scholar]
- Sambrotto, R.N.; Niebauer, H.J.; Goering, J.J.; Iverson, R.L. Relationships among vertical mixing, nitrate uptake, and phytoplankton growth during spring bloom in the S-E Bering Sea. Cont. Shelf Res. 1986, 5, 161–198. [Google Scholar]
- McRoy, C.P. Global maximum of primary production in the North Bering Sea. E.O.S. Comm. 1987, 68, 172. [Google Scholar]
- Walsh, J.J.; McRoy, C.P.; Coachman, L.K.; Goering, J.J.; Nihoul, J.J.; Whitledge, T.E.; Blackburn, T.H.; Parker, P.L.; Wirick, C.D.; Shuert, P.G.; et al. Carbon and nitrogen cycling within the Bering Chukchi Seas: Source regions for organic matter effecting AOU demands of the Arctic Ocean. Progr. Oceanogr. 1989, 22, 277–359. [Google Scholar]
- Highsmith, R.C.; Coyle, K.O. High productivity of northern Bering Sea benthic amphipods. Nature 1990, 862, 862–864. [Google Scholar]
- Chen, M.; Huang, Y.P.; Cai, P.G.; Guo, L.D. Particulate organic carbon export fluxes in the Canada Basin and Bering Sea as derived from 234Th/238U disequilibria. Arctic 2003, 56, 32–44. [Google Scholar]
- Lovvorn, J.R.; Cooper, L.W.; Brooks, M.L.; de Ruyck, C.C.; Bump, J.K.; Grebmeier, J.M. Organic matter pathways to zooplankton and benthos under pack ice in late winter and open water in late summer in the north-central Bering Sea. MEPS 2005, 291, 135–150. [Google Scholar]
- Wiese, F.K.; Wiseman, W.J., Jr.; van Pelt, T.I. Bering sea linkages introduction. Deep-Sea Res. Pt. II 2012, 65–70, 2–5. [Google Scholar]
- Kan, J.; Hanson, T.E.; Ginter, J.M.; Wang, K.; Chen, F. Metaproteomic analysis of Chesapeake Bay bacterial communities. Saline Syst. 2005, 1, 1–13. [Google Scholar]
- Shevchenko, A.; Wilm, M.; Vorm, O.; Mann, M. Mass spectrometric sequencing of proteins from silver stained polyacrylamide gels. Anal. Chem. 1996, 68, 850–858. [Google Scholar]
- Nunn, B.L.; Aker, J.R.; Shaffer, S.A.; Tsai, Y.; Strzepek, R.F.; Boyd, P.W.; Freeman, T.L.; Brittnacher, M.; Malmström, L.; Goodlett, D.R. Deciphering diatom biochemical pathways via whole-cell proteomics. Aquat. Microb. Ecol. 2009, 55, 241–253. [Google Scholar]
- Eng, J.K.; McCormack, A.L.; Yates, J.R. An approach to correlate tandem mass spectral data of peptides with amino acid-sequences in a protein database. J. Am. Soc. Mass Spectrom. 1994, 5, 976–989. [Google Scholar]
- Eng, J.K.; Fischer, B.; Grossmann, J.; MacCoss, M.J. A fast SEQUEST cross correlation algorithm. J. Proteome. Res. 2008, 7, 4598–4602. [Google Scholar]
- Keller, A.; Nesvizhskii, A.I.; Kolker, E.; Aebersold, R. Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal. Chem. 2002, 74, 5383–5392. [Google Scholar]
- Nesvizhskii, A.I.; Keller, A.; Kolker, E.; Aebersold, R. A statistical model for identifying proteins by tandem mass spectrometry. Anal. Chem. 2003, 75, 4646–4658. [Google Scholar]
- Rusch, D.B.; Halpern, A.L.; Sutton, G.; Heidelberg, K.B.; Williamson, S.; Yooseph, S.; Wu, D.; Eisen, J.A.; Hoffman, J.M.; Remington, K.; et al. The Sorcerer II Global Ocean Sampling expedition: Northwest Atlantic through Eastern Tropical Pacific. PLoS Biol. 2007, 5, e77. [Google Scholar]
- Yooseph, S.; Sutton, G.; Rusch, D.B.; Halpern, A.L.; Williamson, S.J.; Remington, K.; Eisen, J.A.; Heidelberg, K.B.; Manning, G.; Li, W.; et al. The Sorcerer II Global Ocean Sampling expedition: Expanding the universe of protein families. PLoS Biol. 2007, 5, 432–466. [Google Scholar]
- Waldhier, M.C.; Dettmer, K.; Gruber, M.A.; Oefner, P.J. Comparison of derivatization and chromatographic methods for GC–MS analysis of amino acid enantiomers in physiological samples. J. Chromatogr. B 2010, 878, 1103–1112. [Google Scholar]
- Cheng, C.N.; Shufeldt, R.C.; Stevenson, F.J. Amino acid analysis of soils and sediments: Extraction and desalting. Soil Biol. Biochem. 1975, 7, 143–151. [Google Scholar]
- Cowie, G.L.; Hedges, J.I. Improved amino acid quantification in environmental-samples —charge-matched recovery standards and reduced analysis time. Mar. Chem. 1992b, 37, 223–238. [Google Scholar]
- Mass spectra data files can be downloaded online at: https://chorusproject.org/anonymous/download/experiment/52779a6b839e496e803de596edbff228
- Pantoja, S.; Lee, C. Molecular weight distribution of proteinaceous material in Long Island Sound sediments. Limnol. Oceanogr. 1999, 44, 1323–1330. [Google Scholar]
- Cronin, J.R.; Morris, R.J. Rapid formation of humic material from diatom debris. In Coastal Upwelling, Its Sediment Record; Suess, E.T., Ed.; Plenum: New York, NY, USA, 1981; pp. 485–496. [Google Scholar]
- Benner, R.; Pakulski, J.D.; McCarthy, M.; Hedges, J.I.; Hatcher, P.G. Bulk chemical characteristics of dissolved organic matter in the Ocean. Science 1992, 255, 1561–1564. [Google Scholar]
- Bennett, R.H.; Hulbert, M.H.; Curry, K.J.; Curry, A.; Douglas, J. Organic matter sequestered in potential energy fields predicted by 3-D clay microstructure model: Direct observations of organo-clay micro- and nanofabric. Marine Geol. 2012, 315, 108–114. [Google Scholar]
- Knicker, H.; Hatcher, P.G. Survival of protein in an organic-rich sediment; possible protection by encapsulation in organic matter. Naturwissenschaften 1997, 84, 231–234. [Google Scholar]
- Hedges, J.I.; Keil, R.G. Organic geochemical perspectives on estuarine processes: Sorption reactions and consequences. Mar. Chem. 1999, 65, 55–65. [Google Scholar]
- Arnarson, T.S.; Keil, R.G. Influence of organic-mineral aggregates on microbial degradation of the dinoflagellate Scrippsiella. trochoidea. Geochim. Cosmochim. Acta 2005, 69, 2111–2117. [Google Scholar]
- De Leeuw, J.W.; Versteegh, G.J.M.; van Bergen, P.F. Biomacromolecules of plants and algae and their fossil analogues. Plant Ecol. 2006, 189, 209–233. [Google Scholar]
- Meysman, F.J.R.; Middelburg, J.J.; Heip, C.H.R. Bioturbation: A fresh look at Darwin’s last idea. Trends Ecol. Evol. 2006, 21, 688–695. [Google Scholar]
- Waldbusser, G.G.; Marinelli, R.L. Evidence of infaunal effects on porewater advection and biogeochemistry in permeable sediments: A proposed infaunal functional group framework. J. Mar. Res. 2009, 67, 503–532. [Google Scholar]
- Zonneveld, K.A.F.; Versteegh, G.J.M.; Kasten, S.; Eglinton, T.I.; Emeis, K.C.; Huguet, C.; Koch, B.P.; de Lange, G.J.; de Leeuw, J.W.; Middelburg, J.J.; et al. Selective preservation of organic matter in marine environments; processes and impact on the sedimentary record. Biogeosciences 2010, 7, 483–511. [Google Scholar]
- Oguri, K.; Harada, N.; Tadai, O. Excess 21°Pb and 137Cs concentrations, mass accumulation rates, and sedimentary processes on the Bering Sea continental shelf. Deep-Sea Res. II 2012, 61–64, 193–204. [Google Scholar]
- Gershanovich, D.E. New data on geomorphology and recent sediments of the bering sea and gulf of Alaska. Marin. Geol. 1968, 6, 281–296. [Google Scholar]
- Moore, E.K.; Nunn, B.L.; Faux, J.F.; Goodlett, D.R.; Harvey, H.R. Protein recycling in the Bering Sea algal incubations. MEPS 2014. in review. [Google Scholar]
- Suess, E. Particulate organic carbon flux in the oceans–surface productivity and oxygen utilization. Nature 1980, 288, 260–263. [Google Scholar]
- Hedges, J.I.; Keil, R.G. Sedimentary organic matter preservation: An assessment and speculative synthesis. Mar. Chem. 1995, 49, 81–115. [Google Scholar]
- Middelburg, J.J.; Meysman, F.J.R. Burial at Sea. Science 2007, 316, 1294–1294. [Google Scholar]
- Grossman, A.R.; Bhaya, D.; Apt, K.E.; Kehoe, D.M. Light-Harvesting complexes in oxygenic photosynthesis: Diversity, control, and evolution. Annu. Rev. Genet. 1995, 29, 231–288. [Google Scholar]
- Lang, M.; Kroth, P.G. Diatom fucoxanthin chlorophyll a/c-binding protein (FCP) and land plant light-harvesting proteins use a similar pathway for thylakoid membrane insertion. J. Biol. Chem. 2001, 276, 7985–7991. [Google Scholar]
- Kallmeyer, J.; Pockalny, R.; Adhikari, R.R.; Smith, D.C.; D’Hondt, S. Global distribution of microbial abundance and biomass in subseafloor sediment. Proc. Natl. Acad. Sci. USA 2012, 109, 16213–16216. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moore, E.K.; Harvey, H.R.; Faux, J.F.; Goodlett, D.R.; Nunn, B.L. Electrophoretic Extraction and Proteomic Characterization of Proteins Buried in Marine Sediments. Chromatography 2014, 1, 176-193. https://doi.org/10.3390/chromatography1040176
Moore EK, Harvey HR, Faux JF, Goodlett DR, Nunn BL. Electrophoretic Extraction and Proteomic Characterization of Proteins Buried in Marine Sediments. Chromatography. 2014; 1(4):176-193. https://doi.org/10.3390/chromatography1040176
Chicago/Turabian StyleMoore, Eli K., H. Rodger Harvey, Jessica F. Faux, David R. Goodlett, and Brook L. Nunn. 2014. "Electrophoretic Extraction and Proteomic Characterization of Proteins Buried in Marine Sediments" Chromatography 1, no. 4: 176-193. https://doi.org/10.3390/chromatography1040176