Dampened Slow Oscillation Connectivity Anticipates Amyloid Deposition in the PS2APP Mouse Model of Alzheimer’s Disease

 ,
, 
 and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Animal Preparation and Surgery
2.3. Signal Acquisition
2.4. Data Processing and Analysis
2.5. Statistical Analyses
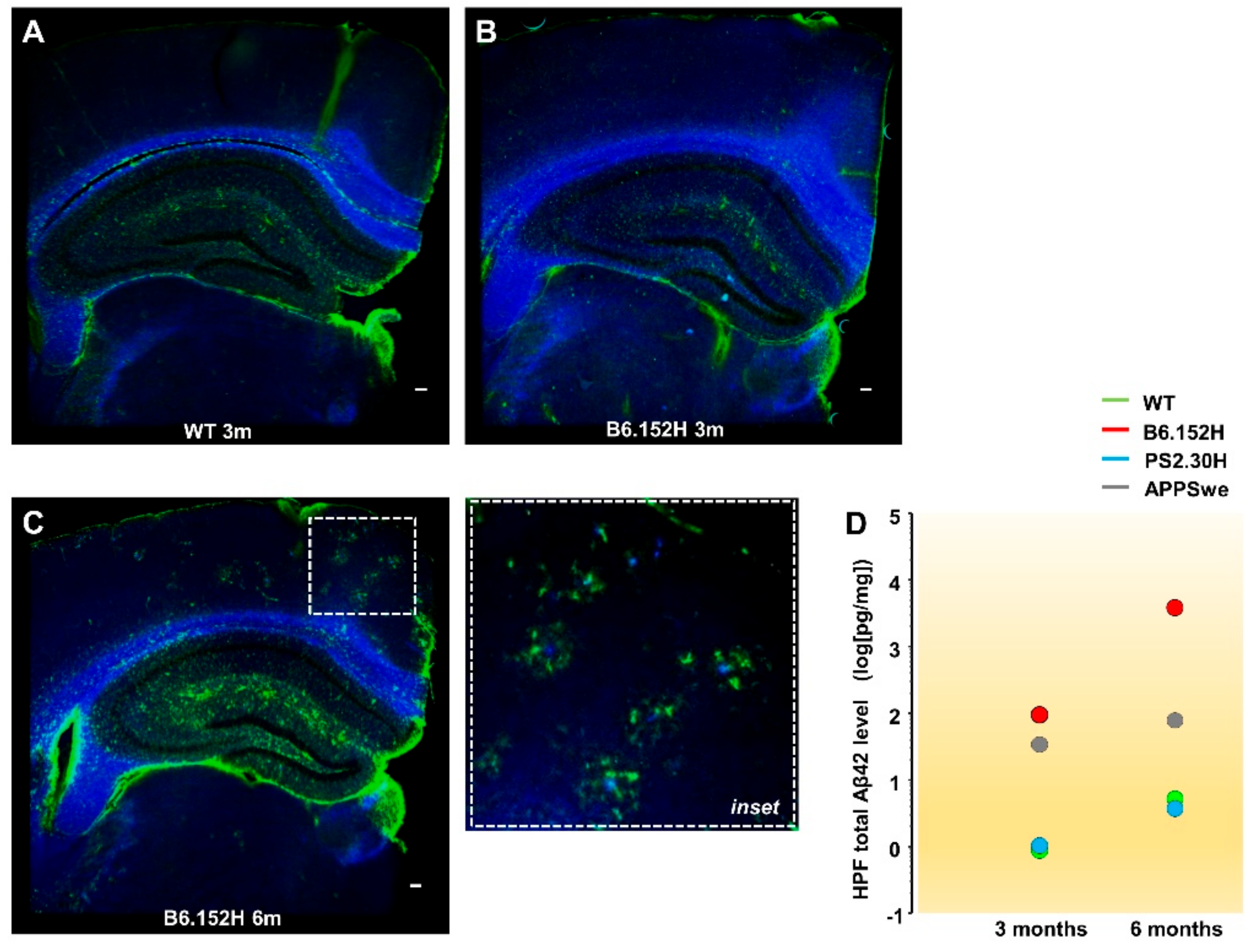
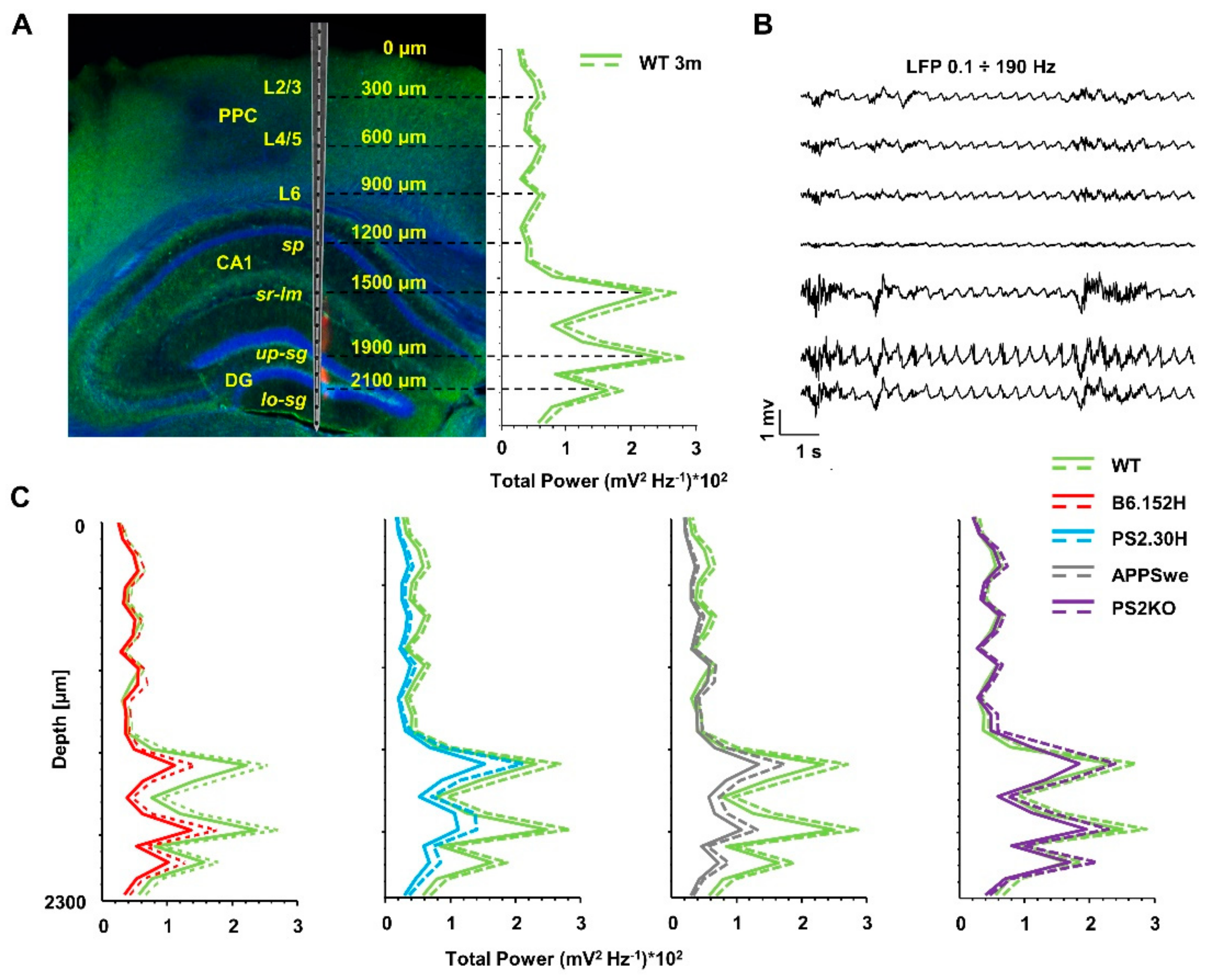
3. Results
3.1. In Vivo Multi-Site LFP Recordings from WT and Tg Mice
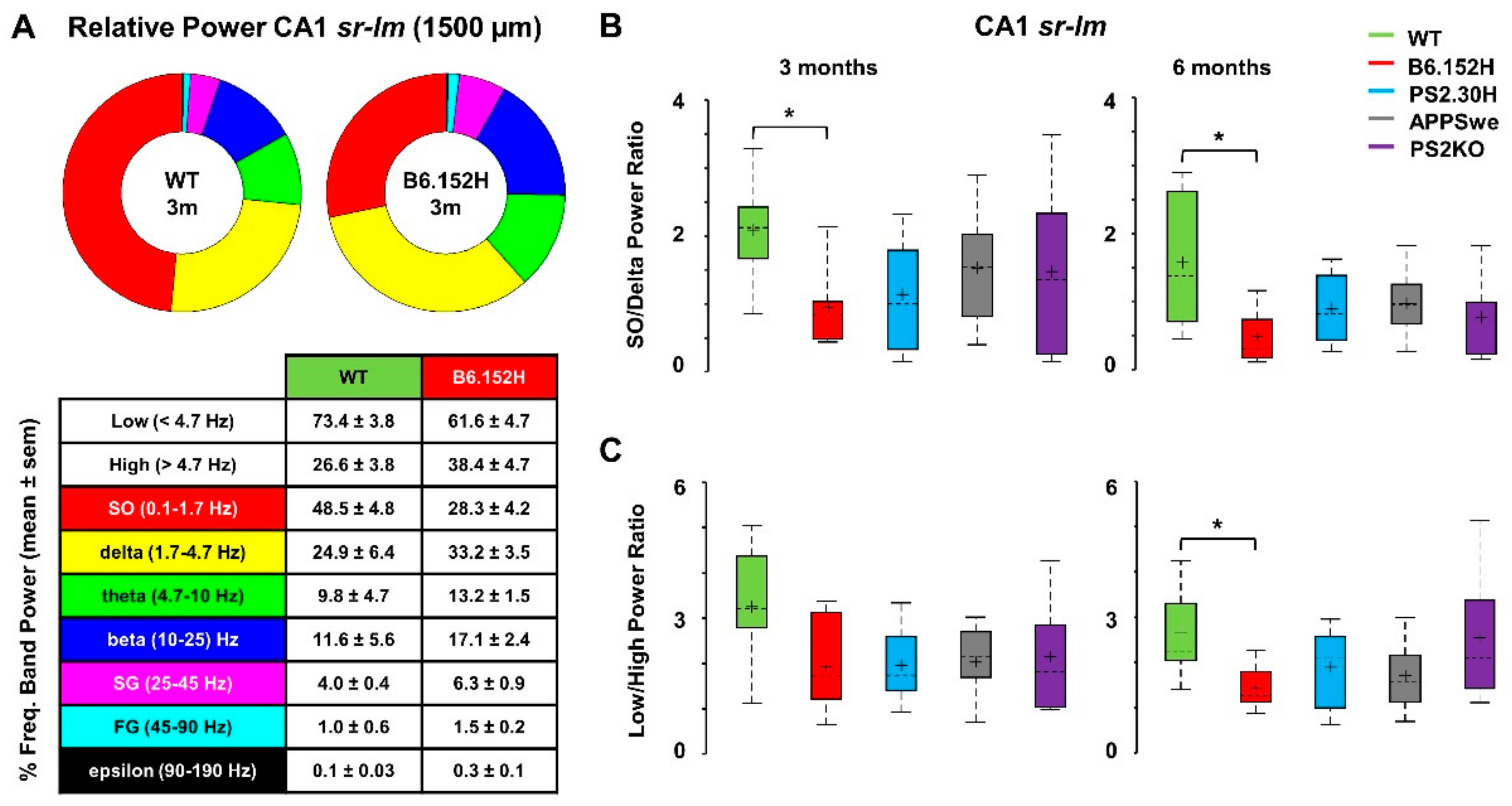
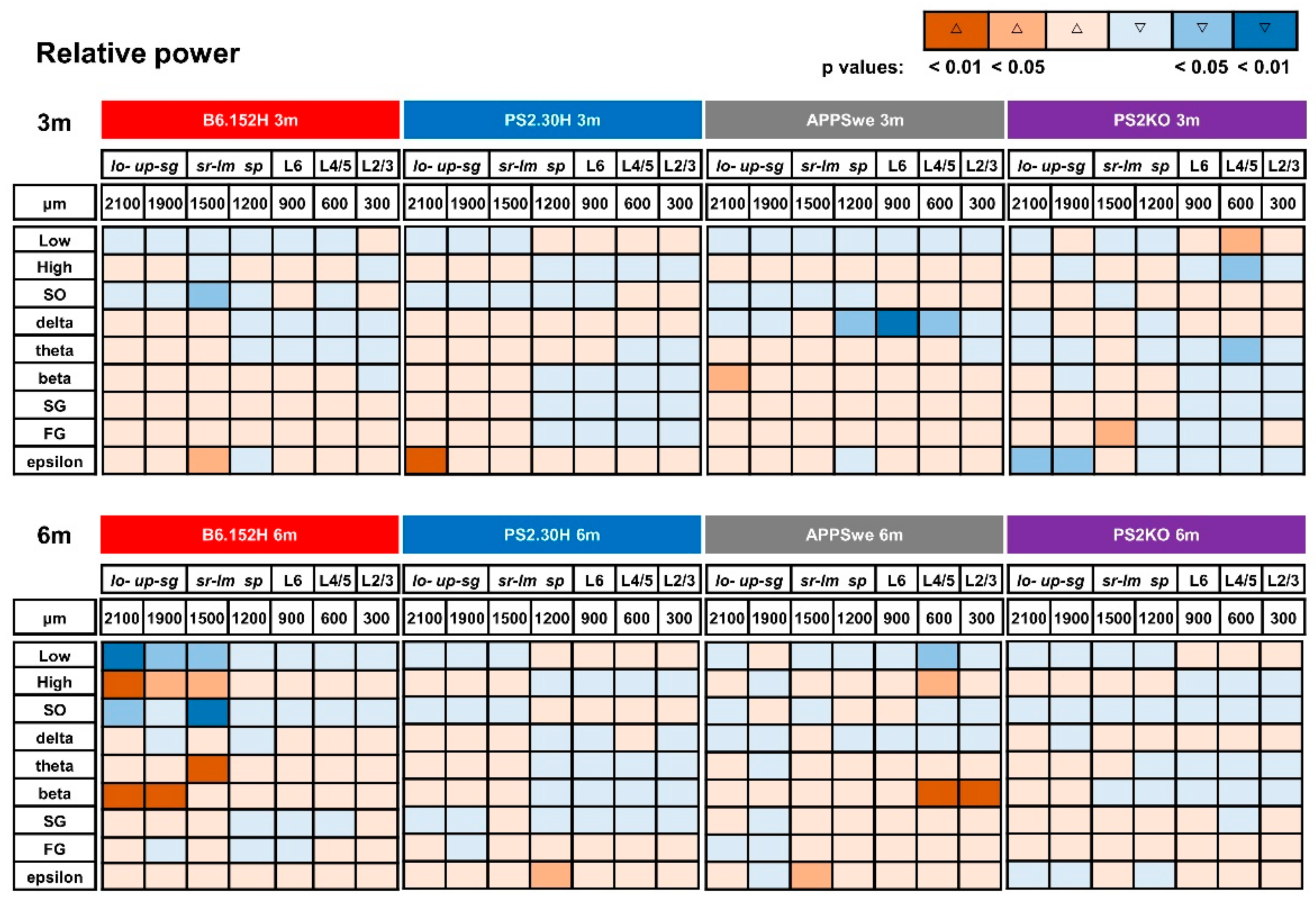
3.2. Power Imbalances Characterize B6.152H Mice
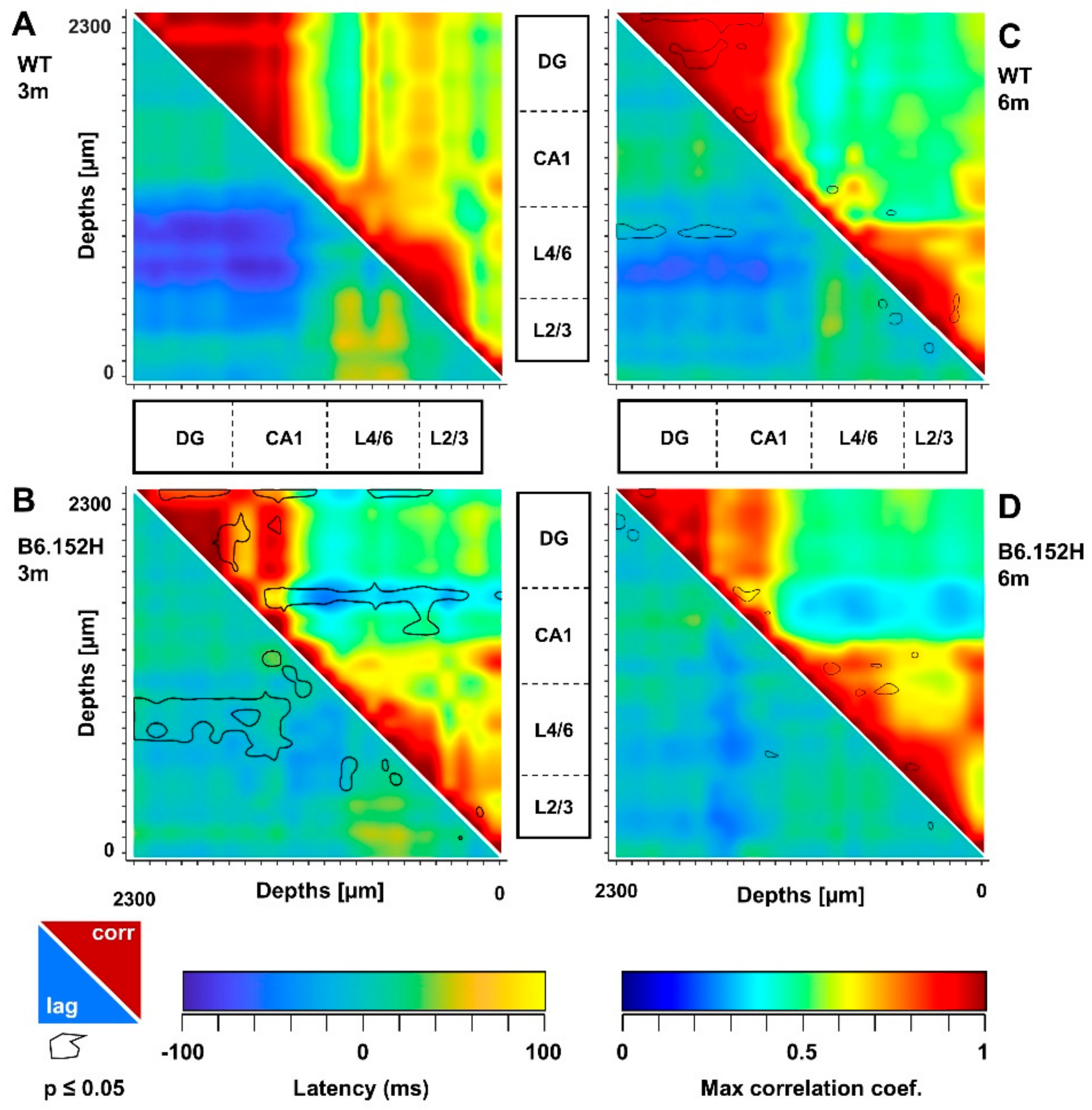
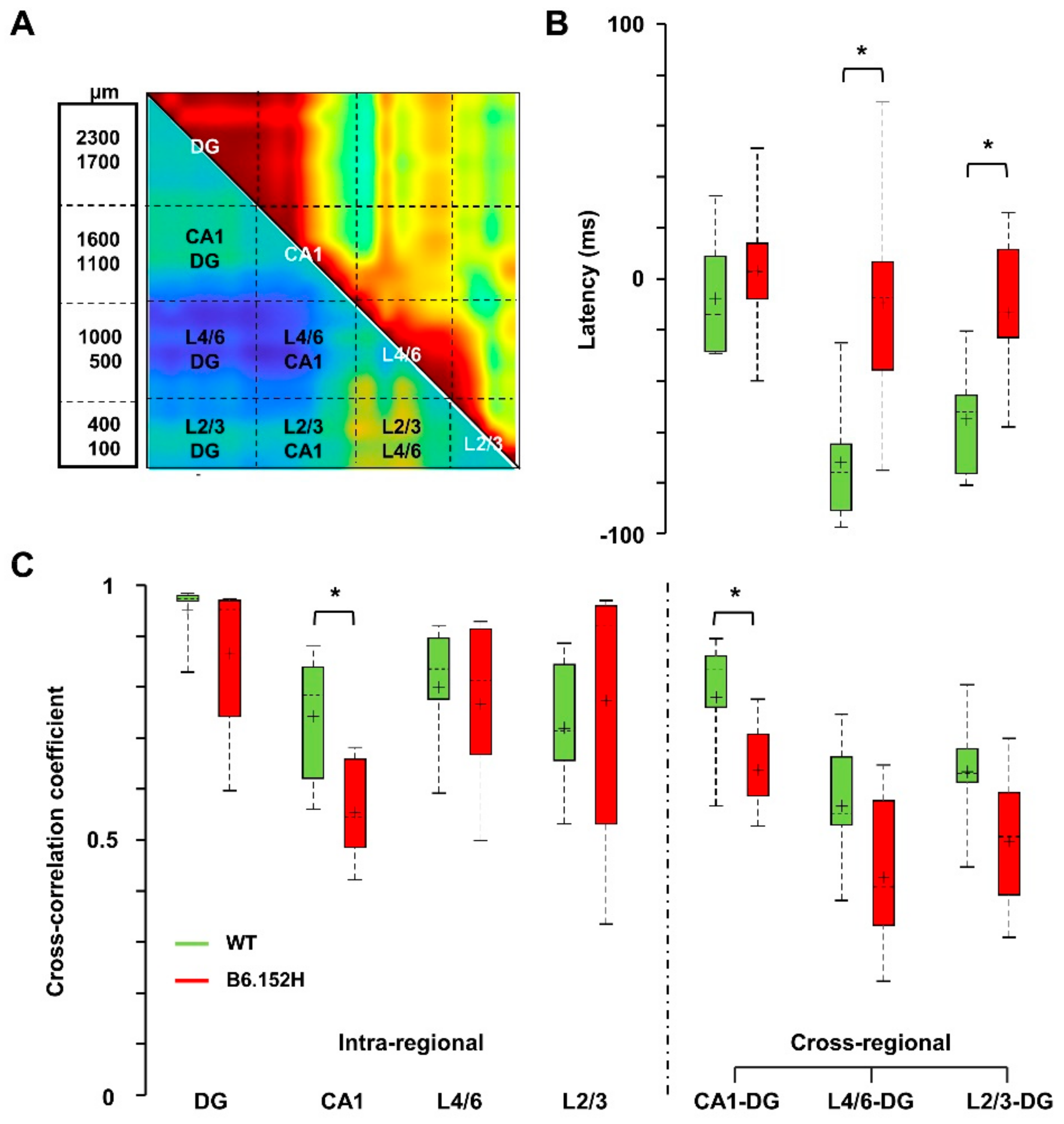
3.3. Altered SO Functional Connectivity in Predeposition B6.152H Mice
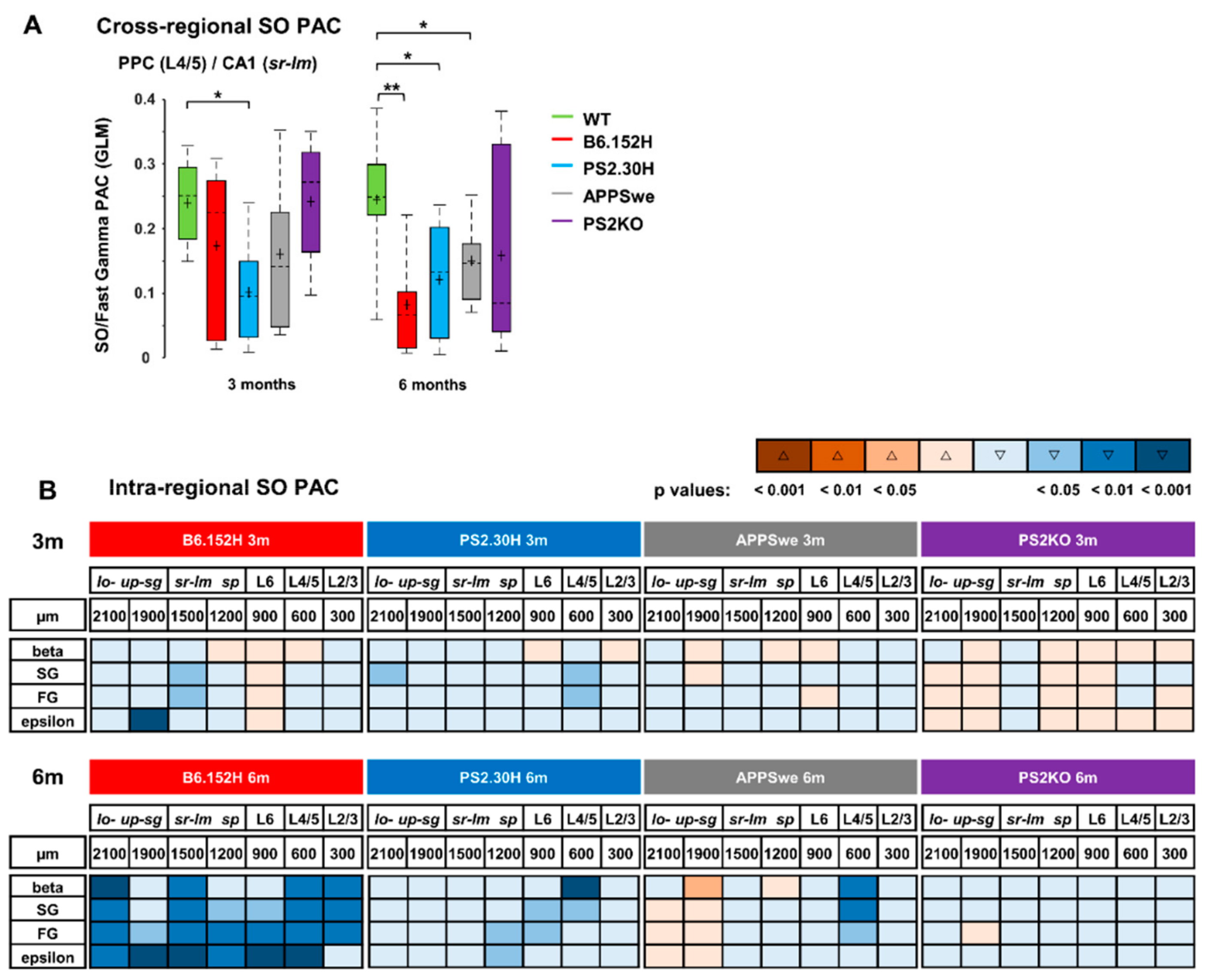
3.4. Loss of Cortico-Hippocampal Phase-Amplitude Coupling in PS2-Based AD Mice
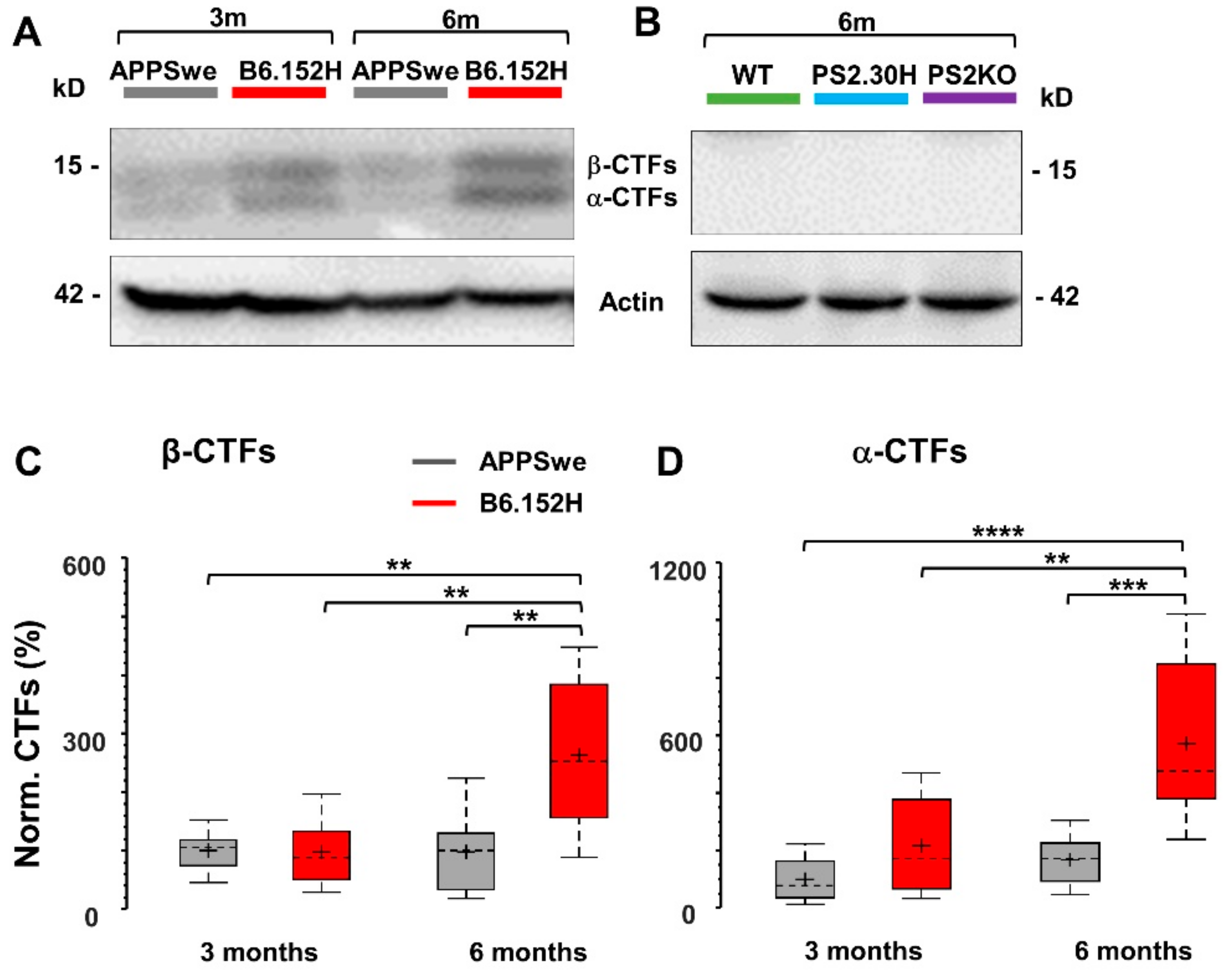
3.5. α- and β-APP Carboxy-Terminal Fragments are Not Involved in Reduced SO Connectivity
4. Discussion
4.1. Loss of SO Connectivity Distinguishes Predeposition B6.152H Mice
4.2. Expression of the PS2 Mutation Causes Loss of SO PAC Independently of Amyloidosis
4.3. Amyloidosis Worsens the Loss of SO PAC
4.4. Low/High Frequency Power Imbalances Mark the Progression of Amyloidosis in B6.152H Mice
4.5. Molecular and Cellular Pathways Involved in SO Changes
4.6. Study Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- McDade, E.; Bateman, R.J. Stop Alzheimer’s before it starts. Nature 2017, 547, 153–155. [Google Scholar] [CrossRef] [PubMed]
- Götz, J.; Bodea, L.G.; Goedert, M. Rodent models for Alzheimer disease. Nat. Rev. Neurosci. 2018, 19, 583–598. [Google Scholar] [CrossRef] [PubMed]
- Puzzo, D.; Gulisano, W.; Palmeri, A.; Arancio, O. Rodent models for Alzheimer’s disease drug discovery. Expert Opin. Drug Discov. 2015, 10, 703–711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Pascoal, T.A.; Mathotaarachchi, S.; Kang, M.S.; Mohaddes, S.; Shin, M.; Park, A.Y.; Parent, M.J.; Benedet, A.L.; Chamoun, M.; Therriault, J.; et al. Abeta-induced vulnerability propagates via the brain’s default mode network. Nat. Commun. 2019, 10, 2353. [Google Scholar] [CrossRef]
- Harris, J.A.; Devidze, N.; Verret, L.; Ho, K.; Halabisky, B.; Thwin, M.T.; Kim, D.; Hamto, P.; Lo, I.; Yu, G.-Q.; et al. Transsynaptic Progression of Amyloid-β-Induced Neuronal Dysfunction within the Entorhinal-Hippocampal Network. Neuron 2010, 68, 428–441. [Google Scholar] [CrossRef] [Green Version]
- Jacobs, H.I.L.; Hedden, T.; Schultz, A.P.; Sepulcre, J.; Perea, R.D.; Amariglio, R.E.; Papp, K.V.; Rentz, D.M.; Sperling, R.A.; Johnson, K.A. Structural tract alterations predict downstream tau accumulation in amyloid-positive older individuals. Nat. Neurosci. 2018, 21, 424–431. [Google Scholar] [CrossRef]
- Khan, U.A.; Liu, L.; Provenzano, F.A.; Berman, D.E.; Profaci, C.P.; Sloan, R.; Mayeux, R.; Duff, K.E.; Small, S.A. Molecular drivers and cortical spread of lateral entorhinal cortex dysfunction in preclinical Alzheimer’s disease. Nat. Neurosci. 2014, 17, 304–311. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.-H.; Siddoway, B.; Kaeser, G.E.; Segota, I.; Rivera, R.; Romanow, W.J.; Liu, C.S.; Park, C.; Kennedy, G.; Long, T.; et al. Somatic APP gene recombination in Alzheimer’s disease and normal neurons. Nature 2018, 563, 639–645. [Google Scholar] [CrossRef]
- Minkeviciene, R.; Rheims, S.; Dobszay, M.B.; Zilberter, M.; Hartikainen, J.; Fulop, L.; Penke, B.; Zilberter, Y.; Harkany, T.; Pitkanen, A.; et al. Amyloid -Induced Neuronal Hyperexcitability Triggers Progressive Epilepsy. J. Neurosci. 2009, 29, 3453–3462. [Google Scholar] [CrossRef]
- Busche, M.A.; Chen, X.; Henning, H.A.; Reichwald, J.; Staufenbiel, M.; Sakmann, B.; Konnerth, A. Critical role of soluble amyloid-β for early hippocampal hyperactivity in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2012, 109, 8740–8745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fontana, R.; Agostini, M.; Murana, E.; Mahmud, M.; Scremin, E.; Rubega, M.; Sparacino, G.; Vassanelli, S.; Fasolato, C. Early hippocampal hyperexcitability in PS2APP mice: Role of mutant PS2 and APP. Neurobiol. Aging 2017, 50, 64–76. [Google Scholar] [CrossRef] [PubMed]
- Ittner, A.A.; Gladbach, A.; Bertz, J.; Suh, L.S.; Ittner, L.M. p38 MAP kinase-mediated NMDA receptor-dependent suppression of hippocampal hypersynchronicity in a mouse model of Alzheimer’s disease. Acta Neuropathol. Commun. 2014, 2, 149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez, P.E.; Zhu, L.; Verret, L.; Vossel, K.A.; Orr, A.G.; Cirrito, J.R.; Devidze, N.; Ho, K.; Yu, G.-Q.; Palop, J.J.; et al. Levetiracetam suppresses neuronal network dysfunction and reverses synaptic and cognitive deficits in an Alzheimer’s disease model. Proc. Natl. Acad. Sci. USA 2012, 109, E2895–E2903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verret, L.; Mann, E.O.; Hang, G.B.; Barth, A.M.I.; Cobos, I.; Ho, K.; Devidze, N.; Masliah, E.; Kreitzer, A.C.; Mody, I.; et al. Inhibitory Interneuron Deficit Links Altered Network Activity and Cognitive Dysfunction in Alzheimer Model. Cell 2012, 149, 708–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palop, J.J.; Mucke, L. Epilepsy and cognitive impairments in Alzheimer disease. Arch. Neurol. 2009, 66, 435–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palop, J.J.; Mucke, L. Network abnormalities and interneuron dysfunction in Alzheimer disease. Nat. Rev. Neurosci. 2016, 17, 777–792. [Google Scholar] [CrossRef]
- Bakker, A.; Albert, M.S.; Krauss, G.; Speck, C.L.; Gallagher, M. Response of the medial temporal lobe network in amnestic mild cognitive impairment to therapeutic intervention assessed by fMRI and memory task performance. NeuroImage Clin. 2015, 7, 688–698. [Google Scholar] [CrossRef] [Green Version]
- Lam, A.D.; Deck, G.; Goldman, A.; Eskandar, E.N.; Noebels, J.; Cole, A.J. Silent hippocampal seizures and spikes identified by foramen ovale electrodes in Alzheimer’s disease. Nat. Med. 2017, 23, 678–680. [Google Scholar] [CrossRef]
- Rutishauser, U.; Ross, I.B.; Mamelak, A.N.; Schuman, E.M. Human memory strength is predicted by theta-frequency phase-locking of single neurons. Nature 2010, 464, 903–907. [Google Scholar] [CrossRef] [Green Version]
- Goutagny, R.; Gu, N.; Cavanagh, C.; Jackson, J.; Chabot, J.G.; Quirion, R.; Krantic, S.; Williams, S. Alterations in hippocampal network oscillations and theta-gamma coupling arise before Aβ overproduction in a mouse model of Alzheimer’s disease. Eur. J. Neurosci. 2013, 37, 1896–1902. [Google Scholar] [CrossRef] [PubMed]
- Mitra, A.; Snyder, A.Z.; Hacker, C.D.; Pahwa, M.; Tagliazucchi, E.; Laufs, H.; Leuthardt, E.C.; Raichle, M.E. Human cortical–hippocampal dialogue in wake and slow-wave sleep. Proc. Natl. Acad. Sci. USA 2016, 113, E6868–E6876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Busche, M.A.; Kekuš, M.; Adelsberger, H.; Noda, T.; Förstl, H.; Nelken, I.; Konnerth, A. Rescue of long-range circuit dysfunction in Alzheimer’s disease models. Nat. Neurosci. 2015, 18, 1623–1630. [Google Scholar] [CrossRef] [PubMed]
- Kastanenka, K.V.; Hou, S.S.; Shakerdge, N.; Logan, R.; Feng, D.; Wegmann, S.; Chopra, V.; Hawkes, J.M.; Chen, X.; Bacskai, B.J. Optogenetic Restoration of Disrupted Slow Oscillations Halts Amyloid Deposition and Restores Calcium Homeostasis in an Animal Model of Alzheimer’s Disease. PLoS ONE 2017, 12, e0170275. [Google Scholar] [CrossRef]
- Ladenbauer, J.; Ladenbauer, J.; Külzow, N.; de Boor, R.; Avramova, E.; Grittner, U.; Flöel, A. Promoting Sleep Oscillations and Their Functional Coupling by Transcranial Stimulation Enhances Memory Consolidation in Mild Cognitive Impairment. J. Neurosci. 2017, 37, 7111–7124. [Google Scholar] [CrossRef] [Green Version]
- Ozmen, L.; Albientz, A.; Czech, C.; Jacobsen, H. Expression of Transgenic APP mRNA Is the Key Determinant for Beta-Amyloid Deposition in PS2APP Transgenic Mice. Neurodegener. Dis. 2009, 6, 29–36. [Google Scholar] [CrossRef]
- Richards, J.G.; Higgins, G.A.; Ouagazzal, A.-M.; Ozmen, L.; Kew, J.N.C.; Bohrmann, B.; Malherbe, P.; Brockhaus, M.; Loetscher, H.; Czech, C.; et al. PS2APP transgenic mice, coexpressing hPS2mut and hAPPswe, show age-related cognitive deficits associated with discrete brain amyloid deposition and inflammation. J. Neurosci. 2003, 23, 8989–9003. [Google Scholar] [CrossRef] [Green Version]
- Brendel, M.; Probst, F.; Jaworska, A.; Overhoff, F.; Korzhova, V.; Albert, N.L.; Beck, R.; Lindner, S.; Gildehaus, F.-J.; Baumann, K.; et al. Glial Activation and Glucose Metabolism in a Transgenic Amyloid Mouse Model: A Triple-Tracer PET Study. J. Nucl. Med. 2016, 57, 954–960. [Google Scholar] [CrossRef] [Green Version]
- Brendel, M.; Kleinberger, G.; Probst, F.; Jaworska, A.; Overhoff, F.; Blume, T.; Albert, N.L.; Carlsen, J.; Lindner, S.; Gildehaus, F.J.; et al. Increase of TREM2 during aging of an Alzheimer’s disease mouse model is paralleled by microglial activation and amyloidosis. Front. Aging Neurosci. 2017, 9, 8. [Google Scholar] [CrossRef] [Green Version]
- Weidensteiner, C.; Metzger, F.; Bruns, A.; Bohrmann, B.; Kuennecke, B.; Von Kienlin, M. Cortical hypoperfusion in the B6.PS2APP mouse model for Alzheimer’s disease: Comprehensive phenotyping of vascular and tissular parameters by MRI. Magn. Reson. Med. 2009, 62, 35–45. [Google Scholar] [CrossRef]
- Hanson, J.E.; Pare, J.F.; Deng, L.; Smith, Y.; Zhou, Q. Altered GluN2B NMDA receptor function and synaptic plasticity during early pathology in the PS2APP mouse model of Alzheimer’s disease. Neurobiol. Dis. 2015, 74, 254–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kipanyula, M.J.; Contreras, L.; Zampese, E.; Lazzari, C.; Wong, A.K.C.; Pizzo, P.; Fasolato, C.; Pozzan, T. Ca2+ dysregulation in neurons from transgenic mice expressing mutant presenilin 2. Aging Cell 2012, 11, 885–893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herreman, A.; Schrijvers, V.; Van Leuven, F.; Serneels, L.; Baekelandt, V.; Zwijsen, A. Presenilin 2 deficiency causes a mild pulmonary phenotype and no changes in amyloid precursor protein processing but enhances the embryonic lethal phenotype of presenilin 1 deficiency. Proc. Natl. Acad. Sci. USA 2002, 96, 11872–11877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, A.; Shen, J. Presenilins in synaptic function and disease. Trends Mol. Med. 2011, 17, 617–624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahmud, M.; Pasqualotto, E.; Bertoldo, A.; Girardi, S.; Maschietto, M.; Vassanelli, S. An automated method for detection of layer activation order in information processing pathway of rat barrel cortex under mechanical whisker stimulation. J. Neurosci. Methods 2011, 196, 141–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.; Yang, S.; Hu, Z.Y.; Liu, G.; Zhou, W.X.; Zhang, Y.X. A new approach to location of the dentate gyrus and perforant path in rats/mice by landmarks on the skull. Acta Neurobiol. Exp. 2012, 72, 468–472. [Google Scholar]
- Wolansky, T.; Clement, E.A.; Peters, S.R.; Palczak, M.A.; Dickson, C.T. Hippocampal Slow Oscillation: A Novel EEG State and Its Coordination with Ongoing Neocortical Activity. J. Neurosci. 2006, 26, 6213–6229. [Google Scholar] [CrossRef] [Green Version]
- Batterink, L.J.; Creery, J.D.; Paller, K.A. Phase of Spontaneous Slow Oscillations during Sleep Influences Memory-Related Processing of Auditory Cues. J. Neurosci. 2016, 36, 1401–1409. [Google Scholar] [CrossRef]
- Born, J. Slow-wave sleep and the consolidation of long-term memory. World J. Biol. Psychiatry 2010, 11, 16–21. [Google Scholar] [CrossRef]
- Volgushev, M. Precise Long-Range Synchronization of Activity and Silence in Neocortical Neurons during Slow-Wave Sleep. J. Neurosci. 2006, 26, 5665–5672. [Google Scholar] [CrossRef] [Green Version]
- Pagliardini, S.; Gosgnach, S.; Dickson, C.T. Spontaneous Sleep-Like Brain State Alternations and Breathing Characteristics in Urethane Anesthetized Mice. PLoS ONE 2013, 8, e70411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Ikonen, S.; Gurevicius, K.; van Groen, T.; Tanila, H. Alteration of cortical EEG in mice carrying mutated human APP transgene. Brain Res. 2002, 943, 181–190. [Google Scholar] [CrossRef]
- Adhikari, A.; Sigurdsson, T.; Topiwala, M.A.; Gordon, J.A. Cross-correlation of instantaneous amplitudes of field potential oscillations: A straightforward method to estimate the directionality and lag between brain areas. J. Neurosci. Methods 2010, 191, 191–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aru, J.J.; Aru, J.J.; Priesemann, V.; Wibral, M.; Lana, L.; Pipa, G.; Singer, W.; Vicente, R. Untangling cross-frequency coupling in neuroscience. Curr. Opin. Neurobiol. 2015, 31, 51–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hyafil, A.; Giraud, A.L.; Fontolan, L.; Gutkin, B. Neural Cross-Frequency Coupling: Connecting Architectures, Mechanisms, and Functions. Trends Neurosci. 2015, 38, 725–740. [Google Scholar] [CrossRef] [PubMed]
- Lega, B.; Burke, J.; Jacobs, J.; Kahana, M.J. Slow-Theta-to-Gamma Phase-Amplitude Coupling in Human Hippocampus Supports the Formation of New Episodic Memories. Cereb. Cortex 2016, 26, 268–278. [Google Scholar] [CrossRef] [Green Version]
- Born, H.A.; Kim, J.-Y.; Savjani, R.R.; Das, P.; Dabaghian, Y.A.; Guo, Q.; Yoo, J.W.; Schuler, D.R.; Cirrito, J.R.; Zheng, H.; et al. Genetic Suppression of Transgenic APP Rescues Hypersynchronous Network Activity in a Mouse Model of Alzeimer’s Disease. J. Neurosci. 2014, 34, 3826–3840. [Google Scholar] [CrossRef]
- Hamm, V.; Héraud, C.; Bott, J.-B.; Herbeaux, K.; Strittmatter, C.; Mathis, C.; Goutagny, R. Differential contribution of APP metabolites to early cognitive deficits in a TgCRND8 mouse model of Alzheimer’s disease. Sci. Adv. 2017, 3, e1601068. [Google Scholar] [CrossRef] [Green Version]
- Mondragón-Rodríguez, S.; Gu, N.; Manseau, F.; Williams, S. Alzheimer’s Transgenic Model Is Characterized by Very Early Brain Network Alterations and β-CTF Fragment Accumulation: Reversal by β-Secretase Inhibition. Front. Cell. Neurosci. 2018, 12, 121. [Google Scholar] [CrossRef]
- Xu, W.; Fitzgerald, S.; Nixon, R.A.; Levy, E.; Wilson, D.A. Early hyperactivity in lateral entorhinal cortex is associated with elevated levels of AβPP metabolites in the Tg2576 mouse model of Alzheimer’s disease. Exp. Neurol. 2015, 264, 82–91. [Google Scholar] [CrossRef] [Green Version]
- Brendel, M.; Jaworska, A.; Grießinger, E.; Rötzer, C.; Burgold, S.; Gildehaus, F.-J.; Carlsen, J.; Cumming, P.; Baumann, K.; Haass, C.; et al. Cross-Sectional Comparison of Small Animal [18F]-Florbetaben Amyloid-PET between Transgenic AD Mouse Models. PLoS ONE 2015, 10, e0116678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grueninger, F.; Bohrmann, B.; Czech, C.; Ballard, T.M.; Frey, J.R.; Weidensteiner, C.; von Kienlin, M.; Ozmen, L. Phosphorylation of Tau at S422 is enhanced by Aβ in TauPS2APP triple transgenic mice. Neurobiol. Dis. 2010, 37, 294–306. [Google Scholar] [CrossRef] [PubMed]
- Mander, B.A.; Marks, S.M.; Vogel, J.W.; Rao, V.; Lu, B.; Saletin, J.M.; Ancoli-Israel, S.; Jagust, W.J.; Walker, M.P. β-amyloid disrupts human NREM slow waves and related hippocampus-dependent memory consolidation. Nat. Neurosci. 2015, 18, 1051–1057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Gulati, T.; Ganguly, K. Competing Roles of Slow Oscillations and Delta Waves in Memory Consolidation versus Forgetting. Cell 2019, 179, 514–526.e13. [Google Scholar] [CrossRef] [PubMed]
- Helfrich, R.F.; Mander, B.A.; Jagust, W.J.; Knight, R.T.; Walker, M.P. Old Brains Come Uncoupled in Sleep: Slow Wave-Spindle Synchrony, Brain Atrophy, and Forgetting. Neuron 2018, 97, 221–230. [Google Scholar] [CrossRef] [Green Version]
- Poskanzer, K.E.; Yuste, R. Astrocytic regulation of cortical UP states. Proc. Natl. Acad. Sci. USA 2011, 108, 18453–18458. [Google Scholar] [CrossRef] [Green Version]
- Fellin, T.; Pascual, O.; Gobbo, S.; Pozzan, T.; Haydon, P.G.; Carmignoto, G. Neuronal Synchrony Mediated by Astrocytic Glutamate through Activation of Extrasynaptic NMDA Receptors. Neuron 2004, 43, 729–743. [Google Scholar] [CrossRef] [Green Version]
- Fellin, T.; Halassa, M.M.; Terunuma, M.; Succol, F.; Takano, H.; Frank, M.; Moss, S.J.; Haydon, P.G. Endogenous nonneuronal modulators of synaptic transmission control cortical slow oscillations in vivo. Proc. Natl. Acad. Sci. USA 2009, 106, 15037–15042. [Google Scholar] [CrossRef] [Green Version]
- Halassa, M.M.; Florian, C.; Fellin, T.; Munoz, J.R.; Lee, S.Y.; Abel, T.; Haydon, P.G.; Frank, M.G. Astrocytic Modulation of Sleep Homeostasis and Cognitive Consequences of Sleep Loss. Neuron 2009, 61, 213–219. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.S.; Ghetti, A.; Pinto-Duarte, A.; Wang, X.; Dziewczapolski, G.; Galimi, F.; Huitron-Resendiz, S.; Pina-Crespo, J.C.; Roberts, A.J.; Verma, I.M.; et al. Astrocytes contribute to gamma oscillations and recognition memory. Proc. Natl. Acad. Sci. USA 2014, 111, E3343–E3352. [Google Scholar] [CrossRef] [Green Version]
- Vignoli, B.; Battistini, G.; Melani, R.; Blum, R.; Santi, S.; Berardi, N.; Canossa, M. Peri-Synaptic Glia Recycles Brain-Derived Neurotrophic Factor for LTP Stabilization and Memory Retention. Neuron 2016, 92, 873–887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheline, Y.I.; Raichle, M.E.; Snyder, A.Z.; Morris, J.C.; Head, D.; Wang, S.; Mintun, M.A. Amyloid Plaques Disrupt Resting State Default Mode Network Connectivity in Cognitively Normal Elderly. Biol. Psychiatry 2010, 67, 584–587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palmqvist, S.; Schöll, M.; Strandberg, O.; Mattsson, N.; Stomrud, E.; Zetterberg, H.; Blennow, K.; Landau, S.; Jagust, W.; Hansson, O. Earliest accumulation of β-amyloid occurs within the default-mode network and concurrently affects brain connectivity. Nat. Commun. 2017, 8, 1214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winer, J.R.; Mander, B.A.; Helfrich, R.F.; Maass, A.; Harrison, T.M.; Baker, S.L.; Knight, R.T.; Jagust, W.J.; Walker, M.P. Sleep as a Potential Biomarker of Tau and β-Amyloid Burden in the Human Brain. J. Neurosci. 2019, 39, 6315–6324. [Google Scholar] [CrossRef] [Green Version]
- Pendin, D.; Fasolato, C.; Basso, E.; Filadi, R.; Greotti, E.; Galla, L.; Gomiero, C.; Leparulo, A.; Redolfi, N.; Scremin, E.; et al. Familial Alzheimer’s disease presenilin-2 mutants affect Ca2+ homeostasis and brain network excitability. Aging Clin. Exp. Res. 2019, 1–4. [Google Scholar] [CrossRef]
- Filadi, R.; Greotti, E.; Turacchio, G.; Luini, A.; Pozzan, T.; Pizzo, P. Presenilin 2 Modulates Endoplasmic Reticulum-Mitochondria Coupling by Tuning the Antagonistic Effect of Mitofusin 2. Cell Rep. 2016, 15, 2226–2238. [Google Scholar] [CrossRef] [Green Version]
- Greotti, E.; Capitanio, P.; Wong, A.; Pozzan, T.; Pizzo, P.; Pendin, D. Familial Alzheimer’s disease-linked presenilin mutants and intracellular Ca2+ handling: A single-organelle, FRET-based analysis. Cell Calcium 2019, 74, 44–56. [Google Scholar] [CrossRef]
- Theurey, P.; Connolly, N.M.C.; Fortunati, I.; Basso, E.; Lauwen, S.; Ferrante, C.; Moreira Pinho, C.; Joselin, A.; Gioran, A.; Bano, D.; et al. Systems biology identifies preserved integrity but impaired metabolism of mitochondria due to a glycolytic defect in Alzheimer’s disease neurons. Aging Cell 2019, 18, e12924. [Google Scholar] [CrossRef] [Green Version]
- Zampese, E.; Fasolato, C.; Kipanyula, M.J.; Bortolozzi, M.; Pozzan, T.; Pizzo, P. Presenilin 2 modulates endoplasmic reticulum (ER)-mitochondria interactions and Ca2+ cross-talk. Proc. Natl. Acad. Sci. USA 2011, 108, 2777–2782. [Google Scholar] [CrossRef] [Green Version]
- Hedskog, L.; Pinho, C.M.; Filadi, R.; Ronnback, A.; Hertwig, L.; Wiehager, B.; Larssen, P.; Gellhaar, S.; Sandebring, A.; Westerlund, M.; et al. Modulation of the endoplasmic reticulum-mitochondria interface in Alzheimer’s disease and related models. Proc. Natl. Acad. Sci. USA 2013, 110, 7916–7921. [Google Scholar] [CrossRef] [Green Version]
- Nixon, R.A. Amyloid precursor protein & endosomal-lysosomal dysfunction in Alzheimer’s disease: Inseparable partners in a multifactorial disease. FASEB J. 2017, 31, 2729–2743. [Google Scholar] [PubMed] [Green Version]
- Beltramo, R.; D’Urso, G.; Dal Maschio, M.; Farisello, P.; Bovetti, S.; Clovis, Y.; Lassi, G.; Tucci, V.; De Pietri Tonelli, D.; Fellin, T. Layer-specific excitatory circuits differentially control recurrent network dynamics in the neocortex. Nat. Neurosci. 2013, 16, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Chauvette, S.; Volgushev, M.; Timofeev, I. Origin of Active States in Local Neocortical Networks during Slow Sleep Oscillation. Cereb. Cortex 2010, 20, 2660–2674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez-Vives, M.V.; McCormick, D.A. Cellular and network mechanisms of rhytmic recurrent activity in neocortex. Nat. Neurosci. 2000, 3, 1027–1034. [Google Scholar] [CrossRef] [PubMed]
- Lorincz, M.L.; Gunner, D.; Bao, Y.; Connelly, W.M.; Isaac, J.T.R.; Hughes, S.W.; Crunelli, V. A distinct class of slow (~0.2–2 Hz) intrinsically bursting layer 5 pyramidal neurons determines UP/DOWN state dynamics in the neocortex. J. Neurosci. 2015, 35, 5442–5458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lison, H.; Happel, M.F.K.; Schneider, F.; Baldauf, K.; Kerbstat, S.; Seelbinder, B.; Schneeberg, J.; Zappe, M.; Goldschmidt, J.; Budinger, E.; et al. Disrupted cross-laminar cortical processing in β amyloid pathology precedes cell death. Neurobiol. Dis. 2014, 63, 62–73. [Google Scholar] [CrossRef] [PubMed]
- Bokde, A.L.W.; Ewers, M.; Hampel, H. Assessing neuronal networks: Understanding Alzheimer’s disease. Prog. Neurobiol. 2009, 89, 125–133. [Google Scholar] [CrossRef]
- Hahn, T.T.G.; Sakmann, B.; Mehta, M.R. Phase-locking of hippocampal interneurons’ membrane potential to neocortical up-down states. Nat. Neurosci. 2006, 9, 1359–1361. [Google Scholar] [CrossRef]
- Chauvette, S.; Crochet, S.; Volgushev, M.; Timofeev, I. Properties of slow oscillation during slow-wave sleep and anesthesia in cats. J. Neurosci. 2011, 31, 14998–15008. [Google Scholar] [CrossRef]
- Lazarewicz, M.T.; Ehrlichman, R.S.; Maxwell, C.R.; Gandal, M.J.; Finkel, L.H.; Siegel, S.J. Ketamine Modulates Theta and Gamma Oscillations. J. Cogn. Neurosci. 2009, 22, 1452–1464. [Google Scholar] [CrossRef] [Green Version]









© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leparulo, A.; Mahmud, M.; Scremin, E.; Pozzan, T.; Vassanelli, S.; Fasolato, C. Dampened Slow Oscillation Connectivity Anticipates Amyloid Deposition in the PS2APP Mouse Model of Alzheimer’s Disease. Cells 2020, 9, 54. https://doi.org/10.3390/cells9010054
Leparulo A, Mahmud M, Scremin E, Pozzan T, Vassanelli S, Fasolato C. Dampened Slow Oscillation Connectivity Anticipates Amyloid Deposition in the PS2APP Mouse Model of Alzheimer’s Disease. Cells. 2020; 9(1):54. https://doi.org/10.3390/cells9010054
Chicago/Turabian StyleLeparulo, Alessandro, Mufti Mahmud, Elena Scremin, Tullio Pozzan, Stefano Vassanelli, and Cristina Fasolato. 2020. "Dampened Slow Oscillation Connectivity Anticipates Amyloid Deposition in the PS2APP Mouse Model of Alzheimer’s Disease" Cells 9, no. 1: 54. https://doi.org/10.3390/cells9010054