Cellular Plasmalogen Content Does Not Influence Arachidonic Acid Levels or Distribution in Macrophages: A Role for Cytosolic Phospholipase A2γ in Phospholipid Remodeling

 , ,
, , 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Cell Culture
2.3. Quantitative PCR
2.4. Small Interfering RNA (siRNA) Transfection
2.5. Gas Chromatography/Mass Spectrometry (GC/MS) Analyses
2.6. Liquid Chromatography/Mass Spectrometry (LC/MS) Analyses of Phospholipids
2.7. Liquid Chromatography/Mass Spectrometry (LC/MS) Analyses of Eicosanoids
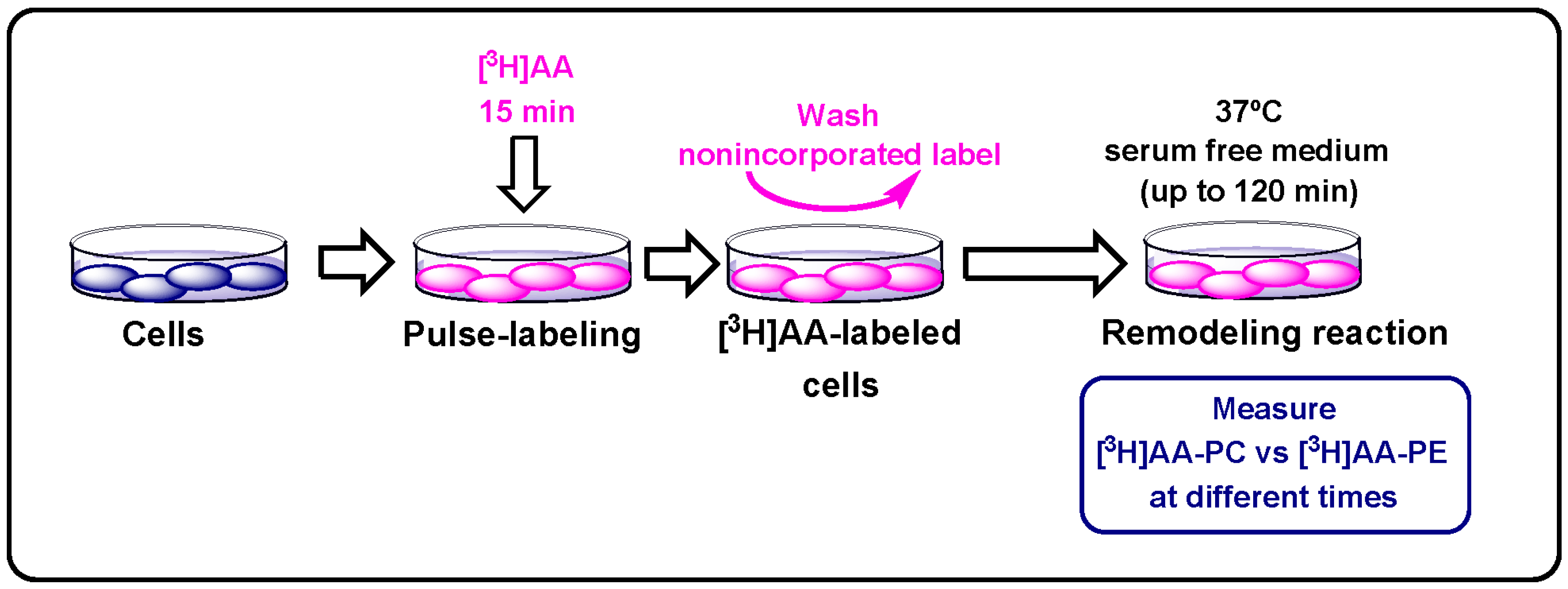
2.8. Measurement of Phospholipid Arachidonate Remodeling
2.9. Statistical Analysis
3. Results
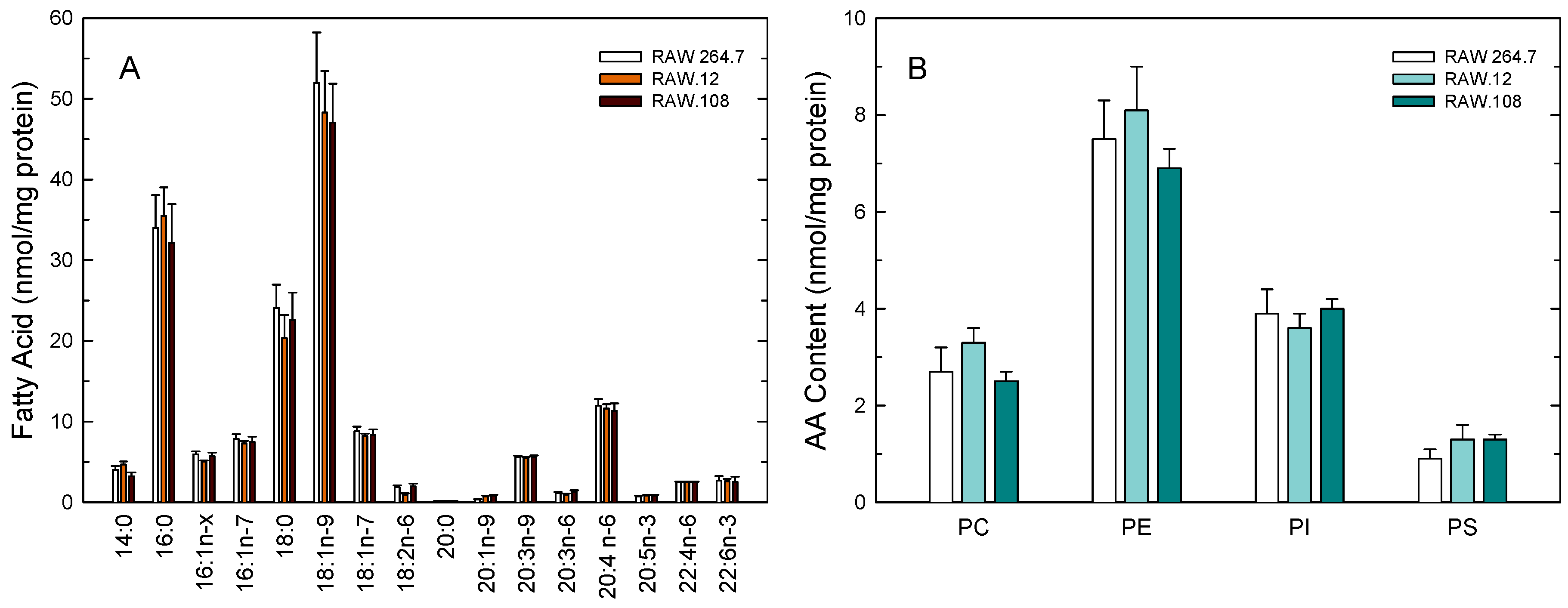
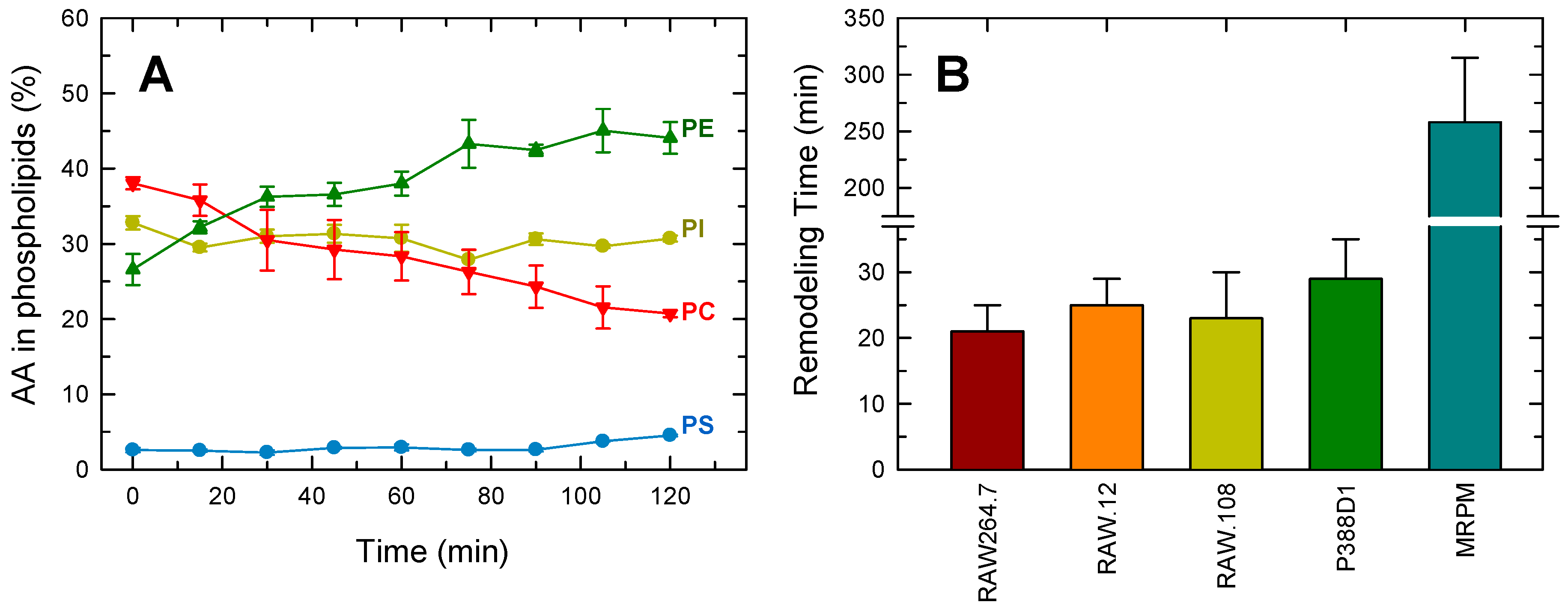
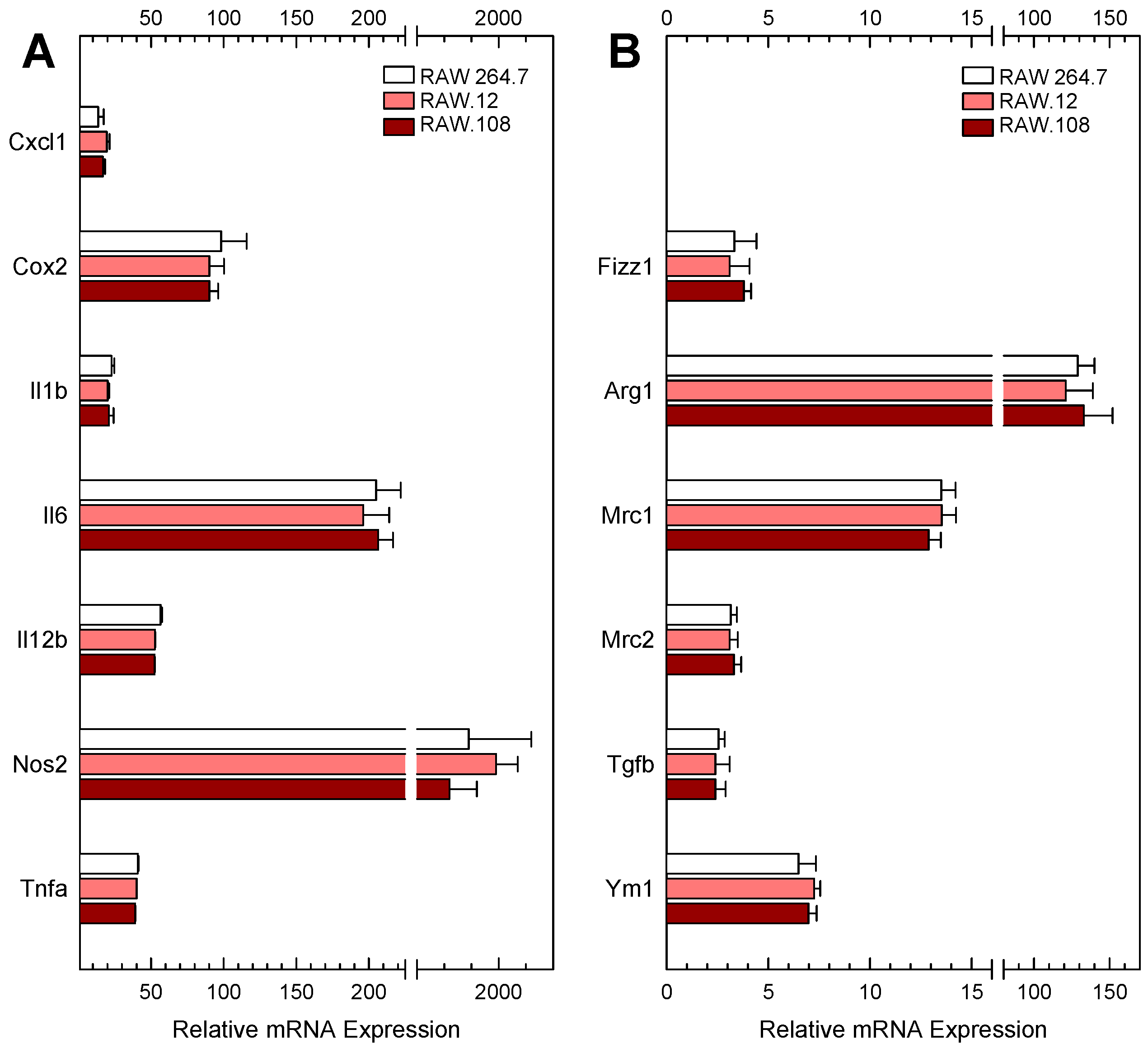
3.1. AA Distribution in RAW264.7 Cells and Plasmalogen-Deficient Variants
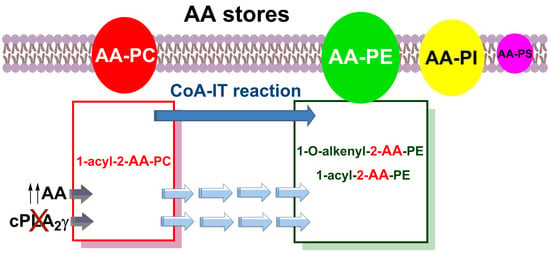
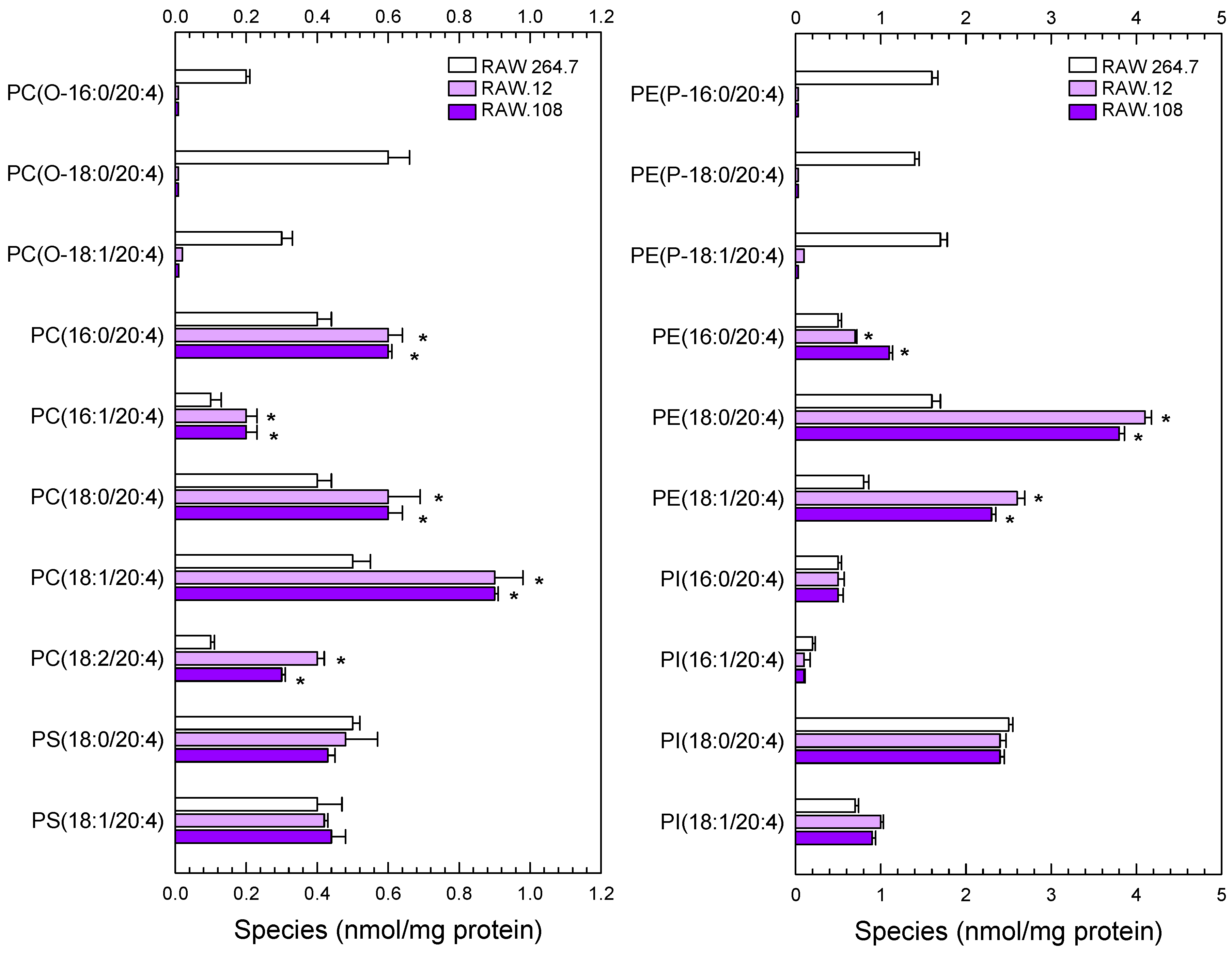
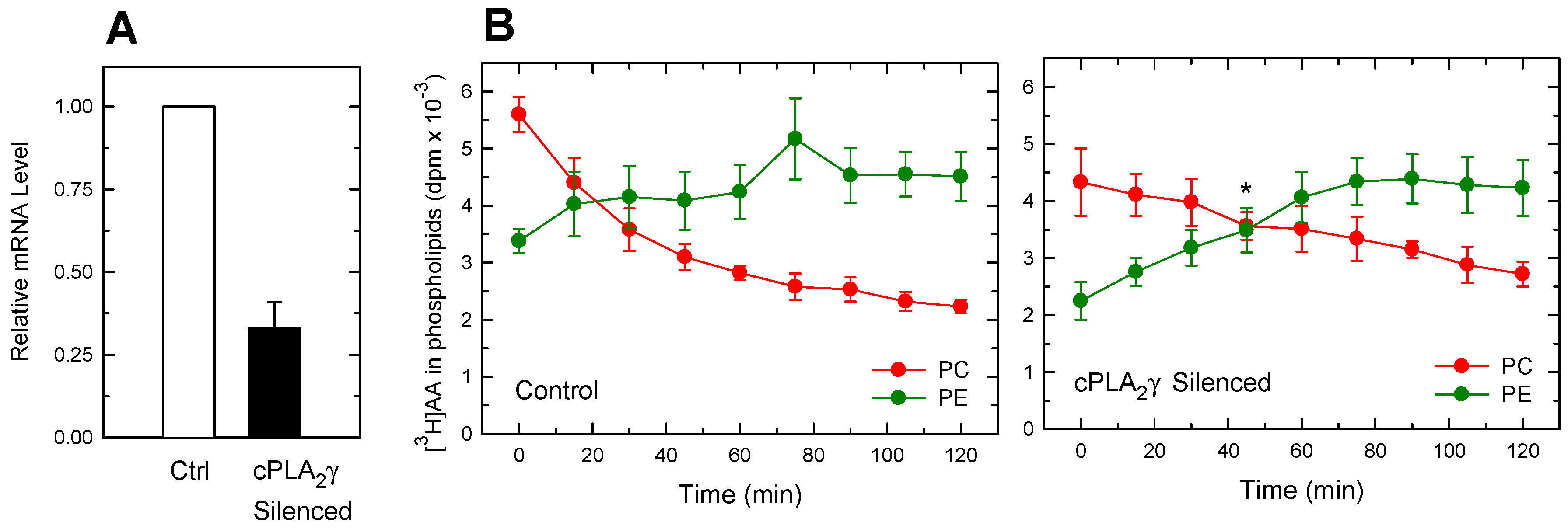
3.2. Importance of Plasmalogen Content for Phosphospholipid AA Remodeling
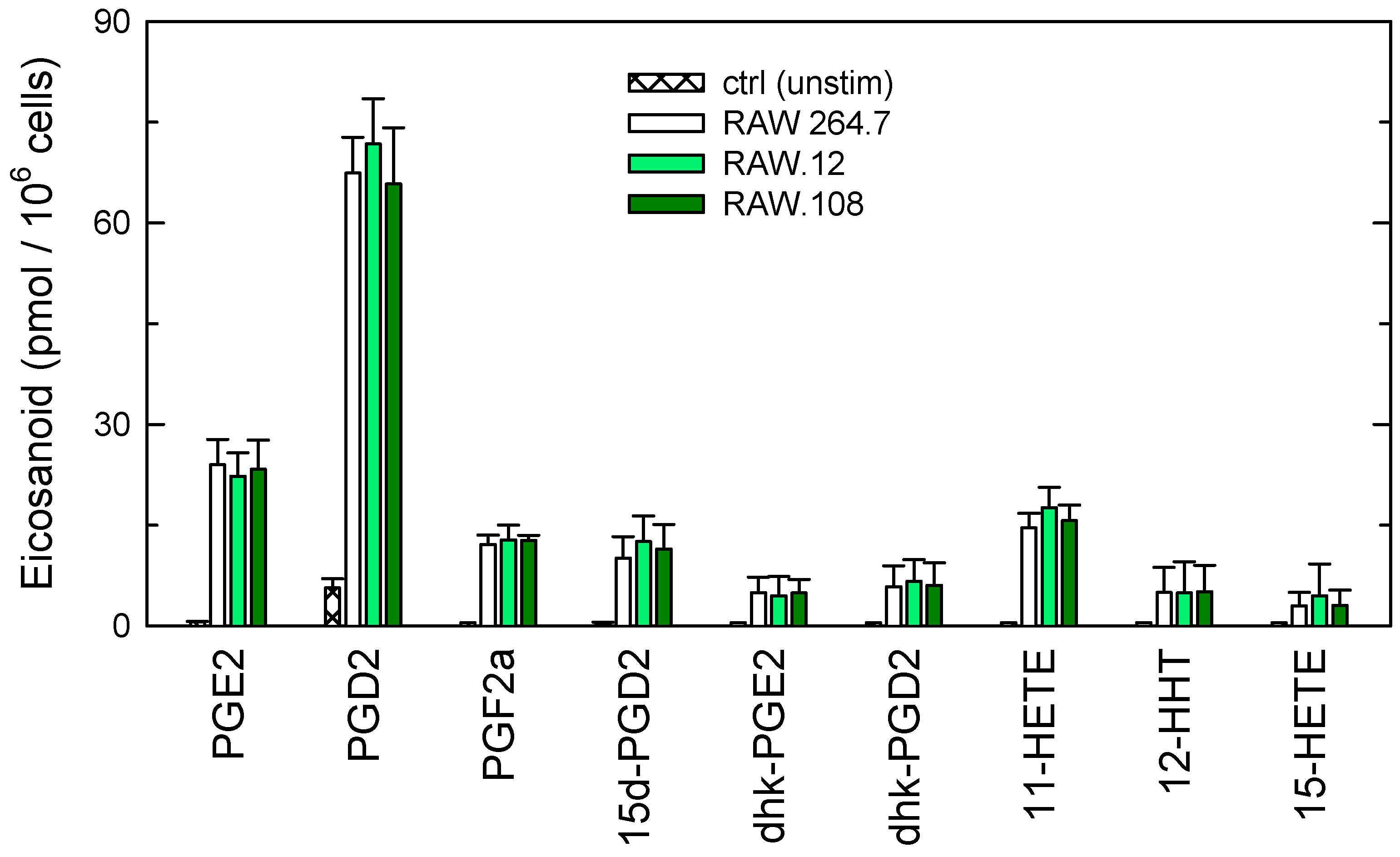
3.3. Role of Plasmalogens in Functional Responses of Macrophages to Receptor Stimulation
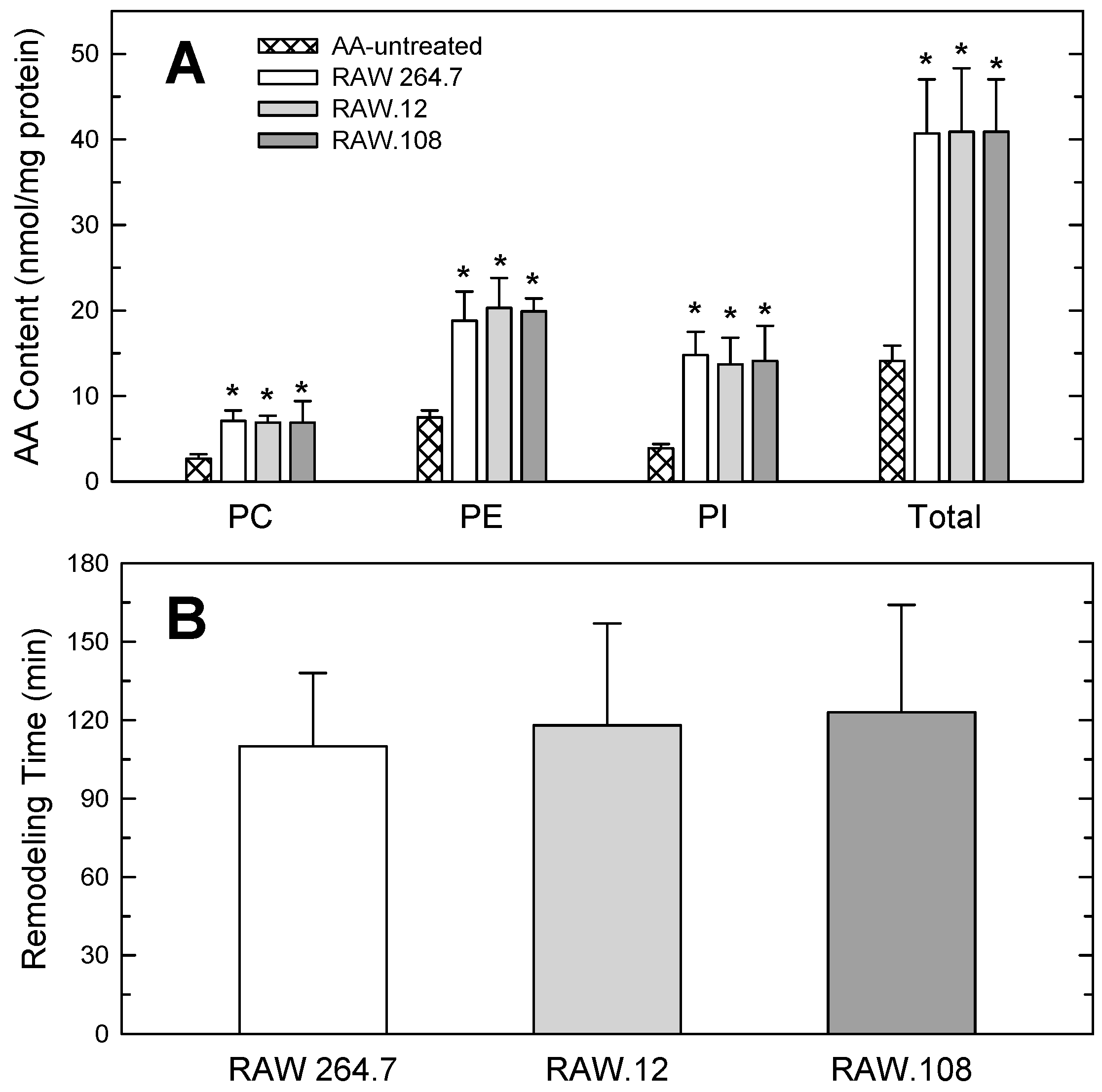
3.4. Studies Utilizing AA-enriched Cells
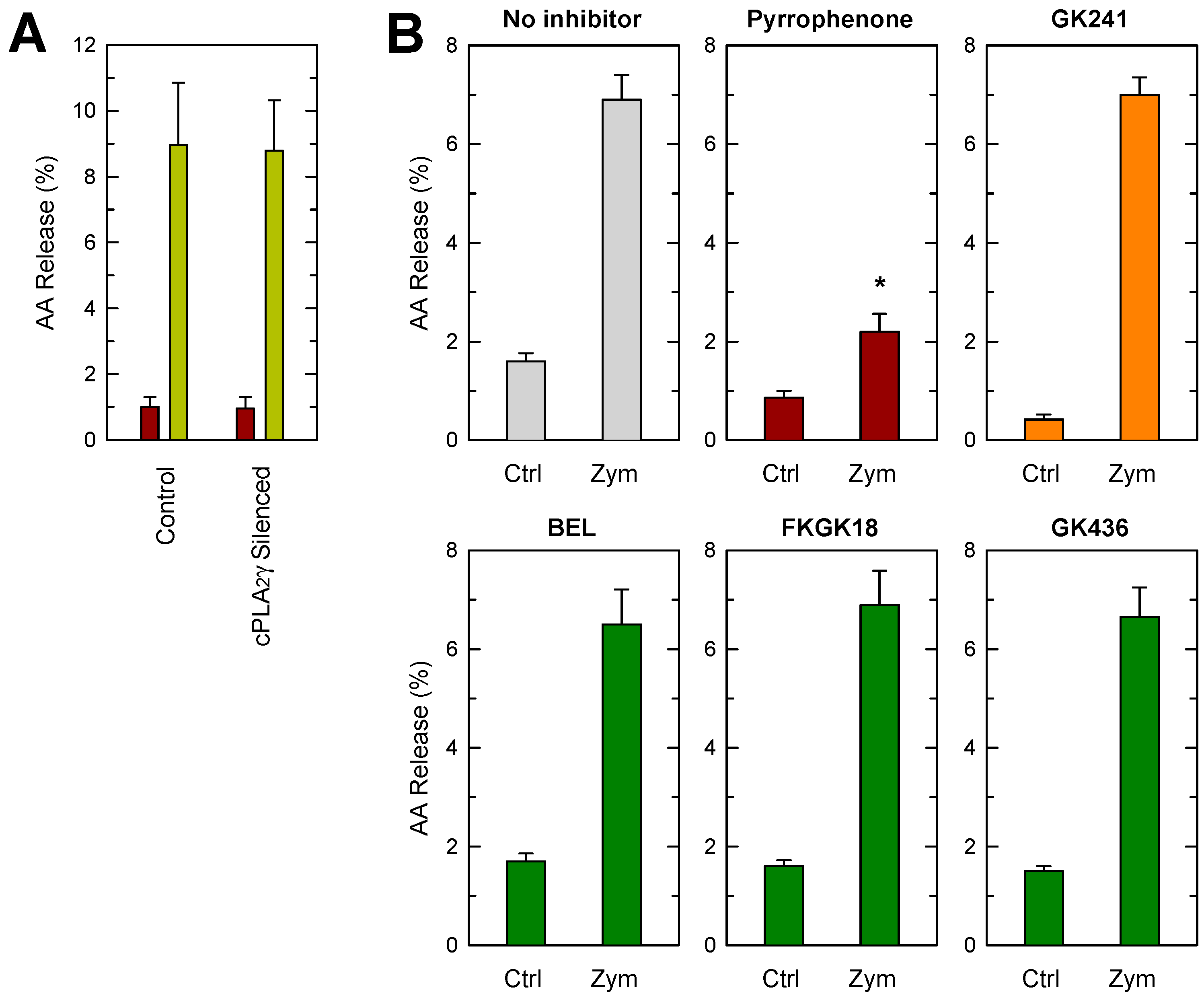
3.5. Phospholipase A2 Inhibition Studies
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Dennis, E.A.; Norris, P.C. Eicosanoid storm in infection and inflammation. Nat. Rev. Immunol. 2015, 15, 511–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Astudillo, A.M.; Balboa, M.A.; Balsinde, J. Selectivity of phospholipid hydrolysis by phospholipase A2 enzymes in activated cells leading to polyunsaturated fatty acid mobilization. Biochim. Biophys. Acta 2019, 1864, 772–783. [Google Scholar] [CrossRef] [PubMed]
- Guijas, C.; Rodríguez, J.P.; Rubio, J.M.; Balboa, M.A.; Balsinde, J. Phospholipase A2 regulation of lipid droplet formation. Biochim. Biophys. Acta 2014, 1841, 1661–1671. [Google Scholar] [CrossRef] [PubMed]
- Astudillo, A.M.; Balgoma, D.; Balboa, M.A.; Balsinde, J. Dynamics of arachidonic acid mobilization by inflammatory cells. Biochim. Biophys. Acta 2012, 1821, 249–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pérez-Chacón, G.; Astudillo, A.M.; Balgoma, D.; Balboa, M.A.; Balsinde, J. 2009. Control of free arachidonic acid levels by phospholipases A2 and lysophospholipid acyltransferases. Biochim. Biophys. Acta 2009, 1791, 1103–1113. [Google Scholar] [CrossRef] [PubMed]
- Balgoma, D.; Astudillo, A.M.; Pérez-Chacón, G.; Montero, O.; Balboa, M.A.; Balsinde, J. Markers of monocyte activation revealed by lipidomic profiling of arachidonic acid-containing phospholipids. J. Immunol. 2010, 184, 3857–3865. [Google Scholar] [CrossRef] [PubMed]
- Gil-de-Gómez, L.; Astudillo, A.M.; Meana, C.; Rubio, J.M.; Guijas, C.; Balboa, M.A.; Balsinde, J. A phosphatidylinositol species acutely generated by activated macrophages regulates innate immune responses. J. Immunol. 2013, 190, 5169–5177. [Google Scholar] [CrossRef]
- Gil-de-Gómez, L.; Astudillo, A.M.; Guijas, C.; Magrioti, V.; Kokotos, G.; Balboa, M.A.; Balsinde, J. Cytosolic group IVA and calcium-independent group VIA phospholipase A2s act on distinct phospholipid pools in zymosan-stimulated mouse peritoneal macrophages. J. Immunol. 2014, 192, 752–762. [Google Scholar] [CrossRef]
- Sugiura, T.; Nakajima, M.; Sekiguchi, N.; Nakagawa, Y.; Waku, K. Different fatty chain compositions of alkenylacyl, alkylacyl and diacyl phospholipids in rabbit alveolar macrophages: High amounts of arachidonic acid in ether phospholipids. Lipids 1983, 18, 125–129. [Google Scholar] [CrossRef]
- Blank, M.L.; Smith, Z.L.; Cress, E.A.; Snyder, F. Molecular species of ethanolamine plasmalogens and transacylase activity in rat tissues are altered by fish oil diets. Biochim. Biophys. Acta 1994, 1214, 295–302. [Google Scholar] [CrossRef]
- Chilton, F.H.; Fonteh, A.N.; Surette, M.E.; Triggiani, M.; Winkler, J.D. Control of arachidonate levels within inflammatory cells. Biochim. Biophys. Acta 1996, 1299, 1–15. [Google Scholar] [CrossRef]
- Chilton, F.H. Potential phospholipid source(s) of arachidonate used for the synthesis of leukotrienes by the human neutrophil. Biochem. J. 1989, 258, 327–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamashita, A.; Hayashi, Y.; Nemoto-Sasaki, Y.; Ito, M.; Oka, S.; Tanikawa, T.; Waku, K.; Sugiura, T. Acyltransferases and transacylases that determine the fatty acid composition of glycerolipids and the metabolism of bioactive lipid mediators in mammalian cells and model organisms. Prog. Lipid Res. 2014, 53, 18–81. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, A.; Hayashi, Y.; Matsumoto, N.; Nemoto-Sasaki, Y.; Koizumi, T.; Inagaki, Y.; Oka, S.; Tanikawa, T.; Sugiura, T. Coenzyme-A-independent transacylation system; possible involvement of phospholipase A2 in transacylation. Biology 2017, 6, 23. [Google Scholar] [CrossRef] [PubMed]
- Murphy, R.C.; Folco, G. Lysophospholipid acyltransferases and leukotriene biosynthesis: Intersection of the Lands cycle and the arachidonate PI cycle. J. Lipid Res. 2019, 60, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Dennis, E.A.; Cao, J.; Hsu, Y.H.; Magrioti, V.; Kokotos, G. Phospholipase A2 enzymes: Physical structure, biological function, disease implication, chemical inhibition, and therapeutic intervention. Chem. Rev. 2011, 111, 6130–6185. [Google Scholar]
- Balboa, M.A.; Sáez, Y.; Balsinde, J. Calcium-independent phospholipase A2 is required for lysozyme secretion in U937 promonocytes. J. Immunol. 2003, 170, 5276–5280. [Google Scholar] [CrossRef] [PubMed]
- Pérez, R.; Melero, R.; Balboa, M.A.; Balsinde, J. Role of group VIA calcium-independent phospholipase A2 in arachidonic acid release, phospholipid fatty acid incorporation, and apoptosis in U937 cells responding to hydrogen peroxide. J. Biol. Chem. 2004, 279, 40385–40391. [Google Scholar] [CrossRef] [PubMed]
- Pérez, R.; Balboa, M.A.; Balsinde, J. Involvement of group VIA calcium-independent phospholipase A2 in macrophage engulfment of hydrogen peroxide-treated U937 cells. J. Immunol. 2006, 176, 2555–2561. [Google Scholar] [CrossRef]
- Balsinde, J.; Balboa, M.A. Cellular regulation and proposed biological functions of group VIA calcium-independent phospholipase A2 in activated cells. Cell. Signal. 2005, 17, 1052–1062. [Google Scholar] [CrossRef]
- Winkler, J.D.; Sung, C.M.; Bennett, C.F.; Chilton, F.H. Characterization of CoA-independent transacylase activity in U937 cells. Biochim. Biophys. Acta 1991, 1081, 339–346. [Google Scholar] [CrossRef]
- Winkler, J.D.; Sung, C.M.; Chabot-Fletcher, M.; Griswold, D.E.; Marshall, L.A.; Chilton, F.H.; Bondinell, W.; Mayer, R.J. β-lactams SB 212047 and SB 216754 are irreversible, time-dependent inhibitors of coenzyme A-independent transacylase. Mol. Pharmacol. 1998, 53, 322–329. [Google Scholar] [CrossRef] [PubMed]
- Winkler, J.D.; Fonteh, A.N.; Sung, C.M.; Heravi, J.D.; Nixon, A.B.; Chabot-Fletcher, M.; Griswold, D.E.; Marshall, L.A.; Chilton, F.H. Effects of CoA-independent transacylase inhibitors on the production of lipid inflammatory mediators. J. Pharmacol. Exp. Ther. 1995, 274, 1338–1347. [Google Scholar] [PubMed]
- Asai, K.; Hirabayashi, T.; Houjou, T.; Uozumi, N.; Taguchi, R.; Shimizu, T. Human group IVC phospholipase A2 (cPLA2γ). Roles in the membrane remodeling and activation induced by oxidative stress. J. Biol. Chem. 2003, 278, 8809–8814. [Google Scholar] [CrossRef] [PubMed]
- Astudillo, A.M.; Pérez-Chacón, G.; Meana, C.; Balgoma, D.; Pol, A.; del Pozo, M.A.; Balboa, M.A.; Balsinde, J. Altered arachidonate distribution in macrophages from caveolin-1 null mice leading to reduced eicosanoid synthesis. J. Biol. Chem. 2011, 286, 35299–35307. [Google Scholar] [CrossRef] [PubMed]
- Valdearcos, M.; Esquinas, E.; Meana, C.; Gil-de-Gómez, L.; Guijas, C.; Balsinde, J.; Balboa, M.A. Subcellular localization and role of lipin-1 in human macrophages. J. Immunol. 2011, 186, 6004–6013. [Google Scholar] [CrossRef] [PubMed]
- Rubio, J.M.; Rodríguez, J.P.; Gil-de-Gómez, L.; Guijas, C.; Balboa, M.A.; Balsinde, J. Group V secreted phospholipase A2 is up-regulated by interleukin-4 in human macrophages and mediates phagocytosis via hydrolysis of ethanolamine phospholipids. J. Immunol. 2015, 194, 3327–3339. [Google Scholar] [CrossRef]
- Guijas, C.; Astudillo, A.M.; Gil-de-Gómez, L.; Rubio, J.M.; Balboa, M.A.; Balsinde, J. 2012. Phospholipid sources for adrenic acid mobilization in RAW 264.7 macrophages: Comparison with arachidonic acid. Biochim. Biophys. Acta 2012, 1821, 1386–1393. [Google Scholar] [CrossRef]
- Gil-de-Gómez, L.; Astudillo, A.M.; Lebrero, P.; Balboa, M.A.; Balsinde, J. Essential role for ethanolamine plasmalogen hydrolysis in bacterial lipopolysaccharide priming of macrophages for enhanced arachidonic acid release. Front. Immunol. 2017, 8, 1251. [Google Scholar] [CrossRef]
- Zoeller, R.A.; Rangaswamy, S.; Herscovitz, H.; Rizzo, W.B.; Hajra, A.K.; Das, A.K.; Moser, H.W.; Moser, A.; Lazarow, P.W.; Santos, M.J. Mutants in a macrophage-like cell line are defective in plasmalogen biosynthesis, but contain functional peroxisomes. J. Biol. Chem. 1992, 267, 8299–8306. [Google Scholar]
- Gaposchkin, D.P.; Zoeller, R.A. Plasmalogen status influences docosahexaenoic acid levels in a macrophage cell line: Insights using ether lipid-deficient variants. J. Lipid Res. 1999, 40, 495–503. [Google Scholar] [PubMed]
- Gaposchkin, D.P.; Farber, H.W.; Zoeller, R.A. On the importance of plasmalogen status in stimulated arachidonic acid release in the macrophage cell line RAW 264.7. Biochim. Biophys. Acta 2008, 1781, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Rubio, J.M.; Astudillo, A.M.; Casas, J.; Balboa, M.A.; Balsinde, J. Regulation of phagocytosis in macrophages by membrane ethanolamine plasmalogens. Front. Immunol. 2018, 9, 1723. [Google Scholar] [CrossRef] [PubMed]
- Ono, T.; Yamada, K.; Chikazawa, Y.; Ueno, M.; Nakamoto, S.; Okuno, T.; Seno, K. Characterization of a novel inhibitor of cytosolic phospholipase A2α, pyrrophenone. Biochem. J. 2002, 363, 727–735. [Google Scholar] [CrossRef] [PubMed]
- Kokotos, G.; Hsu, Y.H.; Burke, J.E.; Baskakis, C.; Kokotos, C.G.; Magrioti, V.; Dennis, E.A. Potent and selective fluoroketone inhibitors of group VIA calcium-independent phospholipase A2. J. Med. Chem. 2010, 53, 3602–3610. [Google Scholar] [CrossRef] [PubMed]
- Dedaki, C.; Kokotou, M.G.; Mouchlis, V.D.; Limnios, D.; Lei, X.; Mu, C.T.; Ramanadham, S.; Magrioti, V.; Dennis, E.A.; Kokotos, G. β-Lactones: A novel class of Ca2+-independent phospholipase A2 (group VIA iPLA2) inhibitors with the ability to inhibit β-cell apoptosis. J. Med. Chem. 2019, 62, 2916–2927. [Google Scholar] [CrossRef] [PubMed]
- Vasilakaki, S.; Barbayianni, E.; Leonis, G.; Papadopoulos, M.G.; Mavromoustakos, T.; Gelb, M.H.; Kokotos, G. Development of a potent 2-oxoamide inhibitor of secreted phospholipase A2 guided by molecular docking calculations and molecular dynamics simulations. Bioorg. Med. Chem. 2016, 24, 1683–1695. [Google Scholar] [CrossRef] [PubMed]
- Shirai, Y.; Balsinde, J.; Dennis, E.A. Localization and functional interrelationships among cytosolic group IV, secreted group V, and Ca2+-independent group VI phospholipase A2s in P388D1 macrophages using GFP/RFP constructs. Biochim. Biophys. Acta 2005, 1735, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Balboa, M.A.; Balsinde, J.; Dillon, D.A.; Carman, G.M.; Dennis, E.A. Proinflammatory macrophage-activating properties of the novel phospholipid diacylglycerol pyrophosphate. J. Biol. Chem. 1999, 274, 522–526. [Google Scholar] [CrossRef] [PubMed]
- Balsinde, J.; Balboa, M.A.; Yedgar, S.; Dennis, E.A. Group V phospholipase A2-mediated oleic acid mobilization in lipopolysaccharide-stimulated P388D1 macrophages. J. Biol. Chem. 2000, 275, 4783–4786. [Google Scholar] [CrossRef]
- Pindado, J.; Balsinde, J.; Balboa, M.A. TLR3-dependent induction of nitric oxide synthase in RAW 264.7 macrophage-like cells via a cytosolic phospholipase A2/cyclooxygenase-2 pathway. J. Immunol. 2007, 179, 4821–4828. [Google Scholar] [CrossRef] [PubMed]
- Ruipérez, V.; Astudillo, M.A.; Balboa, M.A.; Balsinde, J. Coordinate regulation of TLR-mediated arachidonic acid mobilization in macrophages by group IVA and group V phospholipase A2s. J. Immunol. 2009, 182, 3877–3883. [Google Scholar]
- Balsinde, J.; Fernández, B.; Diez, E. Regulation of arachidonic acid release in mouse peritoneal macrophages. The role of extracellular calcium and protein kinase C. J. Immunol. 1990, 144, 4298–4304. [Google Scholar] [PubMed]
- Balsinde, J.; Fernández, B.; Solís-Herruzo, J.A.; Diez, E. Pathways for arachidonic acid mobilization in zymosan-stimulated mouse peritoneal macrophages. Biochim. Biophys. Acta 1992, 1136, 75–82. [Google Scholar] [CrossRef]
- Balboa, M.A.; Balsinde, J. Involvement of calcium-independent phospholipase A2 in hydrogen peroxide-induced accumulation of free fatty acids in human U937 cells. J. Biol. Chem. 2002, 277, 40384–40389. [Google Scholar] [CrossRef]
- Balboa, M.A.; Pérez, R.; Balsinde, J. Amplification mechanisms of inflammation: Paracrine stimulation of arachidonic acid mobilization by secreted phospholipase A2 is regulated by cytosolic phospholipase A2-derived hydroperoxyeicosatetraenoic acid. J. Immunol. 2003, 171, 989–994. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Rodríguez-Prados, J.C.; Través, P.G.; Cuenca, J.; Rico, D.; Aragonés, J.; Martín-Sanz, P.; Cascante, M.; Boscá, L. Substrate fate in activated macrophages: A comparison between innate, classic, and alternative activation. J. Immunol. 2010, 185, 605–614. [Google Scholar] [CrossRef]
- Balboa, M.A.; Balsinde, J.; Dennis, E.A. Involvement of phosphatidate phosphohydrolase in arachidonic acid mobilization in human amnionic WISH cells. J. Biol. Chem. 1998, 273, 7684–7690. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔ Ct method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Valdearcos, M.; Esquinas, E.; Meana, C.; Peña, L.; Gil-de-Gómez, L.; Balsinde, J.; Balboa, M.A. Lipin-2 reduces proinflammatory signaling induced by saturated fatty acids in macrophages. J. Biol. Chem. 2012, 287, 10894–10904. [Google Scholar] [CrossRef] [PubMed]
- Bligh, E.G.; Dyer, W.J. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 1959, 37, 911–917. [Google Scholar] [CrossRef] [PubMed]
- Diez, E.; Balsinde, J.; Aracil, M.; Schüller, A. Ethanol induces release of arachidonic acid but not synthesis of eicosanoids in mouse peritoneal macrophages. Biochim. Biophys. Acta 1987, 921, 82–89. [Google Scholar] [CrossRef]
- Fine, J.B.; Sprecher, H. Unidimensional thin-layer chromatography of phospholipids on boric acid-impregnated plates. J. Lipid Res. 1982, 23, 660–663. [Google Scholar] [PubMed]
- Astudillo, A.M.; Pérez-Chacón, G.; Balgoma, D.; Gil-de-Gómez, L.; Ruipérez, V.; Guijas, C.; Balboa, M.A.; Balsinde, J. Influence of cellular arachidonic acid levels on phospholipid remodeling and CoA-independent transacylase activity in human monocytes and U937 cells. Biochim. Biophys. Acta 2011, 1811, 97–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guijas, C.; Pérez-Chacón, G.; Astudillo, A.M.; Rubio, J.M.; Gil-de-Gómez, L.; Balboa, M.A.; Balsinde, J. Simultaneous activation of p38 and JNK by arachidonic acid stimulates the cytosolic phospholipase A2-dependent synthesis of lipid droplets in human monocytes. J. Lipid Res. 2012, 53, 2343–2354. [Google Scholar] [CrossRef]
- Guijas, C.; Meana, C.; Astudillo, A.M.; Balboa, M.A.; Balsinde, J. Foamy monocytes are enriched in cis-7-hexadecenoic fatty acid (16:1n-9), a possible biomarker for early detection of cardiovascular disease. Cell Chem. Biol. 2016, 23, 689–699. [Google Scholar] [CrossRef]
- Astudillo, A.M.; Meana, C.; Guijas, C.; Pereira, L.; Lebrero, R.; Balboa, M.A.; Balsinde, J. Occurrence and biological activity of palmitoleic acid isomers in phagocytic cells. J. Lipid Res. 2018, 59, 237–249. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez, J.P.; Guijas, C.; Astudillo, A.M.; Rubio, J.M.; Balboa, M.A.; Balsinde, J. Sequestration of 9-hydroxystearic acid in FAHFA (fatty acid esters of hydroxy fatty acids) as a protective mechanism for colon carcinoma cells to avoid apoptotic cell death. Cancers 2019, 11, 524. [Google Scholar] [CrossRef]
- Pérez, R.; Matabosch, X.; Llebaria, A.; Balboa, M.A.; Balsinde, J. Blockade of arachidonic acid incorporation into phospholipids induces apoptosis in U937 promonocytic cells. J. Lipid Res. 2006, 47, 484–491. [Google Scholar] [CrossRef] [Green Version]
- Fahy, E.; Subramaniam, S.; Brown, H.A.; Glass, C.K.; Merrill, A.H., Jr.; Murphy, R.C.; Raetz, C.R.; Russell, D.W.; Seyama, Y.; Shaw, W.; et al. A comprehensive classification system for lipids. J. Lipid Res. 2005, 46, 839–861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chilton, F.H.; Fonteh, A.N.; Sung, C.M.; Hickey, D.M.; Torphy, T.J.; Mayer, R.J.; Marshall, L.A.; Heravi, J.D.; Winkler, J.D. Inhibitors of CoA-independent transacylase block the movement of arachidonate into 1-ether-linked phospholipids of human neutrophils. Biochemistry 1995, 34, 5403–5410. [Google Scholar] [CrossRef] [PubMed]
- Balsinde, J.; Barbour, S.E.; Bianco, I.D.; Dennis, E.A. Arachidonic acid mobilization in P388D1 macrophages is controlled by two distinct Ca2+-dependent phospholipase A2 enzymes. Proc. Natl. Acad. Sci. USA 1994, 91, 11060–11064. [Google Scholar] [CrossRef] [PubMed]
- Balsinde, J.; Balboa, M.A.; Insel, P.A.; Dennis, E.A. Differential regulation of phospholipase D and phospholipase A2 by protein kinase C in P388D1 macrophages. Biochem. J. 1997, 321, 805–809. [Google Scholar] [CrossRef] [PubMed]
- Balsinde, J.; Fernández, B.; Solís-Herruzo, J.A. Increased incorporation of arachidonic acid into phospholipids in zymosan-stimulated mouse peritoneal macrophages. Eur. J. Biochem. 1994, 221, 1013–1018. [Google Scholar] [CrossRef] [PubMed]
- Buczynski, M.W.; Stephens, D.L.; Bowers-Gentry, R.C.; Grkovich, A.; Deems, R.A.; Dennis, E.A. TLR-4 and sustained calcium agonists synergistically produce eicosanoids independent of protein synthesis in RAW 264.7 cells. J. Biol. Chem. 2007, 282, 22834–22847. [Google Scholar] [CrossRef] [PubMed]
- Rouzer, C.A.; Ivanova, P.T.; Byrne, M.O.; Milne, S.B.; Marnett, L.J.; Brown, H.A. Lipid profiling reveals arachidonate deficiency in RAW264.7 cells: Structural and functional implications. Biochemistry 2006, 45, 14795–14808. [Google Scholar] [CrossRef] [PubMed]
- Okuno, T.; Gijón, M.A.; Zarini, S.; Martin, S.A.; Barkley, R.M.; Johnson, C.A.; Ohba, M.; Yokomizo, T.; Murphy, R.C. Altered eicosanoid production and phospholipid remodeling during cell culture. J. Lipid Res. 2018, 59, 542–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emilsson, A.; Sundler, R. Differential activation of phosphatidylinositol deacylation and a pathway via diphosphoinositide in macrophages responding to zymosan and ionophore A23187. J. Biol. Chem. 1984, 259, 3111–3116. [Google Scholar]
- Casas, J.; Gijón, M.A.; Vigo, A.G.; Crespo, M.S.; Balsinde, J.; Balboa, M.A. Phosphatidylinositol 4,5-bisphosphate anchors cytosolic group IVA phospholipase A2 to perinuclear membranes and decreases its calcium requirement for translocation in live cells. Mol. Biol. Cell 2006, 17, 155–162. [Google Scholar] [CrossRef]
- Balsinde, J. Roles of various phospholipases A2 in providing lysophospholipid acceptors for fatty acid phospholipid incorporation and remodelling. Biochem. J. 2002, 364, 695–702. [Google Scholar] [CrossRef] [PubMed]
- Boilard, E.; Surette, M.E. Anti-CD3 and concanavalin A-induced human T cell proliferation is associated with an increased rate of arachidonate-phospholipid remodeling. J. Biol. Chem. 2001, 276, 17568–17575. [Google Scholar] [CrossRef] [PubMed]
- Nikolaou, A.; Kokotou, M.G.; Vasilakaki, S.; Kokotos, G. Small-molecule inhibitors as potential therapeutics and as tools to understand the role of phospholipases A2. Biochim. Biophys. Acta 2019, 1864, 941–956. [Google Scholar] [CrossRef] [PubMed]
- Balsinde, J.; Balboa, M.A.; Dennis, E.A. Identification of a third pathway for arachidonic acid mobilization and prostaglandin production in activated P388D1 macrophage-like cells. J. Biol. Chem. 2000, 275, 22544–22549. [Google Scholar] [CrossRef] [PubMed]
- Marshall, J.; Krump, E.; Lindsay, T.; Downey, G.; Ford, D.A.; Zhu, P.; Walker, P.; Rubin, B. Involvement of cytosolic phospholipase A2 and secretory phospholipase A2 in arachidonic acid release from human neutrophils. J. Immunol. 2000, 164, 2084–2091. [Google Scholar] [CrossRef] [PubMed]
- Degousee, N.; Ghomashchi, F.; Stefanski, E.; Singer, A.G.; Smart, B.P.; Borregaard, N.; Reithmeier, R.; Lindsay, T.F.; Lichtenberger, C.; Reinisch, W.; et al. Groups IV, V, and X phospholipases A2s in human neutrophils: Role in eicosanoid production and gram-negative bacterial phospholipid hydrolysis. J. Biol. Chem. 2002, 277, 5061–5073. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Chacón, G.; Astudillo, A.M.; Ruipérez, V.; Balboa, M.A.; Balsinde, J. Signaling role for lysophosphatidylcholine acyltransferase 3 in receptor-regulated arachidonic acid reacylation reactions in human monocytes. J. Immunol. 2010, 184, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- Ramanadham, S.; Ali, T.; Ashley, J.W.; Bone, R.N.; Hancock, W.D.; Lei, X. Calcium-independent phospholipases A2 and their roles in biological processes and diseases. J. Lipid Res. 2015, 56, 1643–1668. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, C.M.; Mancuso, D.J.; Yan, W.; Sims, H.F.; Gibson, B.; Gross, R.W. Identification, cloning, expression and purification of three novel human calcium-independent phospholipase A2 family members possessing triacylglycerol lipase and acylglycerol transacylase activities. J. Biol. Chem. 2004, 279, 48968–48975. [Google Scholar] [CrossRef] [PubMed]
- Balsinde, J.; Pérez, R.; Balboa, M.A. Calcium-independent phospholipase A2 and apoptosis. Biochim. Biophys. Acta 2006, 1761, 1344–1350. [Google Scholar] [CrossRef] [PubMed]
- Fuentes, L.; Pérez, R.; Nieto, M.L.; Balsinde, J.; Balboa, M.A. Bromoenol lactone promotes cell death by a mechanism involving phosphatidate phosphohydrolase-1 rather than calcium-independent phospholipase A2. J. Biol. Chem. 2003, 278, 44683–44690. [Google Scholar] [CrossRef] [PubMed]
- Brown, H.A.; Murphy, R.C. Working towards an exegesis for lipids in biology. Nat. Chem. Biol. 2009, 5, 602–606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dean, J.M.; Lodhi, I.J. Structural and functional roles of ether lipids. Protein Cell 2018, 9, 196–206. [Google Scholar] [CrossRef] [PubMed]
- Koivuniemi, A. The biophysical properties of plasmalogens originating from their unique molecular architecture. FEBS Lett. 2017, 591, 2700–2713. [Google Scholar] [CrossRef] [PubMed]
- Honsho, M.; Fujiki, Y. Plasmalogen homeostasis. Regulation of plasmalogen biosynthesis and its physiological consequence in mammals. FEBS Lett. 2017, 591, 2720–2729. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, A.; Kamata, R.; Kawagishi, N.; Nakanishi, H.; Suzuki, H.; Sugiura, T.; Waku, K. Roles of C-terminal processing, and involvement in transacylation reaction of human group IVC phospholipase A2 (cPLA2γ). J. Biochem. 2005, 137, 557–567. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, A.; Tanaka, K.; Kamata, R.; Kumazawa, T.; Suzuki, N.; Koga, H.; Waku, K.; Sugiura, T. Subcellular localization and lysophospholipase/transacylation activities of human group IVC phospholipase A2 (cPLA2γ). Biochim. Biophys. Acta 2009, 1791, 1011–1022. [Google Scholar] [CrossRef] [PubMed]
- Stewart, A.; Ghosh, M.; Spencer, D.M.; Leslie, C.C. Enzymatic properties of human cytosolic phospholipase A2γ. J. Biol. Chem. 2002, 277, 29526–29536. [Google Scholar] [CrossRef]
- Breton, M.; Colard, O. Protein kinase C promotes arachidonate mobilization through enhancement of CoA-independent transacylase activity in platelets. Biochem. J. 1991, 280, 93–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winkler, J.D.; Sung, C.M.; Huang, L.; Chilton, F.H. CoA-independent transacylase activity is increased in human neutrophils after treatment with tumor necrosis factor-α. Biochim. Biophys. Acta 1994, 1215, 133–140. [Google Scholar] [CrossRef]
- Fonteh, A.N.; Chilton, F.H. Rapid remodeling of arachidonate from phosphatidylcholine to phosphatidylethanolamine pools during mast cell activation. J. Immunol. 1992, 148, 1784–1791. [Google Scholar] [PubMed]
- Baker, P.R.; Owen, J.S.; Nixon, A.B.; Thomas, L.; Wooten, R.; Daniel, L.W.; O’Flaherty, J.T.; Wykle, R.L. Regulation of platelet-activating factor synthesis in human neutrophils by MAP kinases. Biochim. Biophys. Acta 2002, 1592, 175–184. [Google Scholar] [CrossRef] [Green Version]









© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lebrero, P.; Astudillo, A.M.; Rubio, J.M.; Fernández-Caballero, L.; Kokotos, G.; Balboa, M.A.; Balsinde, J. Cellular Plasmalogen Content Does Not Influence Arachidonic Acid Levels or Distribution in Macrophages: A Role for Cytosolic Phospholipase A2γ in Phospholipid Remodeling. Cells 2019, 8, 799. https://doi.org/10.3390/cells8080799
Lebrero P, Astudillo AM, Rubio JM, Fernández-Caballero L, Kokotos G, Balboa MA, Balsinde J. Cellular Plasmalogen Content Does Not Influence Arachidonic Acid Levels or Distribution in Macrophages: A Role for Cytosolic Phospholipase A2γ in Phospholipid Remodeling. Cells. 2019; 8(8):799. https://doi.org/10.3390/cells8080799
Chicago/Turabian StyleLebrero, Patricia, Alma M. Astudillo, Julio M. Rubio, Lidia Fernández-Caballero, George Kokotos, María A. Balboa, and Jesús Balsinde. 2019. "Cellular Plasmalogen Content Does Not Influence Arachidonic Acid Levels or Distribution in Macrophages: A Role for Cytosolic Phospholipase A2γ in Phospholipid Remodeling" Cells 8, no. 8: 799. https://doi.org/10.3390/cells8080799