Loss of Function of the Gene Encoding the Histone Methyltransferase KMT2D Leads to Deregulation of Mitochondrial Respiration

 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Samples
2.2. Laser Scanning Confocal Microscopy Live Imaging
2.3. Immunoblotting
2.4. RNA Isolation and Quantitative RT-PCR (RT-qPCR)
2.5. Metabolites Extraction
2.6. LC–MS/MS Analysis
2.7. Measurements of Metabolic Fluxes
2.8. Measurement of the Steady-State Content and Rates of Production of ATP
2.9. Mitochondrial Respiratory Complex Enzymatic Activity
2.10. Statistical Analysis
3. Results
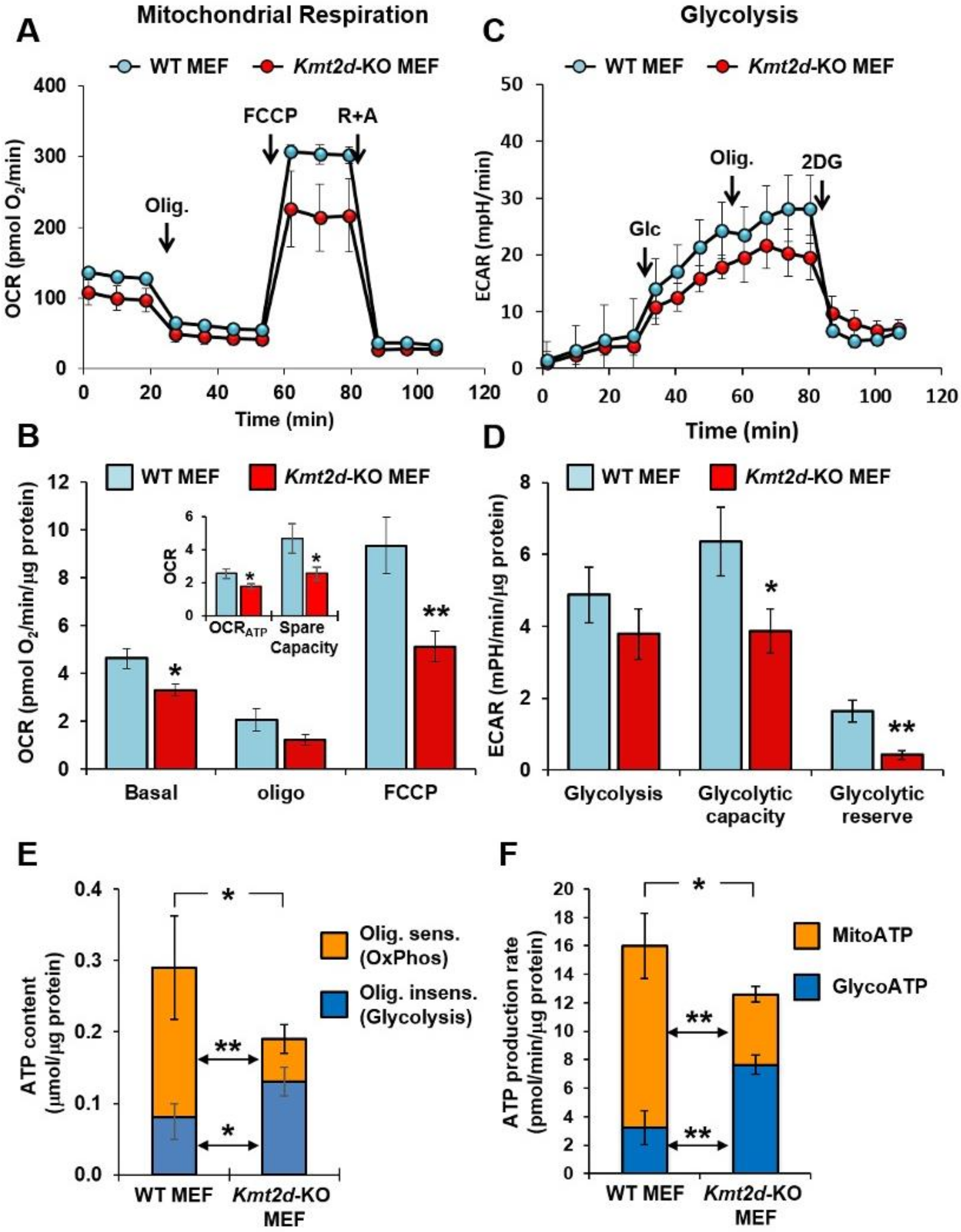
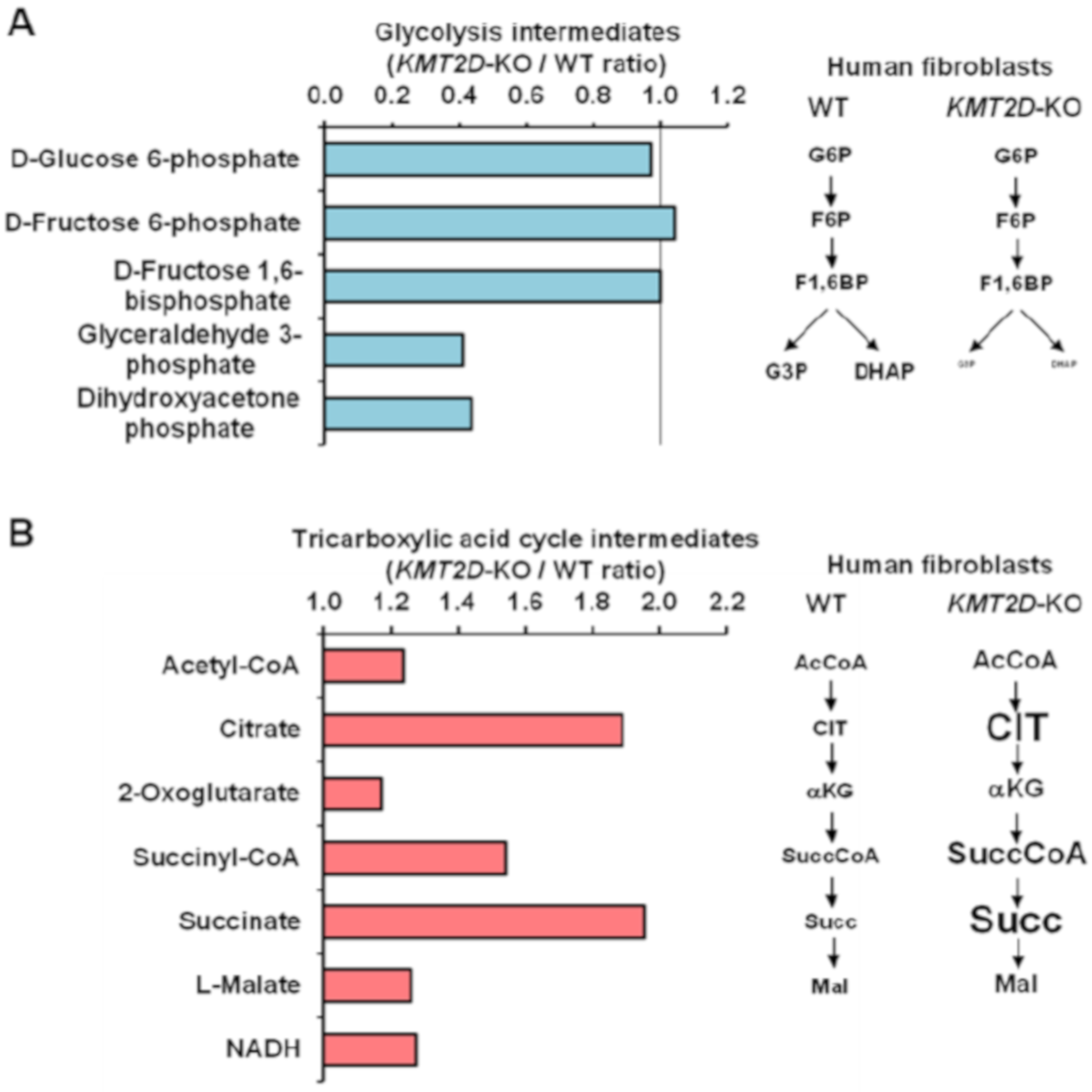
3.1. Loss of Kmt2d Causes Deregulation of the Respiratory and Glycolytic Metabolic Fluxes
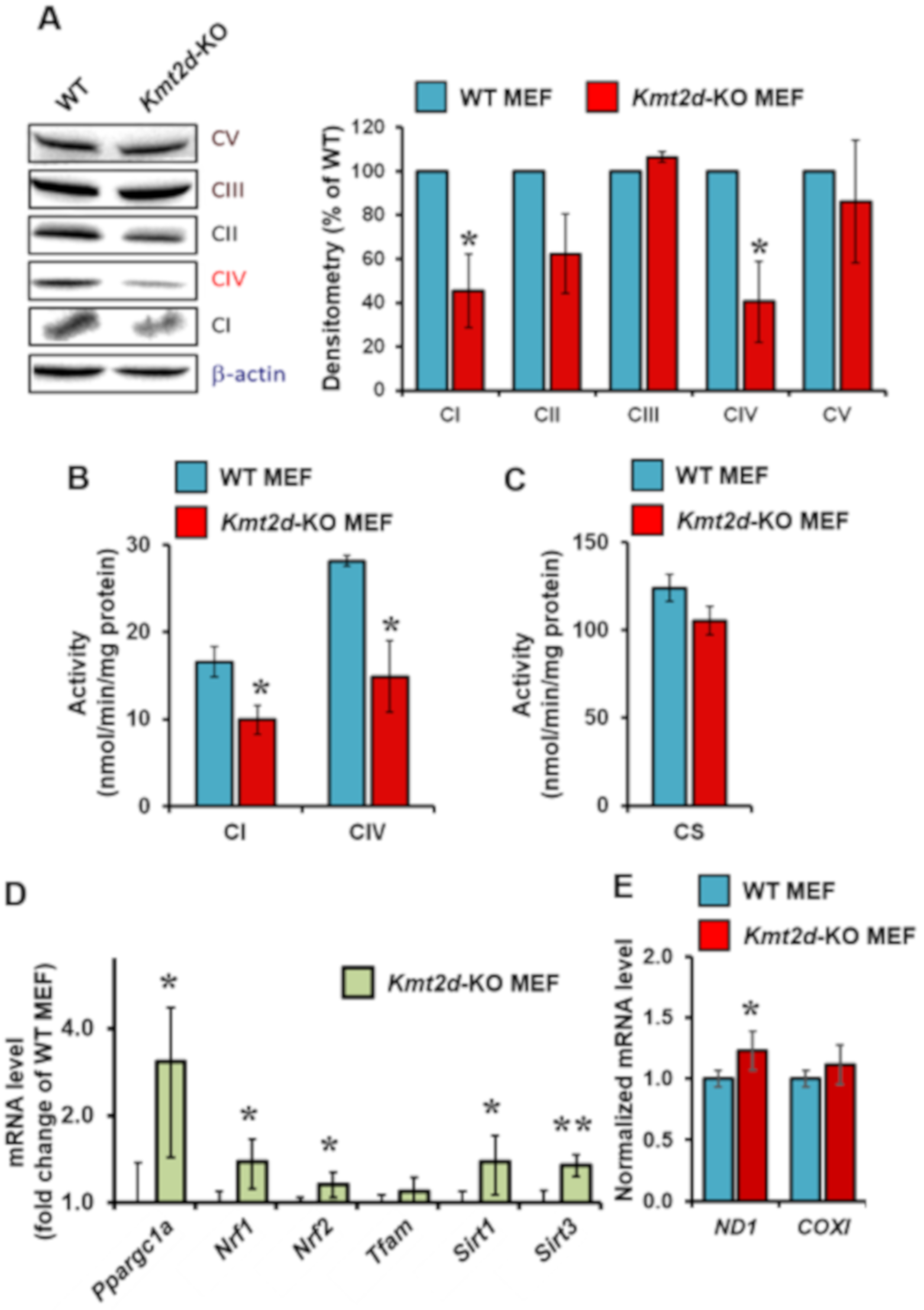
3.2. Loss of Kmt2d Causes Specific Downregulation of the Respiratory Chain Complexes I and IV
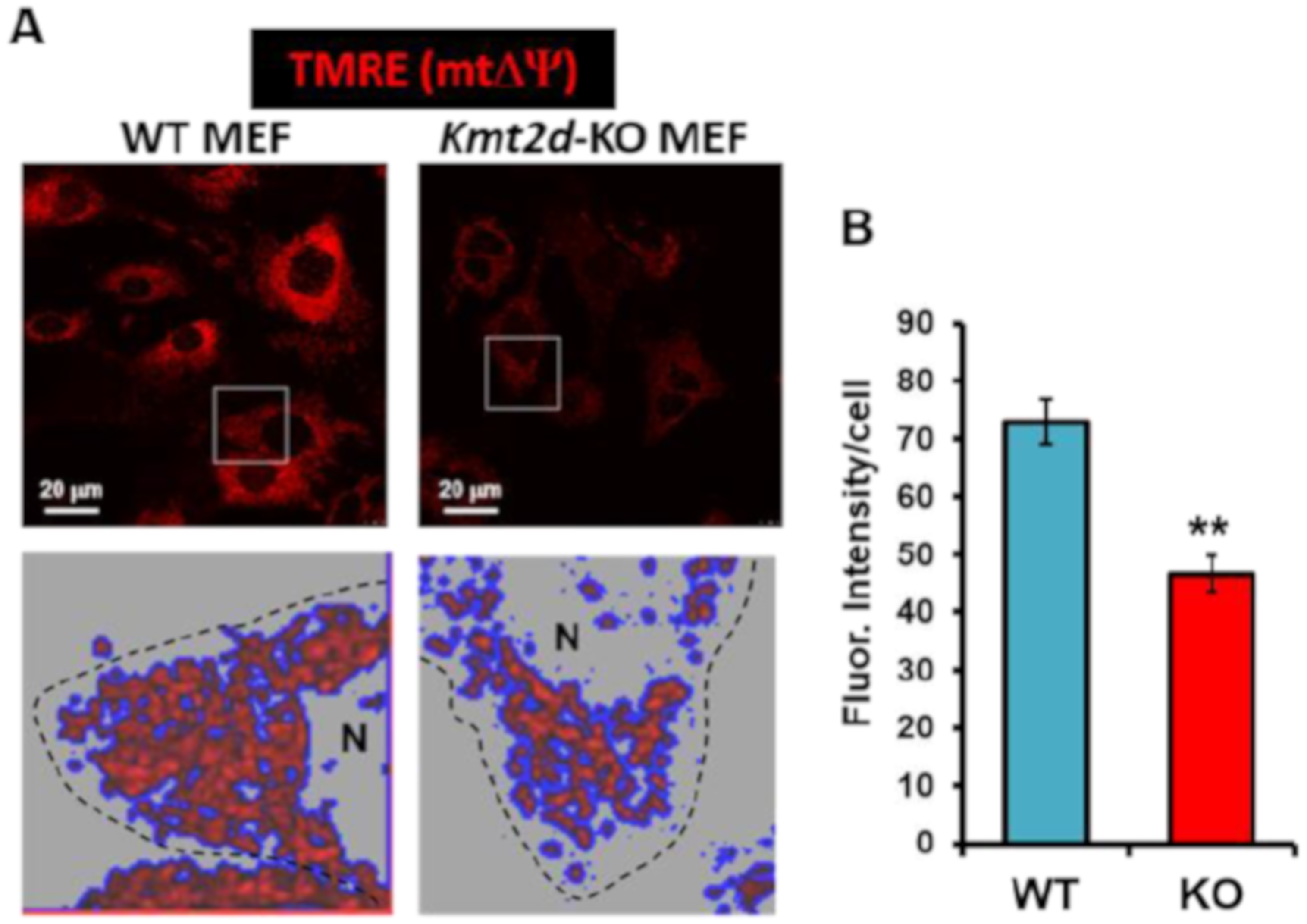
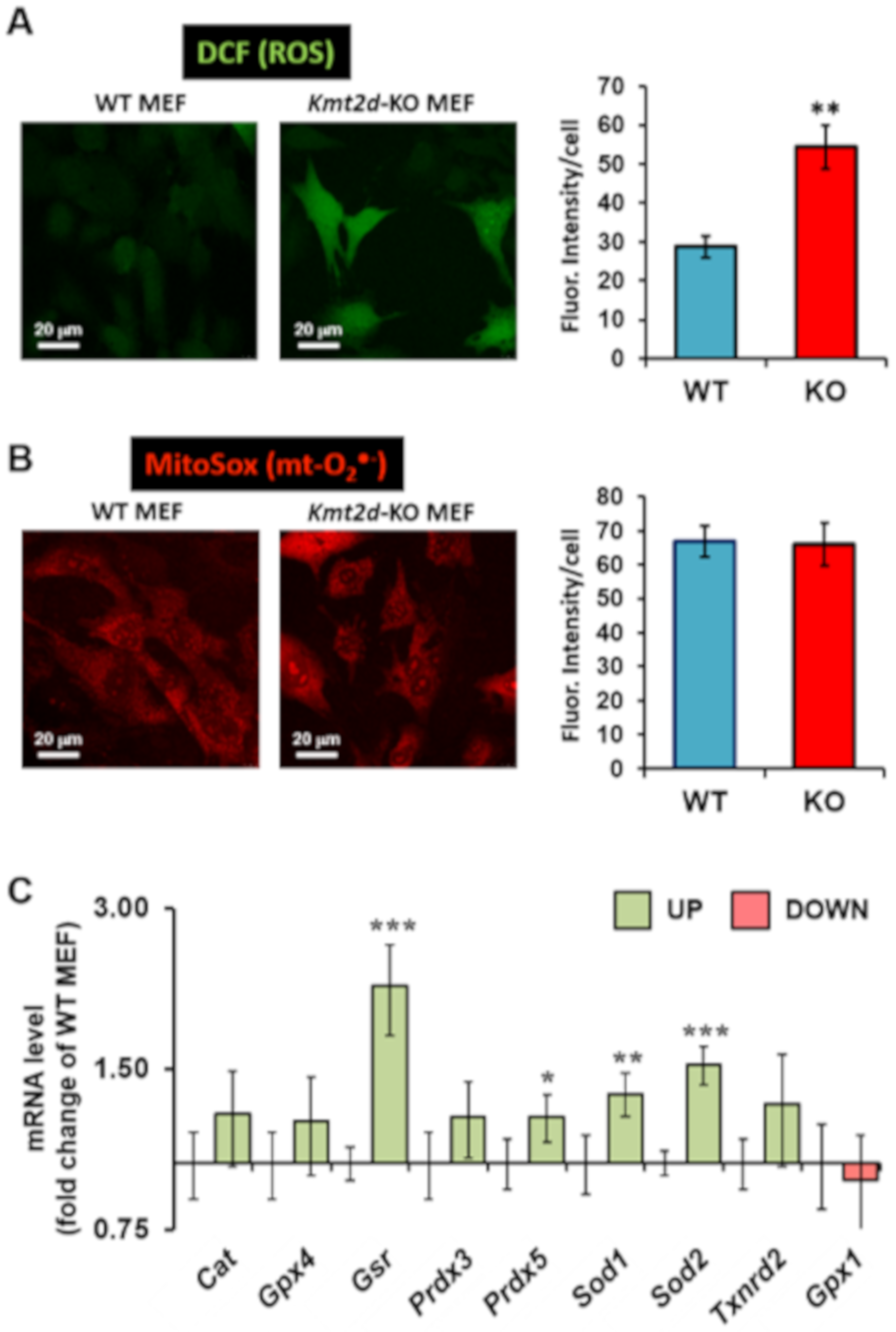
3.3. Loss of Kmt2d Causes Unbalance in the Reactive Oxygen Species Homeostasis
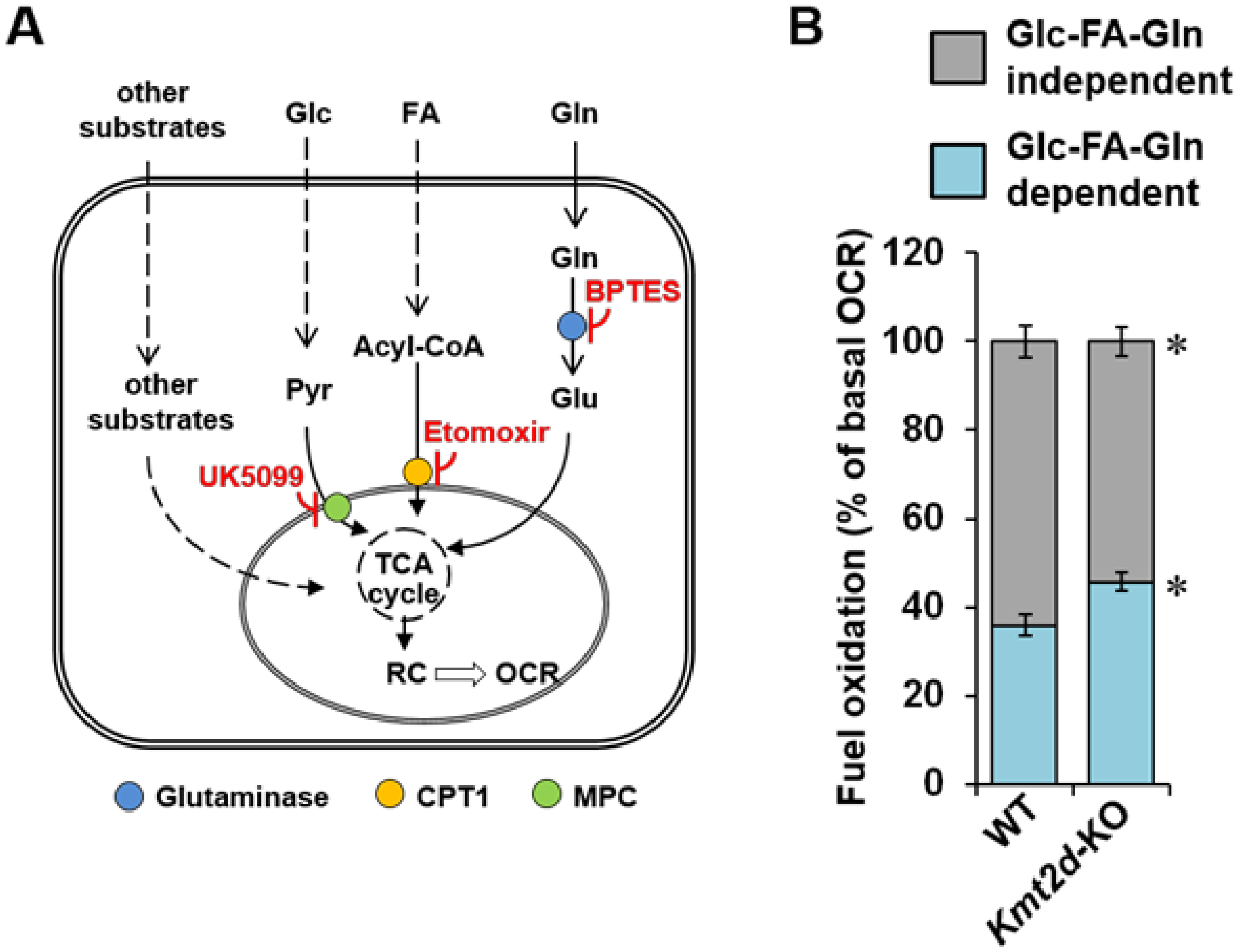
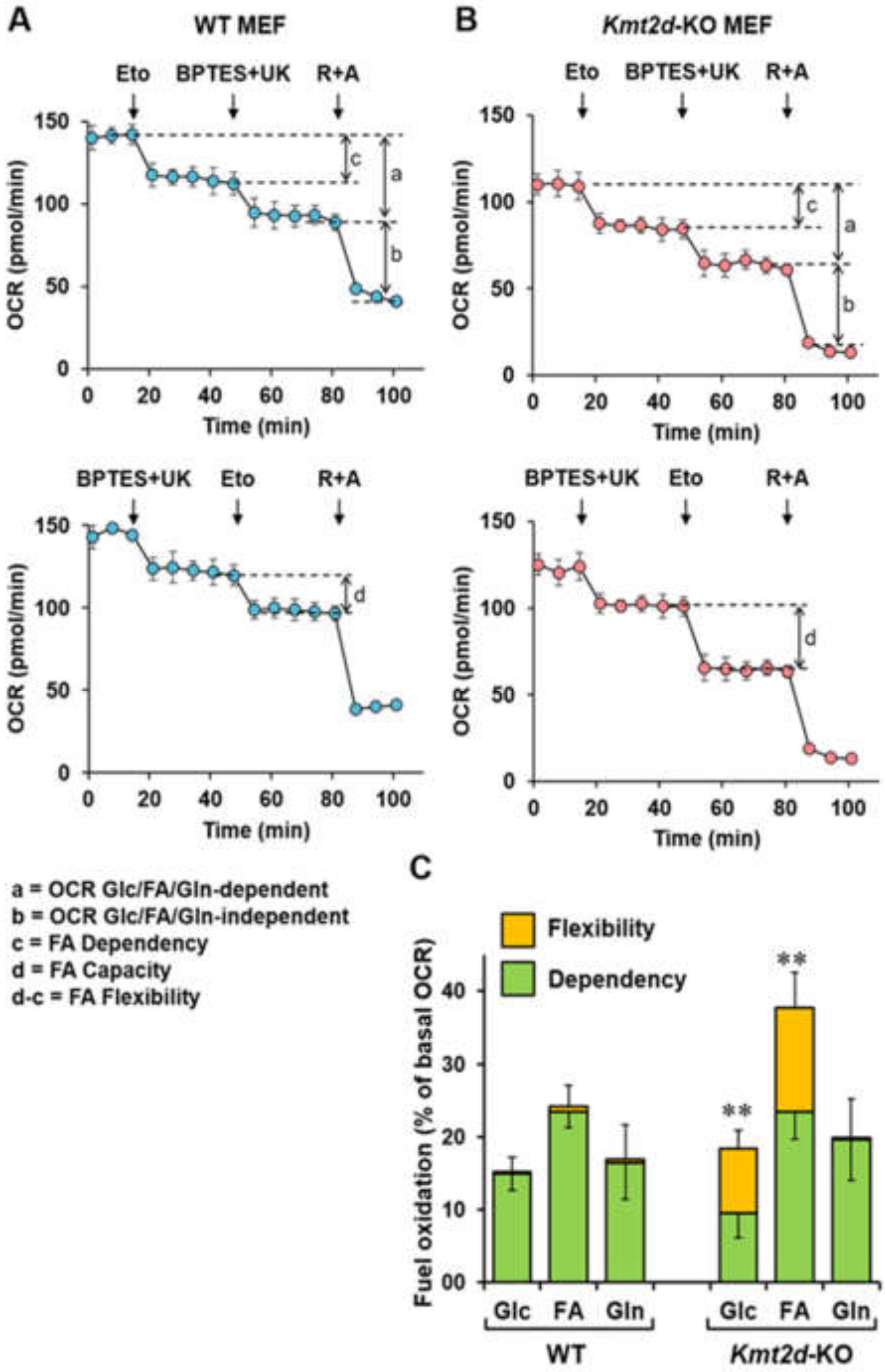
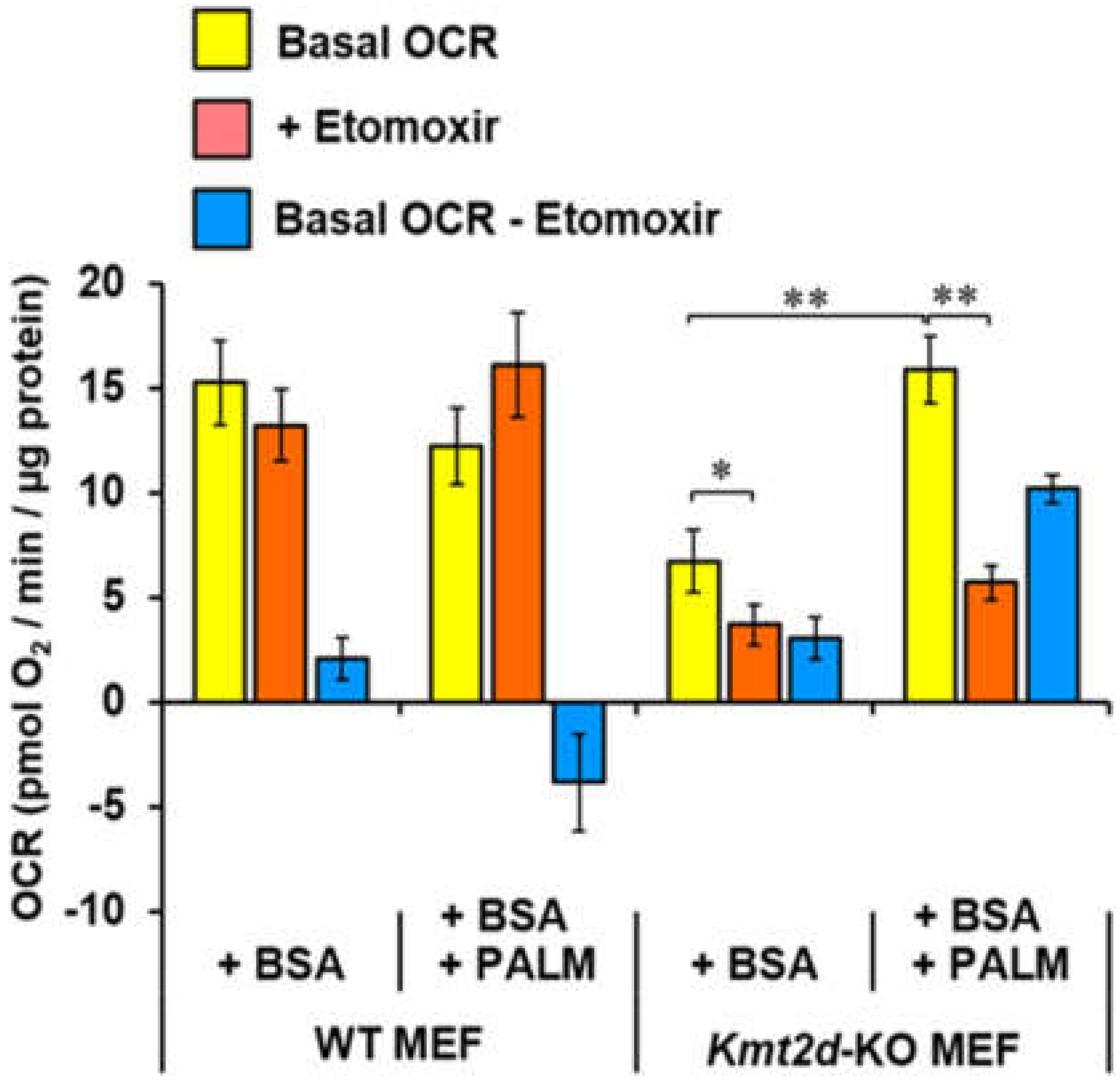
3.4. Loss of Kmt2d Changes the Glucose- and Fatty Acid-Related Metabolic Flexibility
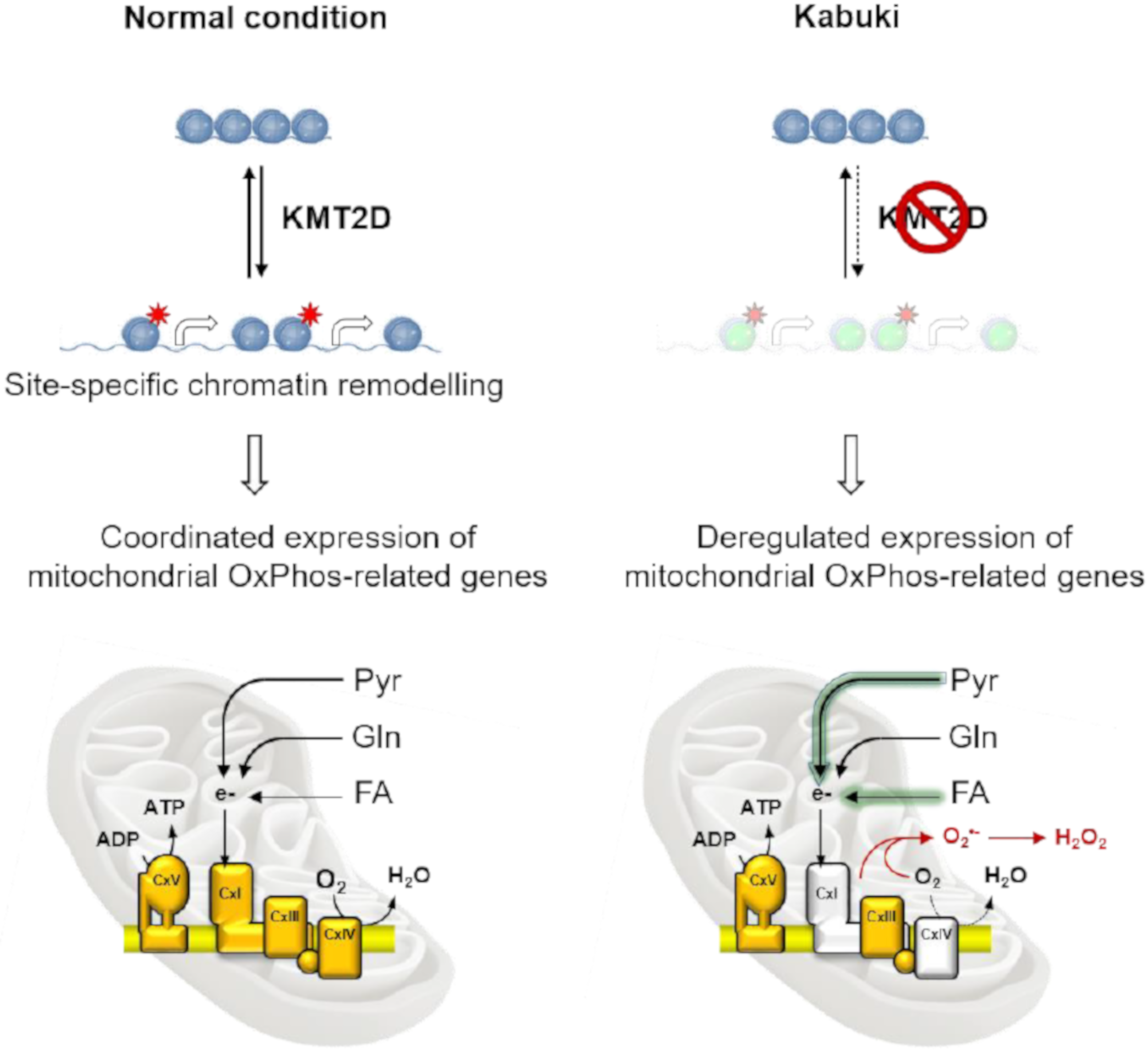
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Reid, M.A.; Dai, Z.; Locasale, J.W. The impact of cellular metabolism on chromatin dynamics and epigenetics. Nat. Cell Biol. 2017, 19, 1298–1306. [Google Scholar] [CrossRef] [PubMed]
- Froimchuk, E.; Jang, Y.; Ge, K. Histone H3 lysine 4 methyltransferase KMT2D. Gene 2017, 627, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Chen, L.H.; Huang, Y.; Chang, C.C.; Wang, P.; Pirozzi, C.J.; Qin, X.; Bao, X.; Greer, P.K.; McLendon, R.E.; et al. KMT2D maintains neoplastic cell proliferation and global histone H3 lysine 4 monomethylation. Oncotarget 2013, 4, 2144–2153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, D.; Gao, X.; Morgan, M.A.; Herz, H.M.; Smith, E.R.; Shilatifard, A. The MLL3/MLL4 branches of the COMPASS family function as major histone H3K4 monomethylases at enhancers. Mol. Cell Biol. 2013, 33, 4745–4754. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.E.; Wang, C.; Xu, S.; Cho, Y.W.; Wang, L.; Feng, X.; Baldridge, A.; Sartorelli, V.; Zhuang, L.; Peng, W.; et al. H3K4 mono- and di-methyltransferase MLL4 is required for enhancer activation during cell differentiation. Elife (Cambridge) 2013, 2, e01503. [Google Scholar]
- Squeo, G.M.; Augello, B.; Massa, V.; Milani, D.; Colombo, E.A.; Mazza, T.; Castellana, S.; Piccione, M.; Maitz, S.; Petracca, A.; et al. Customised next-generation sequencing multigene panel to screen a large cohort of individuals with chromatin-related disorder. J. Med. Genet. 2020. [Google Scholar] [CrossRef]
- Adam, M.P.; Hudgins, L.; Hannibal, M. Kabuki Syndrome. 1993. [Google Scholar]
- Wang, Y.R.; Xu, N.X.; Wang, J.; Wang, X.M. Kabuki syndrome: Review of the clinical features, diagnosis and epigenetic mechanisms. World J. Pediatr. 2019, 15, 528–535. [Google Scholar] [CrossRef]
- Lederer, D.; Grisart, B.; Digilio, M.C.; Benoit, V.; Crespin, M.; Ghariani, S.C.; Maystadt, I.; Dallapiccola, B.; Verellen-Dumoulin, C. Deletion of KDM6A, a Histone Demethylase Interacting with MLL2, in Three Patients with Kabuki Syndrome. Am. J. Hum. Genet. 2012, 90, 119–124. [Google Scholar] [CrossRef] [Green Version]
- Kaikkonen, M.U.; Spann, N.J.; Heinz, S.; Romanoski, C.E.; Allison, K.A.; Stender, J.D.; Chun, H.B.; Tough, D.F.; Prinjha, R.K.; Benner, C.; et al. Remodeling of the enhancer landscape during macrophage activation is coupled to enhancer transcription. Mol. Cell 2013, 51, 310–325. [Google Scholar] [CrossRef] [Green Version]
- Ortega-Molina, A.; Boss, I.W.; Canela, A.; Pan, H.; Jiang, Y.; Zhao, C.; Jiang, M.; Hu, D.; Agirre, X.; Niesvizky, I.; et al. The histone lysine methyltransferase KMT2D sustains a gene expression program that represses B cell lymphoma development. Nat. Med. 2015, 21, 1199–1208. [Google Scholar] [CrossRef]
- Kim, D.H.; Rhee, J.C.; Yeo, S.; Shen, R.; Lee, S.K.; Lee, J.W.; Lee, S. Crucial roles of mixed-lineage leukemia 3 and 4 as epigenetic switches of the hepatic circadian clock controlling bile acid homeostasis in mice. Hepatology 2015, 61, 1012–1023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.H.; Kim, J.; Kwon, J.S.; Sandhu, J.; Tontonoz, P.; Lee, S.K.; Lee, S.; Lee, J.W. Critical Roles of the Histone Methyltransferase MLL4/KMT2D in Murine Hepatic Steatosis Directed by ABL1 and PPARgamma2. Cell Rep. 2016, 17, 1671–1682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matilainen, O.; Quiros, P.M.; Auwerx, J. Mitochondria and Epigenetics - Crosstalk in Homeostasis and Stress. Trends Cell Biol. 2017, 27, 453–463. [Google Scholar] [CrossRef]
- Mora, M.; Angelini, C.; Bignami, F.; Bodin, A.M.; Crimi, M.; Di Donato, J.H.; Felice, A.; Jaeger, C.; Karcagi, V.; LeCam, Y.; et al. The EuroBioBank Network: 10 years of hands-on experience of collaborative, transnational biobanking for rare diseases. Eur. J. Hum. Genet. 2015, 23, 1116–1123. [Google Scholar] [CrossRef] [PubMed]
- Bae, S.; Park, J.; Kim, J.S. Cas-OFFinder: A fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics 2014, 30, 1473–1475. [Google Scholar] [CrossRef] [Green Version]
- McCloskey, D.; Ubhi, B.K. Quantitative and Qualitative Metabolomics for the Investigation of the Intracellular Matabolism. SCIEX TECH Note 2014, 1–11. [Google Scholar]
- Mookerjee, S.A.; Gerencser, A.A.; Nicholls, D.G.; Brand, M.D. Quantifying intracellular rates of glycolytic and oxidative ATP production and consumption using extracellular flux measurements. J. Biol. Chem. 2017, 292, 7189–7207. [Google Scholar] [CrossRef] [Green Version]
- Cela, O.; Piccoli, C.; Scrima, R.; Quarato, G.; Marolla, A.; Cinnella, G.; Dambrosio, M.; Capitanio, N. Bupivacaine uncouples the mitochondrial oxidative phosphorylation, inhibits respiratory chain complexes I and III and enhances ROS production: Results of a study on cell cultures. Mitochondrion 2010, 10, 487–496. [Google Scholar] [CrossRef]
- Pacelli, C.; De Rasmo, D.; Signorile, A.; Grattagliano, I.; di Tullio, G.; D’Orazio, A.; Nico, B.; Comi, G.P.; Ronchi, D.; Ferranini, E.; et al. Mitochondrial defect and PGC-1alpha dysfunction in parkin-associated familial Parkinson’s disease. Biochim. Biophys. Acta 2011, 1812, 1041–1053. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Zhang, Q. Using Seahorse Machine to Measure OCR and ECAR in Cancer Cells. Methods Mol. Biol. 2019, 1928, 353–363. [Google Scholar] [CrossRef]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Zhong, Z.; Xu, Z.; Chen, L.; Wang, Y. 2’,7’-Dichlorodihydrofluorescein as a fluorescent probe for reactive oxygen species measurement: Forty years of application and controversy. Free Radic. Res. 2010, 44, 587–604. [Google Scholar] [CrossRef] [PubMed]
- Schvartzman, J.M.; Thompson, C.B.; Finley, L.W.S. Metabolic regulation of chromatin modifications and gene expression. J. Cell Biol. 2018, 217, 2247–2259. [Google Scholar] [CrossRef] [PubMed]
- Letts, J.A.; Sazanov, L.A. Clarifying the supercomplex: The higher-order organization of the mitochondrial electron transport chain. Nat. Struct Mol. Biol. 2017, 24, 800–808. [Google Scholar] [CrossRef] [PubMed]
- Genova, M.L.; Lenaz, G. The Interplay Between Respiratory Supercomplexes and ROS in Aging. Antioxid. Redox Signal. 2015, 23, 208–238. [Google Scholar] [CrossRef] [PubMed]
- Scarpulla, R.C. Transcriptional paradigms in mammalian mitochondrial biogenesis and function. Physiol. Rev. 2008, 88, 611–638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Handschin, C.; Spiegelman, B.M. Peroxisome proliferator-activated receptor gamma coactivator 1 coactivators, energy homeostasis, and metabolism. Endocr. Rev. 2006, 27, 728–735. [Google Scholar] [CrossRef]
- Wenz, T. Regulation of mitochondrial biogenesis and PGC-1alpha under cellular stress. Mitochondrion 2013, 13, 134–142. [Google Scholar] [CrossRef]
- Fan, W.; Evans, R. PPARs and ERRs: Molecular mediators of mitochondrial metabolism. Curr. Opin. Cell Biol. 2015, 33, 49–54. [Google Scholar] [CrossRef] [Green Version]
- Suthammarak, W.; Yang, Y.Y.; Morgan, P.G.; Sedensky, M.M. Complex I function is defective in complex IV-deficient Caenorhabditis elegans. J. Biol. Chem. 2009, 284, 6425–6435. [Google Scholar] [CrossRef] [Green Version]
- Protasoni, M.; Perez-Perez, R.; Lobo-Jarne, T.; Harbour, M.E.; Ding, S.; Penas, A.; Diaz, F.; Moraes, C.T.; Fearnley, I.M.; Zeviani, M.; et al. Respiratory supercomplexes act as a platform for complex III-mediated maturation of human mitochondrial complexes I and IV. EMBO J. 2020, 39, e102817. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Saha, P.K.; Yang, Q.H.; Lee, S.; Park, J.Y.; Suh, Y.; Lee, S.K.; Chan, L.; Roeder, R.G.; Lee, J.W. Targeted inactivation of MLL3 histone H3-Lys-4 methyltransferase activity in the mouse reveals vital roles for MLL3 in adipogenesis. Proc. Natl. Acad. Sci. USA 2008, 105, 19229–19234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, C.; Chang, C.C.; Wortham, M.; Chen, L.H.; Kernagis, D.N.; Qin, X.; Cho, Y.W.; Chi, J.T.; Grant, G.A.; McLendon, R.E.; et al. Global identification of MLL2-targeted loci reveals MLL2’s role in diverse signaling pathways. Proc. Natl. Acad. Sci. USA 2012, 109, 17603–17608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.H.; Zhuang, J.; Li, J.; Hwang, P.M. p53 as guardian of the mitochondrial genome. FEBS Lett. 2016, 590, 924–934. [Google Scholar] [CrossRef]
- Park, J.Y.; Wang, P.Y.; Matsumoto, T.; Sung, H.J.; Ma, W.; Choi, J.W.; Anderson, S.A.; Leary, S.C.; Balaban, R.S.; Kang, J.G.; et al. p53 improves aerobic exercise capacity and augments skeletal muscle mitochondrial DNA content. Circ. Res. 2009, 105, 705–712. [Google Scholar] [CrossRef]
- Matoba, S.; Kang, J.G.; Patino, W.D.; Wragg, A.; Boehm, M.; Gavrilova, O.; Hurley, P.J.; Bunz, F.; Hwang, P.M. p53 regulates mitochondrial respiration. Science 2006, 312, 1650–1653. [Google Scholar] [CrossRef]
- Cela, O.; Scrima, R.; Pazienza, V.; Merla, G.; Benegiamo, G.; Augello, B.; Fugetto, S.; Menga, M.; Rubino, R.; Fuhr, L.; et al. Clock genes-dependent acetylation of complex I sets rhythmic activity of mitochondrial OxPhos. Biochim Biophys Acta 2016, 1863, 596–606. [Google Scholar] [CrossRef]
- Scrima, R.; Cela, O.; Merla, G.; Augello, B.; Rubino, R.; Quarato, G.; Fugetto, S.; Menga, M.; Fuhr, L.; Relogio, A.; et al. Clock-genes and mitochondrial respiratory activity: Evidence of a reciprocal interplay. Biochim Biophys Acta 2016, 1857, 1344–1351. [Google Scholar] [CrossRef]
- Koutsioumpa, M.; Hatziapostolou, M.; Polytarchou, C.; Tolosa, E.J.; Almada, L.L.; Mahurkar-Joshi, S.; Williams, J.; Tirado-Rodriguez, A.B.; Huerta-Yepez, S.; Karavias, D.; et al. Lysine methyltransferase 2D regulates pancreatic carcinogenesis through metabolic reprogramming. Gut 2019, 68, 1271–1286. [Google Scholar] [CrossRef]
- Jiang, H.; Shukla, A.; Wang, X.; Chen, W.Y.; Bernstein, B.E.; Roeder, R.G. Role for Dpy-30 in ES cell-fate specification by regulation of H3K4 methylation within bivalent domains. Cell 2011, 144, 513–525. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Augustin, J.; Hu, J.; Jiang, H. Physical Interactions and Functional Coordination between the Core Subunits of Set1/Mll Complexes and the Reprogramming Factors. PLoS ONE 2015, 10, e0145336. [Google Scholar] [CrossRef]
- Yang, Z.; Shah, K.; Khodadadi-Jamayran, A.; Jiang, H. Control of Hematopoietic Stem and Progenitor Cell Function through Epigenetic Regulation of Energy Metabolism and Genome Integrity. Stem Cell Reports 2019, 13, 61–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carosso, G.A.; Boukas, L.; Augustin, J.J.; Nguyen, H.N.; Winer, B.L.; Cannon, G.H.; Robertson, J.D.; Zhang, L.; Hansen, K.D.; Goff, L.A.; et al. Precocious neuronal differentiation and disrupted oxygen responses in Kabuki syndrome. JCI Insight 2019, 4. [Google Scholar]
- Benjamin, J.S.; Pilarowski, G.O.; Carosso, G.A.; Zhang, L.; Huso, D.L.; Goff, L.A.; Vernon, H.J.; Hansen, K.D.; Bjornsson, H.T. A ketogenic diet rescues hippocampal memory defects in a mouse model of Kabuki syndrome. Proc. Natl. Acad. Sci. USA 2017, 114, 125–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]









© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pacelli, C.; Adipietro, I.; Malerba, N.; Squeo, G.M.; Piccoli, C.; Amoresano, A.; Pinto, G.; Pucci, P.; Lee, J.-E.; Ge, K.; et al. Loss of Function of the Gene Encoding the Histone Methyltransferase KMT2D Leads to Deregulation of Mitochondrial Respiration. Cells 2020, 9, 1685. https://doi.org/10.3390/cells9071685
Pacelli C, Adipietro I, Malerba N, Squeo GM, Piccoli C, Amoresano A, Pinto G, Pucci P, Lee J-E, Ge K, et al. Loss of Function of the Gene Encoding the Histone Methyltransferase KMT2D Leads to Deregulation of Mitochondrial Respiration. Cells. 2020; 9(7):1685. https://doi.org/10.3390/cells9071685
Chicago/Turabian StylePacelli, Consiglia, Iolanda Adipietro, Natascia Malerba, Gabriella Maria Squeo, Claudia Piccoli, Angela Amoresano, Gabriella Pinto, Pietro Pucci, Ji-Eun Lee, Kai Ge, and et al. 2020. "Loss of Function of the Gene Encoding the Histone Methyltransferase KMT2D Leads to Deregulation of Mitochondrial Respiration" Cells 9, no. 7: 1685. https://doi.org/10.3390/cells9071685