Graphene-Based Heterogeneous Catalysis: Role of Graphene


1
Department of Physics and Centre for 2D Advanced Materials, National University of Singapore, 2 Science Drive 3, Singapore 117551, Singapore
2
Department of Chemistry, National University of Singapore, 3 Science Drive 3, Singapore 117543, Singapore
*
Authors to whom correspondence should be addressed.
Catalysts 2020, 10(1), 53; https://doi.org/10.3390/catal10010053
Submission received: 6 December 2019
/
Revised: 20 December 2019
/
Accepted: 20 December 2019
/
Published: 1 January 2020
(This article belongs to the Special Issue Graphene-based Catalysis)
Abstract
:Graphene, the reincarnation of a surface, offers new opportunities in catalytic applications, not only because of its peculiar electronic structure, but also because of the ease of modulating it. A vast number of proposals have been made to support this point, but there has been a lack of a systematic understanding of the different roles of graphene, as many other reviews published have focused on the synthesis and characterization of the various graphene-based catalysts. In this review, we surveyed the vast literature related to various theoretical proposals and experimental realizations of graphene-based catalysts to first classify and then elucidate the different roles played by graphene in solid-state heterogeneous catalysis. Owing to its one-atom thickness and zero bandgap with low density of states around Fermi level, graphene has great potential in catalysis applications. In general, graphene can function as a support for catalysts, a cover to protect catalysts, or the catalytic center itself. Understanding these functions is important in the design of catalysts in terms of how to optimize the electronic structure of the active sites for particular applications, a few case studies of which will be presented for each role.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
1.1. Graphene in Solid-State Catalysis
Graphene, a single-atom-thick two-dimensional (2d) material, has attracted enormous interest because of its great potential in electronics originating from its high electrical conductivity and unique electronic structure with zero bandgap [1,2,3,4,5]. However, over the years, this ultra-stable and inert material has gradually found its way as a rising star in catalysis [6,7,8,9,10,11]. In general, a catalyst can be thought of as being composed of two parts, namely, the active site, where the reaction takes place, and the surrounding environment, which plays the role of tuning the electronic structure of the active center to optimize its catalytic activity while maintaining structural integrity during the harsh operating conditions. There have been many theoretical proposals and experimental realizations of the application of graphene in heterogeneous catalysis, but the roles of graphene in catalysis can be easily classified according to this dichotomy, i.e., graphene either forms part of the environment in which the catalytic center is, functioning as a support or a cover, or it is the catalytic center.
1.2. Why Graphene Is Attractive in Solid-State Catalysis
The performance of a conventional catalyst can often be controlled by its surface-to-volume (STV) ratio. Graphene can be attractive in catalytic applications, as it has an extremely large STV ratio, owing to its 2d nature and single-atom thickness. Also, its high chemical stability and ultrahigh thermal conductivity [12] could potentially facilitate the high loading of catalytically active sites, and its high electrical conductivity renders it suitable for electrochemical processes. These properties allow graphene to stay intact and resistant to degradation under harsh operating conditions, e.g., under high temperature, application of high bias voltage, and in either strongly acidic or alkaline media. If precious noble metals are used, they can usually be recovered easily by combusting the carbon support.
More importantly, the peculiar atomic and electronic structure of graphene offers new opportunities in catalysis. Graphene is a single atom thick, which means its chemical properties are very sensitive to the change of chemical environment. Graphene has zero bandgap and very low density of states (DOS) around the Fermi level (Ef). Integrating the DOS of graphene from Ef to Ef + 0.5 eV produces less than 0.01 electrons, suggesting that a very small amount of charge transfer would induce a significant degree of shift in the Ef of graphene. All of these make graphene stand out from other 2d materials in catalysis applications, because the interactions between graphene and the supported catalyst or the substrate underneath are highly tunable via physical or chemical methods.
Noting that pristine graphene is chemically inert, as demonstrated from the small adsorption energies and limited charge transfer of the simulated adsorptions of various clusters and small gaseous molecules on it [13,14,15,16], the biggest issue in graphene-based catalysis is to find effective yet practical ways of activating graphene. This can be achieved via the introduction of defects (by creating vacancies or by doping with foreign atoms [17,18,19,20]) or by either doping the substrate underneath epitaxial graphene [11,21]. In the latter case, graphene can actually function as the catalytic center. Nevertheless, this chemical inertness does not necessarily translate into a drawback. Epitaxial graphene can be an effective overlay over precious and nonprecious metal catalysts against harsh reaction media, modulating the reactivity of the active sites while offering spatially confined environment for reactions to occur [22,23].
2. Different Roles of Graphene in Solid-State Catalysis
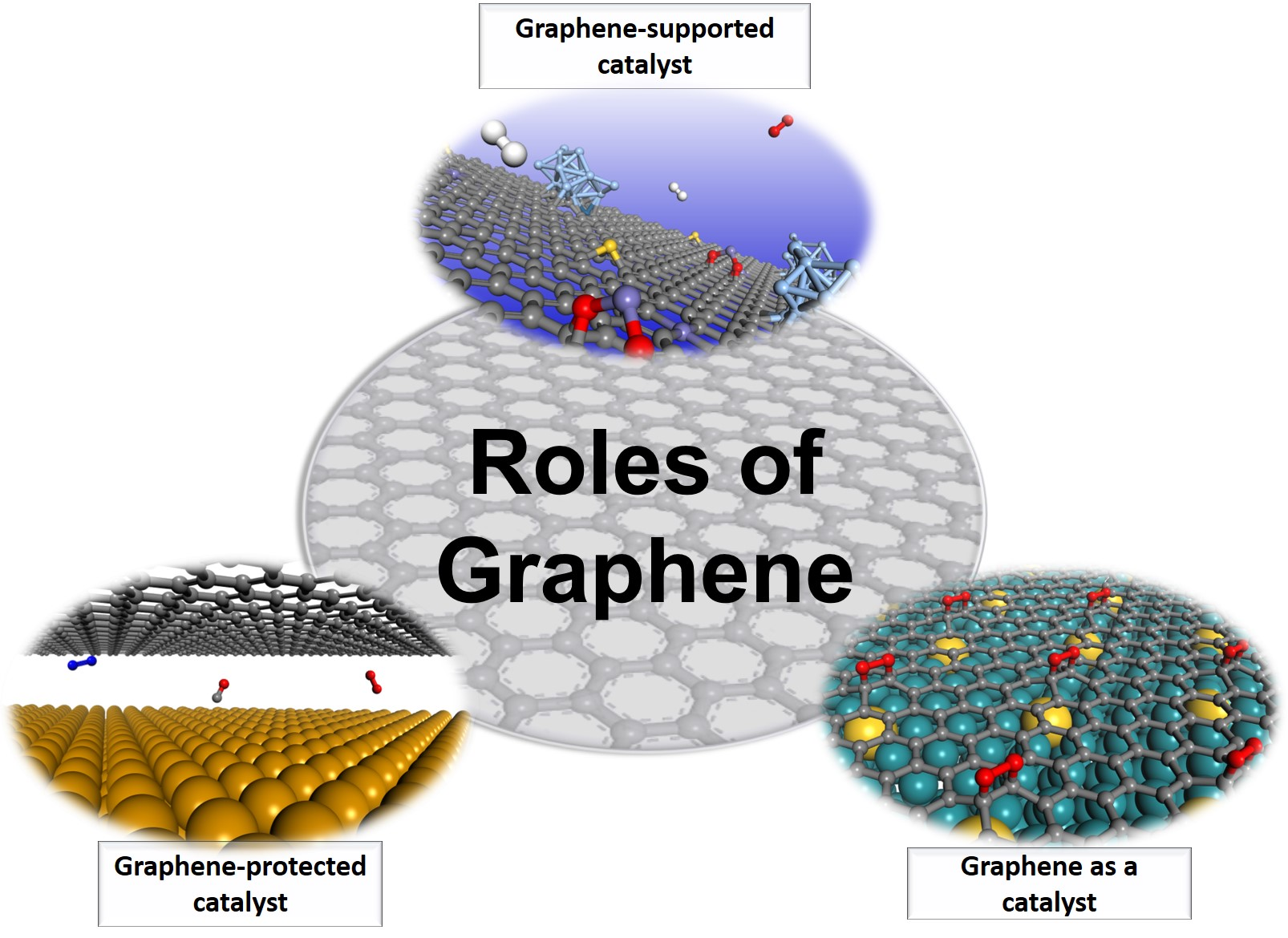
One can essentially identify three distinct roles that graphene can assume in solid-state catalysis when surveying the literature, namely, as a support, a cover, or the catalytic center, as shown in Figure 1. Graphene can be an excellent support for catalysts because of the highly tunable interaction between graphene and the supported catalysts. Graphene itself can be a surprisingly good catalyst due to the highly tunable graphene-substrate interaction. Thanks to its stable structure and chemical inertness, graphene has also been used a wonderful protective cover in many catalysis applications. In the following subsections, each role of graphene will be clearly elucidated with specific examples. By and large, there are many more theoretical proposals and experimental realizations of graphene-supported catalysts than the other two. This may be partially attributed to the burgeoning field of single-atom catalysis, which have attracted considerable attention recently.
2.1. Graphene as a Support
When functioning as a support, graphene can support either nano- or atomic-scale catalysts, as depicted in Figure 2. There are essentially four classes of graphene-supported catalysts based on the foreign atoms introduced: graphene confining (1) metallic nanoclusters/nanoparticles (Mx/G); (2) single atoms including both metallic and non-metallic ones (M1/G); (3) single atoms-Nx moieties embedded in the graphene matrix (M1Nx/G); and (4) single atoms or clusters supported by functionalized graphene such as graphene oxide (Mx/GO) or reduced graphene oxide (Mx/rGO). Regardless of the catalytic active species, graphene should anchor them well while functioning as a support.
It is well known that metal atoms catalyze the growth of carbon nanotubes, and that was the starting point when metal-carbon interactions were studied [24]. Hence, research in metallic species supported by graphene came as a natural extension from the studies that were first started with carbon nanotubes and fullerenes because they were discovered earlier. It has now been accepted that the adsorption of metallic atoms on pristine graphene, i.e., having adatoms on pristine graphene, can be used to tune the electronic structure of graphene [25,26]. As an example, transition metal atoms adsorbing on graphene can turn the material magnetic, which is particularly interesting for graphene-based electronic and spintronic devices [27]. It was found that the bonding between metal adatoms and graphene can be either ionic or covalent. In the case of those from groups 1, 2 and 13 of the Periodic Table, ionic bonding dominates the interaction, which is characterized by the significant charge transfer from the adatoms to graphene, while leaving the electronic states of graphene largely unperturbed [28,29,30]. Such adatoms make good agents for the electron-doping of graphene. On the other hand, it was found that the bonding between graphene and adatoms of transition, noble and group 14 metals is covalent due to strong hybridization of their electronic states upon adsorption [25,26,28,30,31].
Regardless of the bonding, generally, metal-graphene interactions are ‘weak’ and will thus lead to aggregation and formation of metal nanoclusters, as was first pointed out in 2005 by Sun et al., in their theoretical study [32]. This clustering of the metal atoms was subsequently validated in experiments [33,34]. Notice that when we describe the interactions between adatoms on pristine graphene, we put the word ‘weak’ in inverted commas, because to say that such interactions are ‘weak’ can be misnomer as the calculated adsorption energies of certain metallic atoms such as Li, Co and Ni can be larger than 1.0 eV [26,30,31,35]. Therefore, the origin of the instability of such atomically dispersed species on graphenic surfaces is actually due to the much greater cohesive energy of the bulk metal of the adatom being considered, as is evident from the fact that the adsorption energy is only a fraction of the corresponding cohesive energy [28,30,36,37]. In addition, the low to moderate migration energies [38] confer high mobility to the adsorbates on the graphene, such that they tend to aggregate, which is undesirable. As such, the challenge here for graphene to be viable as a catalyst support is the tuning of the adsorbate-graphene interaction. To better anchor the foreign atoms, pristine graphene must therefore undergo specific treatment to increase its chemical reactivity, which will then improve the interaction between the active species and graphene, simultaneously stabilizing them and tuning their catalytic performance.
2.1.1. Vacancy Enhanced Graphene Reactivity
Structural or chemical defects are natural ways of modifying the local chemical properties of graphene and thus tuning the graphene-adsorbate interaction. While the most common intrinsic point structural defect occurring in graphene is a double vacancy (DV), as it is more thermodynamically preferred [39], the simplest one is a single vacancy (SV). Vacancies can serve as a trap for incoming atoms and nanoclusters. Various clusters and single atoms, including non-metallic ones, have been shown to adsorb more exothermically in the vacancies. The effect of a vacancy is that it disrupts both the σ bonding and π bonding of the surrounding carbon atoms, leaving them undercoordinated with dangling bonds. This dangling bond effect gives rise to localized states near the Fermi level, as observed in the local density of states probed by scanning tunneling spectroscopy, which corresponds to protrusions around the SV when surveying the topography of the surface using a scanning tunneling microscope [40]. Hybridization between these states and the electronic states of the adsorbates results in larger adsorption energies of the adsorbates compared to the cohesive energy on graphene which can reduce the mobility of adsorbates on graphene, thus decreasing the diffusion barrier and the probability of the aggregation.
This was supported by the theoretical study by Okamoto in 2006, which reported that the metal nanoclusters could be made less labile when anchored on defective graphene, although without further analysis [13]. Later, the dynamics of atomically dispersed Au and Pt were studied experimentally by Banhart and co-workers [41,42]. From their in-plane diffusion measurements, they deduced that the migration energies of Au and Pt on defective graphene at 600 °C were both approximately 2.5 eV, with the value of migration energy of Au being supported by theoretical calculations [43]. In addition, the binding energy of an Au atom to the defective graphene is enhanced by at least 20 times [27]. Such results are indicative that the stability of adsorbates on defective graphene is dramatically improved. Significant charge redistribution takes place between the adsorbed cluster or atom and defective graphene at the interface, altering the electronic structure of the adsorbates, and thus making them suitable catalysts.
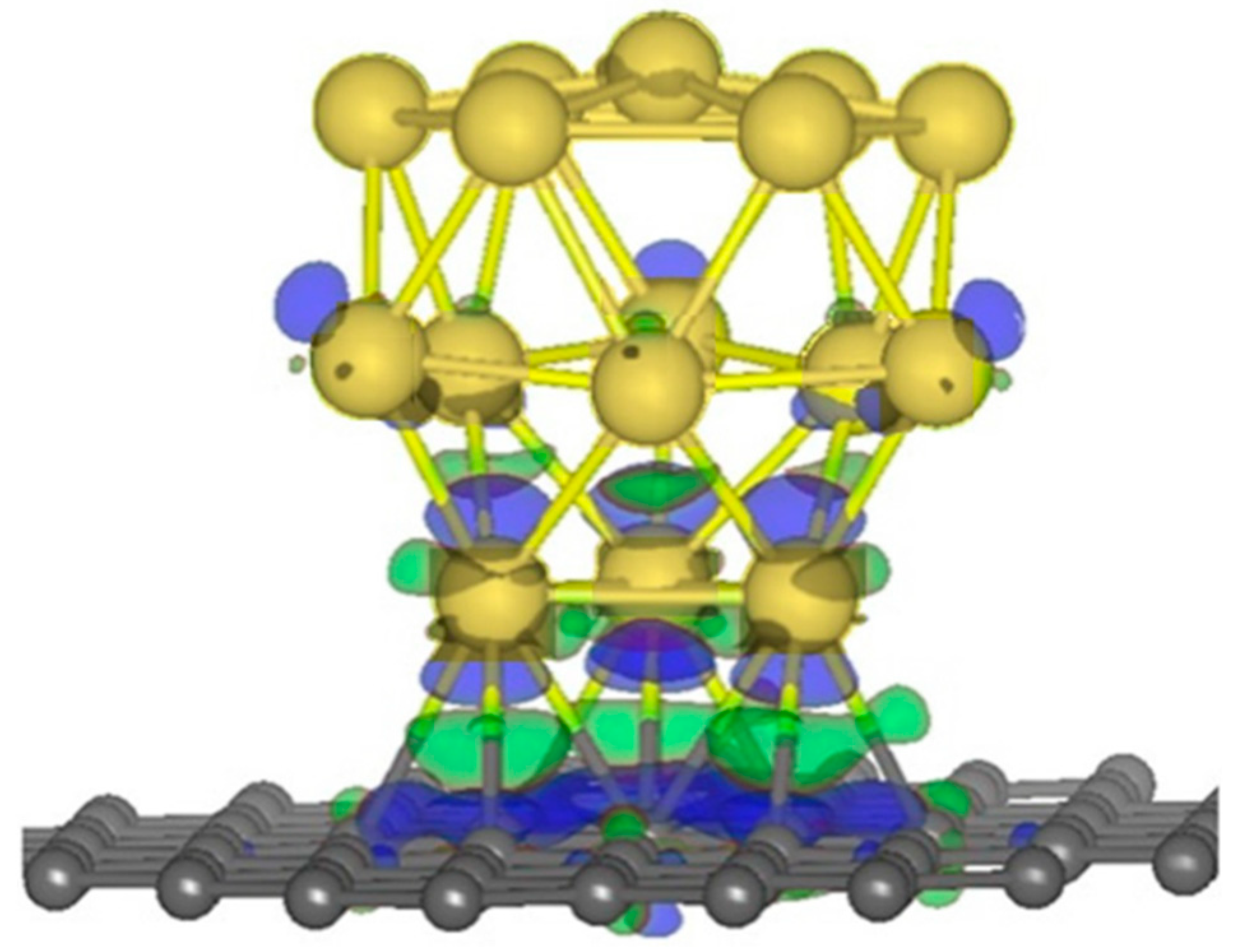
Nanoscale catalysts. Perhaps the first experimental work on graphene-supported nanocluster catalysis came in 2009 reporting on Pt subnanoclusters electrocatalyzing methanol oxidation reaction with an unusual high activity [44]. This then spurred more studies investigating the reason behind the enhanced catalytic activity adsorbates supported by defective graphene. Zhou et al. was the earliest systematic computational study on the catalytic properties of supported nanoclusters on defective graphene [45]. It was pointed out that the origin of the greatly enhanced chemisorption of metal clusters is the strong interaction between the SV in graphene and the adsorbed metal clusters (as demonstrated in Figure 3) due to the defect-induced breaking sp2 of the bonding of the graphene, leading to the strong hybridization between carbon 2p and Au/Pt 5d orbital around the defect, inducing interfacial charge transfer and hence a significant change in the electronic structures of the clusters. Such changes then optimize their catalytic properties. The exothermic adsorption of O2 molecules on the metal clusters with elongated O-O bond lengths and the oxidation of CO to CO2 only require an energy barrier smaller than 0.3 eV to be overcome for the four systems. In comparison, without the monovacancy in graphene, O2 does not adsorb on the Au clusters on pristine graphene, and thus the reactivity of the Au clusters supported on defective graphene was concluded to be defect-induced.
Consequently, Ramasubramaniam and co-workers studied various Pt nanoclusters on graphene, with various defects giving similar conclusions with regard to CO oxidation reaction [46,47,48]. The enhanced adsorption of the nanoclusters due to the strong hybridization of the adsorbates with the dangling bonds of neighboring carbon atoms in the vicinity of the vacancy also holds for other nanoclusters, such as Fe13 [49], Al13 [49], Nin (n = 1–6) [50] and Con (n = 2, 4, 6) [51]. The hybridization thus leads to interfacial charge transfer, which is the general explanation of the higher catalytic activities of the supported metallic nanoclusters.
Single-atom catalysts. Lu et al. first carried out a DFT study on how a single atom embedded in graphene with an SV can result in support-induced catalysis. A single gas-phased Au atom has an electronic configuration of 5d106s1. It was found that when an Au atom substitutes for a C atom, the dopant atom transfers the charge to the neighboring atoms, resulting in a positively charged Au dopant with the 6s orbital and the spin-down components of the top 5d orbital become empty. On the other hand, the spin-up components of the top 5d orbital of Au and 2p orbital of neighboring C atoms are partially filled and are localized around the Fermi level, which dominate the magnetic moment of the system, as shown in Figure 4. It was then predicted that the two-step CO oxidation will preferentially proceed via a Langmuir-Hinshelwood (LH) step with an activation energy of only 0.31 eV, followed by Eley-Rideal (ER) step involving an activation energy 0.18 eV. Detailed analysis of the reaction mechanism revealed that the partially occupied 5d states originating from the Au dopant play an important role in the catalytic activity. Upon the co-adsorption of O2 with CO on Au, Au transfers electrons to the initially half-filled 2π* orbital of O2, resulting in the empty 5d orbital of Au and the spin up component of 2π* orbital being populated. The O-O bond in O2 elongates, which facilitates the facile oxidation of CO, for which the activation of O2 is often the rate-limiting step.
Following this work, many theoretical predictions were made with different substitutional dopant metallic atoms, including Fe [52,53,54], Cu [55], Pt [56], Al [57,58,59], Zn [59], Co [59], Ni [59], Sn [60], Mo [61], Pd [62,63], W [64], Mn [65], and Cr [66], and including non-metallic ones such as Si [67,68], N [69], P [69,70], Ge [58], demonstrating that such graphene support atoms are excellent thermocatalysts for CO oxidation, where the catalytic centers are at the dopants. By no means is this list exhaustive. It is also worthy of note that besides O2, other small molecules such as H2, CO, NO2, NH3 and H2O2 have been predicted to be able to chemisorb on graphene by forming covalent interactions with the doping atom [15,17,18,37]. This would suggest that such simple M1/G may not be limited only to catalyzing the CO oxidation reaction.
Early experimental realizations of confining single atoms in the graphene matrix were mostly limited to doping graphene with either nitrogen or boron either during graphene synthesis or post-treatment, because of the potential applications of N-doped and B-doped graphene in electronics. In addition, such nonmetal-doped carbon nanotubes have been recognized as promising metal-free catalysts for oxygen reduction reaction (ORR), oxygen evolution reaction (OER), and hydrogen evolution reaction (HER) [23,71]. Nevertheless, most metal-free graphene-based catalysis has concentrated on N-doped systems, probably due to the difficulty in the fabrication of B-doped graphene catalysts [23]. A peculiarity in N-doped graphene is that the active sites are generally the carbon atoms in the vicinity of the dopant. As nitrogen is more electronegative, electrons are depleted from the C atoms, but the single atom-induced activity in N-doped graphene originates from the back-donation of the lone pair of electrons on N to the C atoms surrounding the N atom, increasing the electron density on the C atoms, as reflected as bright regions in STM measurements [72].
When graphene is substitutionally doped with nitrogen, it can result in four major types of nitrogen atoms, embedded in the graphene lattice according to their chemical environment, namely, graphitic, pyridinic, pyrrolic, and pyridine oxide like, depending on the method used in their synthesis [73,74]. In the selective oxidation of benzylic C-H bonds [73] and the epoxidation of trans-stilbene [74], Ma and co-workers synthesized N-doped graphene. It was found that the higher the nitrogen content, the higher both the catalytic activity and the yield of the desired product, and in particular, the catalyst with 8.9% nitrogen content gave a selective oxidation of ethylbenzene a conversion of 98.6% and a 91.3% yield of the product, with the N dopants occupying the graphitic positions preferentially. Some light was shed on the superior catalytic performance of X-ray absorption spectroscopy (XAS) by studying the reaction progress, with the authors concluding that the superior catalytic activity of N-doped graphene compared to the undoped one was due to the promotion of the formation of reactive peroxide-like species in the selective oxidation, inducing a greater degree of C-H activation. It was found from the DFT simulations that the peroxide-like species preferentially adsorbed onto the ortho-carbon over the other two types of carbon and could not be adsorbed onto the N dopant. After adsorption, the ortho-carbon protrudes out of the graphene plane, supported by the decreased π* adsorption in the C K-edge XAS spectra.
On the other hand, doping with metallic atoms into graphene is challenging. Traditional technology, such as ion implantation, a large-scale site-selective doping method employed in semiconductor processing, met with little success, as it was difficult to achieve fine spatial control [75]. Higher efficacy was achieved when Zhang and co-workers devised a two-step process. Vacancies are created when C atoms are first knocked out by bombarding them using high-energy ions, which are then filled up by desired dopants [19]. However, defect formation is only sporadic and lacks spatial control. Warner and co-workers later improved on the method employed in defect creation by using aberration-corrected transmission electron microscope (AC-TEM) in knocking out the C atoms in graphene with a 10 × 10 nm2 spatial control, but also allows in situ monitoring, without the need for removing the sample from vacuum [76,77]. All in all, vacancies indeed function as trap sites filled by dopants that were initially diffusing on the surface of graphene, and the stability of the dopants, at least under room conditions, was ensured due to the much larger binding energies of the metal–vacancy complexes. While these two groups successfully achieved the atomic doping of graphene, the dopants were observed to undergo limited migration to adjacent lattice sites. This means that the doping concentration of these atoms cannot be too high, or the atoms will be too close to each other and may aggregate and sinter to form clusters while undergoing limited migration.
Recently, there have been reports on the fabrication of atomically dispersed metal atoms or metal dimers in the graphene matrix [78,79,80,81,82]. Of these, two groups have reported that the catalytic centers for their reactions were predominantly carbon-coordinated Ni atoms [81,82]. These Ni atoms were found to be anchored in various graphene defects and remained stable after a sustained period of operation without significant reduction in catalytic activities. Some others reported the successful embedding of metal atoms and dimers into the graphene matrix, but advanced characterization techniques reveal that metallic species were embedded with the help of anchor atoms [78,79,80]. The use of foreign atoms as anchors will be covered in the following subsection.
2.1.2. Anchor Atoms to Stabilize the Supported Atomic Scale Catalysts
Use of anchor atoms can further stabilize the dopant due to the formation of stronger dopant–anchor atom bonds than dopant–carbon bonds, and this can effectively immobilize the dopant atoms on the condition that carbon can also form strong bonds with these anchor atoms. As such, the anchor atoms act as ‘mediators’, which not only form strong interactions with carbon atoms, but also the dopant atoms. Oxygen and nitrogen are species that can be used as anchor atoms from the common knowledge that C–O and C–N bonds are particularly strong and stable in organic chemistry, giving rise to various homologous series with compounds that are either oxygen- or nitrogen-containing.
As mentioned earlier, there have been several reports on the successful embedding of metallic species, which is possible with the help of O atoms as anchors [78,79,80]. The O atoms were intentionally introduced during the synthesis process to stabilize the metal single-atom catalysts supported on graphene, as sketched in Figure 5, where O atoms were used to anchor Co single-atom catalysts [74]. By extension, exploiting graphene derivatives may be a good alternative to prevent aggregation of the single atoms or metal clusters since the heteroatoms present may also function as anchoring functional groups for the adsorbates. Taking graphene oxide (GO) as an example, it was earlier demonstrated that GO may serve as better support for iron atoms [83] or metal clusters [84] for CO oxidation, because neither defect needs to be introduced, nor does strain need to be applied.
The possibility of using N-doped graphene to use a substrate to confine the dopants within the layer was proposed as early as 2008 by Kim and Jhi [36]. They claimed that the adsorption energies of Sc, Ti, V atoms exceed the corresponding bulk metal cohesive energy and the dopants can therefore remain atomically dispersed. However, with the exploration of utilizing graphitic carbon nitride (g-C3N4) in catalysis, several groups turned to this particular N-doped graphene to anchor dopant atoms. The first report was probably on Pd embedded in g-C3N4, which showed good catalytic performance for the hydrogenation of alkynes and nitroarenes [85,86]. However, it was recently reported that g-C3N4 generally can support a lower loading than graphene owing to its much stronger interlayer interactions [83]. Readers who are interested in g-C3N4-doped systems for catalysis are advised to consult other reviews found elsewhere [9,87,88,89].
Additionally, g-C3N4, metal-Nx moieties are also commonly found in nature in macrocyclic organic molecules like porphyrin and phthalocyanine. Such compounds have proved to be very versatile and exhibit high tailorability, as the Nx pore can be filled with different metallic ions while maintaining the structural integrity. The design of M1Nx/G catalysts can be interpreted as M1Nx embedded in the graphene framework, experimentally showing that catalytic active species are effectively immobilized [90,91,92]. Deng et al. reported the anchoring of a Fe atom in the graphene matrix by four surrounding N atoms via high-energy ball milling of iron phthalocyanine molecules and graphene [90]. Advanced characterization techniques did not show any aggregation of Fe, and the Fe1N4 moieties seemingly filled in the graphene divacancies, as shown in Figure 6. The catalyst synthesized showed high activity and selectivity for the oxidation of benzene to phenol. From a first-principle study, Si atoms anchored by four surrounding N atoms in the graphene matrix also showed good thermal stabilities in the molecular dynamics simulations and predicted to function as an efficient catalyst for CO oxidation [93]. The use of anchor atoms introduces another degree of freedom in the design of graphene-supported catalysts, which giving rise to different ratios of metal to nitrogen when nitrogen atoms are used [92,94], and possibly modulating the catalytic performance [94].
2.1.3. Strained-Enhanced Graphene Reactivity
Zhou et al. proposed that pristine graphene could achieve the requirements as a good catalytic support for metal clusters just by applying a moderate biaxial tensile strain along the zigzag and armchair directions [95]. Comparing the band structures of pristine graphene and graphene subjected to a 5% strain, graphene under strain remains gapless, as the symmetry between two sublattices is not broken. The width of the bands is noticeably smaller for the graphene under strain, and the decrease in band width is due to the weaker interaction among the carbon atoms, as they are further apart under strain, which was confirmed for the π and π* bands near the Fermi energy, which determines the chemical reactivity of graphene by considering a single-orbital (C 2pz) nearest-neighbor tight-binding model.
Of the five clusters considered—Pt4, Ag7, Pd9, Al13 and Au16—only Pt4 and Pd9 can be anchored on pristine graphene with significant adsorption energies (>1.0 eV), while the rest are only weakly adsorbed. When a strain is applied, the cluster-graphene distance decreases, and the adsorption energy of the clusters increases with increasing amount of strain; the onset of significant energy changes occurs at 5% for all clusters, indicating the strain-enhanced stabilization of the metal clusters. Such stabilization can be understood from the increase in the chemical reactivity of graphene when strain is applied. While the π binding between the carbon atoms in graphene is weakened, it is significant enough to strengthen the interaction between the carbon 2pz orbital and molecular orbital of adsorbed clusters. Such enhancement in adsorption was also observed experimentally [96].
The authors then focused on the analysis of the chemisorption and the catalytic activities of the Au16 clusters, which is intriguing. The moderate amount of strain reverses the charge transfer from the graphene to the Au16 cluster. While supported on graphene without strain being applied, the cluster is slightly negatively charged and chemically inactive towards O2 adsorption. When the strain was applied to graphene, the cluster became positively charged, shifting the HOMO of Au16 above Fermi level, as shown in Figure 7. Furthermore, the band structure of graphene was distorted, breaking the π and π* bands at the Dirac point, inducing a band gap of around 0.4 eV. This is evidence for the significant strain-induced Au16–graphene interaction, allowing chemisorption of O2, decreasing the energy barrier for the CO oxidation reaction from ~3.0 eV in the gas phase to less than 0.2 eV. The strain-induced activity of graphene-supported catalysts has been used to explain experimental findings [96], but the direct experimental evidence of strain effects in graphene catalysis is still missing thus far.
2.2. Graphene as a Cover
As mentioned earlier, the extensive conjugation of the p orbitals and the low density of states as a result of the zero bandgap result in the structural stability and chemical inertness of pristine graphene. The chemical inertness of graphene can in fact be useful in providing an effective overlay over precious and nonprecious metal catalysts against harsh reaction media, modulating the reactivity of the active sites beneath it. The modulation of the reactivity of the active sites in heterogeneous catalysis is similar to a concept in enzymatic systems, where the matrices of the catalytically active atoms play an important role in adjusting the geometric configurations in order to induce a change in the electronic structure of the catalyst atoms such that they are optimal for the catalysis, i.e., the matrix induces the catalyst atoms to be in the entactic state.
Earlier studies on the catalytic properties of the nanocavities in zeolitic systems and metal-organic frameworks (MOFs), which are zero-dimensional (0d), and in carbon nanotubes, which are one-dimensional (1d), reveal that the adsorption of small molecules is modulated by the nest effect and quantum confinement effect. However, in comparison with 2d confined spaces, those with 0d and 1d natures are not structurally well-defined, and it is therefore difficult for them to be aptly theoretically modelled or experimentally investigated with advanced characterization techniques. Firstly, the 2d cover should be able induce the quantum confinement effect, which reduces the chemical reactivity of the catalytically active species, thus achieving optimization of the adsorption of target molecules and simultaneously alleviating the degradation of the catalyst in harsh operating environment. With graphene as the first 2d material discovered, graphene spearheaded a whole new direction of chemistry under 2d cover.
In the studies of CO intercalation involving graphene grown on Pt(111) surface by Bao and coworkers, it was found that upon exposure to CO, the gas intercalates at the interface and adsorbs on Pt [97]. A weaker adsorption of CO was observed with the graphene cover than adsorption on free Pt. This was confirmed by the fact that CO desorbs at a much lower temperature with the graphene cover than without cover. Similarly, oxygen intercalation was studied in graphene in Ru(0001) by Sutter and coworkers, and it was found that the graphene cover can also weaken the adsorption of oxygen on Ru(0001), allowing desorption to take place at a lower temperature [98]. These studies indicate that the space between graphene and the metal substrate can function as a nanoreactor for chemical reactions to take place, as illustrated in Figure 8. Using CO oxidation as a probe, not only does the cover weaken the adsorption of CO on Pt, which alleviates the problem of the CO poisoning effect, thereby inhibiting the catalytic sites, it also induces the weakening of the C–O bond in adsorbed CO and promotes the formation of O–CO bond in the CO oxidation reaction by transferring electrons from the carbon atoms of graphene directly above the CO to the CO and O adsorbates [97].
The typical height of such nanoreactors, i.e., the distance between graphene and the metal substrate, is around 3 Å. This means that the intercalating species should be small enough for graphene to exert a quantum confinement effect in modulating their adsorption and desorption on the metal substrate. This implies that such chemistry under cover is only restricted to catalyzing reactions involving small molecular species. To extend the applicability of graphene-based solid-state catalysts to a greater variety of reactions, the catalytic activity of graphene should be tuned such that reactions can take place directly on the exposed graphene surface.
2.3. Graphene Itself as a Catalyst: Substrate Engineering of Graphene Reactivity
To tune the electronic properties of graphene such that the carbon atoms can directly act as the catalytic center, substrate engineering seems a plausible and promising way, because graphene is only one atomic layer thick. When impurity atoms (IAs) or defects are introduced at the surface of the substrate, charge redistribution occurs, and the local chemical properties of graphene are modulated. One key advantage is that graphene can be grown on an appropriate substrate, and the substrate can be engineered before or after growth for catalytic purposes, avoiding the need for graphene exfoliation and transfer, which retains the high quality of graphene grown. Such facile activation of graphene makes it relevant in industrial applications, as it avoids complicated synthetic processes, while at the same time enabling growth of large areas of graphene with a high concentration of active sites. Today, the two most common approaches for large-area graphene synthesis are chemical vapor deposition over metal crystals and high-temperature annealing of hexagonal silicon carbide (h-SiC), rather than simple mechanical exfoliation, which produces small and irregular graphene flakes.
For the former graphene growth method, there are three classes of three metal substrates that graphene can be grown over, including strongly interacting ones, e.g., Ru(0001) and Rh(111), weakly interacting ones, e.g., Au(111) and Pt(111), and finally metal substrates like Ir(111) and Pd(111), which lie in between the spectrum from the strongly interacting ones to the weakly interacting ones. When graphene interacts strongly with the metallic substrate, significant hybridization occurs, which not only induces buckling and corrugation of the carbon overlay, but also leads to the disappearance of the characteristic Dirac cones in graphene. At the other end of the spectrum, graphene only physisorbs on weakly interacting metal surfaces, while preserving the linear dispersion at Dirac points.
To illustrate the activation of graphene via substrate engineering, Zhang and coworkers turned to graphene on Ru(0001) as the study system [11], as Ru is one of the best substrates for graphene growth, and the system has been clearly elucidated [99,100]. The graphene binds strongly to Ru(0001) and is n-doped, but the significant lattice mismatch of graphene on Ru(0001) gives rise to moiré patterns. To account for the moiré pattern, four possible high-symmetry configurations of gr/Ru(0001), i.e., fcc-top, hcp-top, fcc-hcp, and bridge-top, in which the carbon atoms are stacked differently on top of the Ru atoms, were considered. It was found that graphene in fcc-top, hcp-top and bridge-top configurations bound strongly to the Ru, with significant charge transfer from the substrate to the graphene. On the other hand, graphene only adsorbs very weakly onto Ru in the fcc-hcp configuration, as there is no visible charge redistribution. Nevertheless, the graphene in all four configurations remained inert towards oxygen.
To engineer the substrate, introduction of a point defect in the substrate, in the form of either an IA and SV, was investigated. It was found that Au, Au, Cu and Zn dopants, and a V in the interfacial Ru layer, were able to activate graphene for all configurations except for the fcc-hcp ones. By plotting the charge redistribution plots superimposed on the optimized atomic structures, the defect weakened the bonding between these affected C atoms and the substrate, resulting in a weaker graphene adsorption compared with undoped cases for the fcc-top, hcp-top and bridge-top configurations. For the fcc-hcp case, the introduction of either IA or SV had negligible effects on graphene due to the weak interaction between graphene and the substrate, and hence will not be considered further from this point onwards.
It is expected that the defect induced charge transfer to create a local region in the supported graphene with different chemical properties from other parts. This activates graphene, inducing chemisorption of O2 on graphene only on those local regions. The chemisorption was confirmed by three indicators: the significant adsorption energies of O2 on the activated graphene (Eads), the gaining of a significant amount of charge by the more electronegative O2 molecule from graphene (ΔQ), and the elongation of the O–O bond compared to the calculated gas-phase value of 1.24 Å (dO-O). Furthermore, density of states (DOS) analysis revealed that the electrons gained by O2 filled its 2π* orbital, which is its lowest unoccupied molecular orbital (LUMO). Noting that O2 is magnetic, the electrons transferred from the graphene entered the antibonding LUMO, pulling it down to below Fermi energy, eliminating the magnetism and lengthening the O–O bond after adsorption. The bond elongation implies the activation of the O2 molecule. The catalytic activity is then studied with the oxidation of CO as the probe reaction. In all cases, a complete cycle of the catalyzed oxidation proceeds with a two-step Eley-Rideal (ER) mechanism since the CO molecule does not bind onto the supported graphene. The first step involves the oxidation of one CO to CO2, which approaches the adsorbed O2 from gas phase. CO (g) + O2 (ads) → CO2 (g) + O (ads). Another CO then approaches the leftover adsorbed O atom, forming CO2, regenerating the clean surface. CO (g) + O (ads) → CO2 (g).
From this study, some points can be highlighted. Firstly, the substrate chosen should interact strongly with graphene that is n-doped. Secondly, the dopant (or vacancy) should have a weaker interaction with the graphene than the substrate to induce charge redistribution from the defect to the carbon atoms just above the defect, enabling the donation of electrons to the incoming oxygen molecules from the gas phase. When the substrate underneath is substitutionally doped with an IA, as illustrated in Figure 9, such systems can be considered a novel type of single-atom catalyst, inducing the catalytic activity first by facilitating the charge transfer from the dopant to the carbon atoms.
3. Conclusions
Reactions happen on the surface of traditional three-dimensional catalysts and therefore giving rise to the term, ‘surface catalysis’. The discovery of the first two-dimensional material, graphene, represents the reincarnation of the surface, which, in fact, has widened the horizon of the field of surface catalysis, with graphene being a rising star in terms of its potential applications in catalysis. We pointed out that the vast number of papers related to graphene-based solid-state catalysts found in the literature is no accident, but can be attributed to the peculiar electronic structure of graphene (zero bandgap with low DOS around Fermi level) and the ease of its modulation due to its being only one atom thick. One of the key issues in graphene-based catalysis is to find an effective yet practical way to activate chemically inert graphene. Vacancies, impurity atoms, and substrate engineering have been discussed as tools for activating graphene. Additionally, the concept of single-atom catalysis is another frontier of catalysis, and the marriage of both graphene and single-atom catalysis has achieved significant success. Over the course of the years, as the experimental technique matures, the fabrication of such graphene-supported single-atom catalysts has become plausible, but the lack of fine control is still the limiting factor that prevents its industrial application. As a result, other aspects of graphene have been scrutinized, and it has been found that in addition to acting as a support, graphene can also act as a cover over metallic substrates, modulating the reactivity of the surface beneath the cover. Finally, the local chemical properties of epitaxial graphene can be tuned via substrate engineering, whereby the graphene can be turned as the active center. It is hoped that this is a timely review summarizing the different roles played by graphene in solid-state catalysis.
Author Contributions
All contributed significantly to the writing of this review. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by NUS academic research fund (R-144-000-410-114) and NUS green energy program (R-143-000-A63-114).
Acknowledgments
We acknowledge the support from NUS academic research fund (R-144-000-410-114) and NUS green energy program (R143-000-A63-114). Computational works were performed at the Graphene Research Centre computing cluster facilities.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Geim, A.K.; Novoselov, K.S. The rise of graphene. Nat. Mater. 2007, 6, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Neto, A.H.C.; Guinea, F.; Peres, N.M.R.; Novoselov, K.S.; Geim, A.K. The electronic properties of graphene. Rev. Mod. Phys. 2009, 81, 109–162. [Google Scholar] [CrossRef] [Green Version]
- Das Sarma, S.; Adam, S.; Hwang, E.H.; Rossi, E. Electronic transport in two-dimensional graphene. Rev. Mod. Phys. 2011, 83, 407–470. [Google Scholar] [CrossRef] [Green Version]
- Morozov, S.; Novoselov, K.; Katsnelson, M.I.; Schedin, F.; Elias, D.C.; Jaszczak, J.A.; Geim, A.K. Giant Intrinsic Carrier Mobilities in Graphene and Its Bilayer. Phys. Rev. Lett. 2008, 100, 016602. [Google Scholar] [CrossRef] [Green Version]
- Novoselov, K.; Geim, A.K.; Morozov, S.; Jiang, D.; Zhang, Y.; Dubonos, S.V.; Grigorieva, I.V.; Firsov, A.A. Electric Field Effect in Atomically Thin Carbon Films. Science 2004, 306, 666–669. [Google Scholar] [CrossRef] [Green Version]
- Machado, B.F.; Serp, P. Graphene-based materials for catalysis. Catal. Sci. Technol. 2012, 2, 54–75. [Google Scholar] [CrossRef]
- Fan, X.; Zhang, G.; Zhang, F. Multiple roles of graphene in heterogeneous catalysis. Chem. Soc. Rev. 2015, 44, 3023–3035. [Google Scholar] [CrossRef]
- Navalon, S.; Dhakshinamoorthy, A.; Alvaro, M.; Garcia, H. Metal nanoparticles supported on two-dimensional graphenes as heterogeneous catalysts. Coord. Chem. Rev. 2016, 312, 99–148. [Google Scholar] [CrossRef]
- Wang, Y.; Mao, J.; Meng, X.; Yu, L.; Deng, D.; Bao, X. Catalysis with Two-Dimensional Materials Confining Single Atoms: Concept, Design, and Applications. Chem. Rev. 2019, 119, 1806–1854. [Google Scholar] [CrossRef]
- Lu, Y.-H.; Zhou, M.; Zhang, C.; Feng, Y.-P. Metal-Embedded Graphene: A Possible Catalyst with High Activity. J. Phys. Chem. C 2009, 113, 20156–20160. [Google Scholar] [CrossRef]
- Guo, N.; Yam, K.M.; Zhang, C. Substrate engineering of graphene reactivity: Towards high-performance graphene-based catalysts. npj 2D Mater. Appl. 2018, 2, 1. [Google Scholar] [CrossRef] [Green Version]
- Pop, E.; Varshney, V.; Roy, A.K. Thermal properties of graphene: Fundamentals and applications. MRS Bull. 2012, 37, 1273–1281. [Google Scholar] [CrossRef] [Green Version]
- Okamoto, Y. Density-functional calculations of icosahedral M13 (M=Pt and Au) clusters on graphene sheets and flakes. Chem. Phys. Lett. 2006, 420, 382–386. [Google Scholar] [CrossRef]
- Leenaerts, O.; Partoens, B.; Peeters, F.M. Adsorption of H2O, NH3, CO, NO2, and NO on graphene: A first-principles study. Phys. Rev. B 2008, 77, 125416. [Google Scholar] [CrossRef] [Green Version]
- Zou, Y.; Li, F.; Zhu, Z.H.; Zhao, M.W.; Xu, X.G.; Su, X.Y. An ab initio study on gas sensing properties of graphene and Si-doped graphene. Eur. Phys. J. B 2011, 81, 475–479. [Google Scholar] [CrossRef]
- Antiochia, R.; Tortolini, C.; Tasca, F.; Gorton, L.; Bollella, P. Chapter 1—Graphene and 2D-Like Nanomaterials: Different Biofunctionalization Pathways for Electrochemical Biosensor Developmen. In Graphene Bioelectronics; Tiwari, A., Ed.; Elsevier: Amsterdam, The Netherlands, 2018; pp. 1–35. [Google Scholar]
- Dai, J.; Yuan, J. Adsorption of molecular oxygen on doped graphene: Atomic, electronic, and magnetic properties. Phys. Rev. B 2010, 81, 165414. [Google Scholar] [CrossRef] [Green Version]
- Zhou, M.; Lu, Y.-H.; Cai, Y.-Q.; Zhang, C.; Feng, Y.-P. Adsorption of gas molecules on transition metal embedded graphene: A search for high-performance graphene-based catalysts and gas sensors. Nanotechnology 2011, 22, 385502. [Google Scholar] [CrossRef]
- Wang, H.; Wang, Q.; Cheng, Y.; Li, K.; Yao, Y.; Zhang, Q.; Dong, C.; Wang, P.; Schwingenschlögl, U.; Yang, W.; et al. Doping monolayer graphene with single atom substitutions. Nano Lett. 2012, 12, 141–144. [Google Scholar] [CrossRef]
- Wang, X.; Sun, G.; Routh, P.; Kim, D.-H.; Huang, W.; Chen, P. Heteroatom-doped graphene materials: Syntheses, properties and applications. Chem. Soc. Rev. 2014, 43, 7067–7098. [Google Scholar] [CrossRef] [Green Version]
- Guo, N.; Xi, Y.; Liu, S.; Zhang, C. Greatly Enhancing Catalytic Activity of Graphene by Doping the Underlying Metal Substrate. Sci. Rep. 2015, 5, 12058. [Google Scholar] [CrossRef] [Green Version]
- Fu, Q.; Bao, X. Surface chemistry and catalysis confined under two-dimensional materials. Chem. Soc. Rev. 2017, 46, 1842–1874. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.; Meng, X.; Deng, D.; Bao, X. Confinement Catalysis with 2D Materials for Energy Conversion. Adv. Mater. 2019, 31, e1901996. [Google Scholar] [CrossRef]
- Lee, Y.H.; Kim, S.G.; Tománek, D. Catalytic Growth of Single-Wall Carbon Nanotubes: An Ab Initio Study. Phys. Rev. Lett. 1997, 78, 2393–2396. [Google Scholar] [CrossRef] [Green Version]
- Gierz, I.; Riedl, C.; Starke, U.; Ast, C.R.; Kern, K. Atomic Hole Doping of Graphene. Nano Lett. 2008, 8, 4603–4607. [Google Scholar] [CrossRef] [Green Version]
- Johll, H.; Kang, H.C.; Tok, E.S. Density functional theory study of Fe, Co, and Ni adatoms and dimers adsorbed on graphene. Phys. Rev. B 2009, 79, 245416. [Google Scholar] [CrossRef]
- Krasheninnikov, A.; Lehtinen, P.O.; Foster, A.S.; Pyykkö, P.; Nieminen, R.M. Embedding Transition-Metal Atoms in Graphene: Structure, Bonding, and Magnetism. Phys. Rev. Lett. 2009, 102, 126807. [Google Scholar] [CrossRef] [Green Version]
- Chan, K.T.; Neaton, J.B.; Cohen, M.L. First-principles study of metal adatom adsorption on graphene. Phys. Rev. B 2008, 77, 235430. [Google Scholar] [CrossRef] [Green Version]
- Lugo-Solis, A.; Vasiliev, I. Ab initio study of K adsorption on graphene and carbon nanotubes: Role of long-range ionic forces. Phys. Rev. B 2007, 76, 235431. [Google Scholar] [CrossRef]
- Liu, X.; Wang, C.Z.; Yao, Y.X.; Lu, W.C.; Hupalo, M.; Tringides, M.C.; Ho, K.M. Bonding and charge transfer by metal adatom adsorption on graphene. Phys. Rev. B 2011, 83, 235411. [Google Scholar] [CrossRef]
- Sevinçli, H.; Topsakal, M.; Durgun, E.; Ciraci, S. Electronic and magnetic properties of 3d transition-metal atom adsorbed graphene and graphene nanoribbons. Phys. Rev. B 2008, 77, 195434. [Google Scholar] [CrossRef]
- Sun, Q.; Wang, Q.; Jena, P.; Kawazoe, Y. Clustering of Ti on a C60 Surface and Its Effect on Hydrogen Storage. J. Am. Chem. Soc. 2005, 127, 14582–14583. [Google Scholar] [CrossRef] [PubMed]
- Suarez-Martinez, I.; Felten, A.; Pireaux, J.J.; Bittencourt, C.; Ewels, C.P. Transition metal deposition on graphene and carbon nanotubes. J. Nanosci. Nanotechnol. 2009, 9, 6171–6175. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Feng, Q.; Cheng, Y.; Yao, Y.; Wang, Q.; Li, K.; Schwingenschlögl, U.; Zhang, X.X.; Yang, W. Atomic Bonding between Metal and Graphene. J. Phys. Chem. C 2013, 117, 4632–4638. [Google Scholar] [CrossRef]
- Mao, Y.; Yuan, J.; Zhong, J. Density functional calculation of transition metal adatom adsorption on graphene. J. Phys. Condens. Matter 2008, 20, 115209. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.; Jhi, S.-H.; Park, N. Effective metal dispersion in pyridinelike nitrogen doped graphenes for hydrogen storage. Appl. Phys. Lett. 2008, 92, 013106. [Google Scholar] [CrossRef] [Green Version]
- Kim, G.; Jhi, S.-H.; Lim, S.; Park, N. Effect of vacancy defects in graphene on metal anchoring and hydrogen adsorption. Appl. Phys. Lett. 2009, 94, 173102. [Google Scholar] [CrossRef] [Green Version]
- Nakada, K.; Ishii, A. Migration of adatom adsorption on graphene using DFT calculation. Solid State Commun. 2011, 151, 13–16. [Google Scholar] [CrossRef]
- Banhart, F.; Kotakoski, J.; Krasheninnikov, A.V. Structural Defects in Graphene. ACS Nano 2011, 5, 26–41. [Google Scholar] [CrossRef] [Green Version]
- Ugeda, M.M.; Brihuega, I.; Guinea, F.; Gómez-Rodríguez, J.M. Missing Atom as a Source of Carbon Magnetism. Phys. Rev. Lett. 2010, 104, 096804. [Google Scholar] [CrossRef]
- Gan, Y.; Sun, L.; Banhart, F. One- and Two-Dimensional Diffusion of Metal Atoms in Graphene. Small 2008, 4, 587–591. [Google Scholar] [CrossRef]
- Rodríguez-Manzo, J.A.; Cretu, O.; Banhart, F. Trapping of Metal Atoms in Vacancies of Carbon Nanotubes and Graphene. ACS Nano 2010, 4, 3422–3428. [Google Scholar] [CrossRef] [PubMed]
- Malola, S.; Häkkinen, H.; Koskinen, P. Gold in graphene: In-plane adsorption and diffusion. Appl. Phys. Lett. 2009, 94, 43106. [Google Scholar] [CrossRef] [Green Version]
- Yoo, E.; Okata, T.; Akita, T.; Kohyama, M.; Nakamura, J.; Honma, I. Enhanced Electrocatalytic Activity of Pt Subnanoclusters on Graphene Nanosheet Surface. Nano Lett. 2009, 9, 2255–2259. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Zhang, A.; Dai, Z.; Zhang, C.; Feng, Y.P. Greatly enhanced adsorption and catalytic activity of Au and Pt clusters on defective graphene. J. Chem. Phys. 2010, 132, 194704. [Google Scholar] [CrossRef] [PubMed]
- Fampiou, I.; Ramasubramaniam, A. Binding of Pt Nanoclusters to Point Defects in Graphene: Adsorption, Morphology, and Electronic Structure. J. Phys. Chem. C 2012, 116, 6543–6555. [Google Scholar] [CrossRef]
- Fampiou, I.; Ramasubramaniam, A. CO Adsorption on Defective Graphene-Supported Pt13 Nanoclusters. J. Phys. Chem. C 2013, 117, 19927–19933. [Google Scholar] [CrossRef]
- Fampiou, I.; Ramasubramaniam, A. Influence of Support Effects on CO Oxidation Kinetics on CO-Saturated Graphene-Supported Pt13 Nanoclusters. J. Phys. Chem. C 2015, 119, 8703–8710. [Google Scholar] [CrossRef]
- Lim, D.-H.; Negreira, A.S.; Wilcox, J. DFT Studies on the Interaction of Defective Graphene-Supported Fe and Al Nanoparticles. J. Phys. Chem. C 2011, 115, 8961–8970. [Google Scholar] [CrossRef]
- Song, W.; Wang, J.-L.; Wang, B.; Hu, W.-P.; Wang, Y. First-principles study on the structures and electronic properties of graphene-supported Nin (n = 1–6) clusters. Mol. Simul. 2018, 44, 1529–1538. [Google Scholar] [CrossRef]
- Wang, L.; Ma, S.; Jiao, Z.; Yuan, D. Capability of defective graphene-supported Co4 nanoparticle toward ammonia dehydrogenation. Appl. Surf. Sci. 2019, 465, 1–9. [Google Scholar] [CrossRef]
- Li, Y.; Zhou, Z.; Yu, G.; Chen, W.; Chen, Z. CO Catalytic Oxidation on Iron-Embedded Graphene: Computational Quest for Low-Cost Nanocatalysts. J. Phys. Chem. C 2010, 114, 6250–6254. [Google Scholar] [CrossRef]
- Wannakao, S.; Nongnual, T.; Khongpracha, P.; Maihom, T.; Limtrakul, J. Reaction Mechanisms for CO Catalytic Oxidation by N2O on Fe-Embedded Graphene. J. Phys. Chem. C 2012, 116, 16992–16998. [Google Scholar] [CrossRef]
- Zhou, J.; Tang, Y.; Shen, Z.; Chen, W.; Li, C.; Dai, X. High catalytic activity for CO oxidation on single Fe atom stabilized in graphene vacancies. RSC Adv. 2016, 6, 93985–93996. [Google Scholar] [CrossRef]
- Song, E.H.; Wen, Z.; Jiang, Q. CO Catalytic Oxidation on Copper-Embedded Graphene. J. Phys. Chem. C 2011, 115, 3678–3683. [Google Scholar] [CrossRef]
- Tang, Y.; Yang, Z.; Dai, X. A theoretical simulation on the catalytic oxidation of CO on Pt/graphene. Phys. Chem. Chem. Phys. 2012, 14, 16566–16572. [Google Scholar] [CrossRef]
- Jiang, Q.G.; Ao, Z.M.; Li, S.; Wen, Z. Density functional theory calculations on the CO catalytic oxidation on Al-embedded graphene. RSC Adv. 2014, 4, 20290–20296. [Google Scholar] [CrossRef] [Green Version]
- Esrafili, M.D.; Nematollahi, P.; Abdollahpour, H. A comparative DFT study on the CO oxidation reaction over Al- and Ge-embedded graphene as efficient metal-free catalysts. Appl. Surf. Sci. 2016, 378, 418–425. [Google Scholar] [CrossRef]
- Tang, Y.; Dai, X.; Yang, Z.; Liu, Z.; Pan, L.; Ma, D.; Lu, Z. Tuning the catalytic property of non-noble metallic impurities in graphene. Carbon 2014, 71, 139–149. [Google Scholar] [CrossRef]
- Esrafili, M.D.; Saeidi, N. Sn-embedded graphene: An active catalyst for CO oxidation to CO2? Physica E 2015, 74, 382–387. [Google Scholar] [CrossRef]
- Tang, Y.; Pan, L.; Chen, W.; Li, C.; Shen, Z.; Dai, X. Reaction mechanisms for CO catalytic oxidation on monodisperse Mo atom-embedded graphene. Appl. Phys. A 2015, 119, 475–485. [Google Scholar] [CrossRef]
- Esrafili, M.D.; Nematollahi, P.; Nurazar, R. Pd-embedded graphene: An efficient and highly active catalyst for oxidation of CO. Superlattices Microstruct. 2016, 92, 60–67. [Google Scholar] [CrossRef]
- Xu, G.; Wang, R.; Yang, F.; Ma, D.; Yang, Z.; Lu, Z. CO oxidation on single Pd atom embedded defect-graphene via a new termolecular Eley-Rideal mechanism. Carbon 2017, 118, 35–42. [Google Scholar] [CrossRef]
- Dai, G.; Chen, L.; Zhao, X. Tungsten-Embedded Graphene: Theoretical Study on a Potential High-Activity Catalyst toward CO Oxidation. Materials 2018, 11, 1848. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Q.; Zhang, J.; Ao, Z.; Huang, H.; He, H.; Wu, Y. First Principles Study on the CO Oxidation on Mn-Embedded Divacancy Graphene. Front. Chem. 2018, 6, 187. [Google Scholar] [CrossRef] [Green Version]
- Dai, G.; Chen, L.; Zhao, X. Catalytic oxidation mechanisms of carbon monoxide over single and double vacancy Cr-embedded graphene. J. Mater. Sci. 2019, 54, 1395–1408. [Google Scholar] [CrossRef]
- Zhao, J.-X.; Chen, Y.; Fu, H.-G. Si-embedded graphene: An efficient and metal-free catalyst for CO oxidation by N2O or O2. Theor. Chem. Acc. 2012, 131, 1242. [Google Scholar] [CrossRef]
- Tang, Y.; Liu, Z.; Dai, X.; Yang, Z.; Chen, W.; Ma, D.; Lu, Z. Theoretical study on the Si-doped graphene as an efficient metal-free catalyst for CO oxidation. Appl. Surf. Sci. 2014, 308, 402–407. [Google Scholar] [CrossRef]
- Esrafili, M.D.; Mohammad-Valipour, R.; Mousavi-Khoshdel, S.M.; Nematollahi, P. A Comparative Study of CO Oxidation on Nitrogen- and Phosphorus-Doped Graphene. ChemPhysChem 2015, 16, 3719–3727. [Google Scholar] [CrossRef]
- Esrafili, M.D.; Mousavian, P. Probing reaction pathways for oxidation of CO by O2 molecule over P-doped divacancy graphene: A DFT study. Appl. Surf. Sci. 2018, 440, 580–585. [Google Scholar] [CrossRef]
- Deng, D.; Novoselov, K.S.; Fu, Q.; Zheng, N.; Tian, Z.; Bao, X. Catalysis with two-dimensional materials and their heterostructures. Nat. Nanotechnol. 2016, 11, 218–230. [Google Scholar] [CrossRef]
- Deng, D.; Pan, X.; Yu, L.; Cui, Y.; Jiang, Y.; Qi, J.; Li, W.-X.; Fu, Q.; Ma, X.; Xue, Q.; et al. Toward N-Doped Graphene via Solvothermal Synthesis. Chem. Mater. 2011, 23, 1188–1193. [Google Scholar] [CrossRef]
- Gao, Y.; Hu, G.; Zhong, J.; Shi, Z.; Zhu, Y.; Su, D.S.; Wang, J.; Bao, X.; Ma, D. Nitrogen-Doped sp2-Hybridized Carbon as a Superior Catalyst for Selective Oxidation. Angew. Chem. Int. Ed. Engl. 2013, 52, 2109–2113. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Gao, Y.; Chen, W.; Tang, P.; Li, W.; Shi, Z.; Su, D.; Wang, J.; Ma, D. Catalytic Epoxidation Reaction over N-Containing sp2 Carbon Catalysts. ACS Catal. 2014, 4, 1261–1266. [Google Scholar] [CrossRef]
- Bangert, U.; Bleloch, A.; Gass, M.H.; Seepujak, A.; Berg, J.V.D. Doping of few-layered graphene and carbon nanotubes using ion implantation. Phys. Rev. B 2010, 81, 245423. [Google Scholar] [CrossRef]
- Robertson, A.W.; Allen, C.S.; Wu, Y.A.; He, K.; Olivier, J.; Neethling, J.; Kirkland, A.I.; Warner, J.H. Spatial control of defect creation in graphene at the nanoscale. Nat. Commun. 2012, 3, 1144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robertson, A.W.; Montanari, B.; He, K.; Kim, J.; Allen, C.S.; Wu, Y.A.; Olivier, J.; Neethling, J.; Harrison, N.; Kirkland, A.I.; et al. Dynamics of Single Fe Atoms in Graphene Vacancies. Nano Lett. 2013, 13, 1468–1475. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Cheng, H.; Yi, H.; Lin, Y.; Yao, T.; Wang, C.; Li, J.; Wei, S.; Lu, J. Single-Atom Pd(1)/Graphene Catalyst Achieved by Atomic Layer Deposition: Remarkable Performance in Selective Hydrogenation of 1,3-Butadiene. J. Am. Chem. Soc. 2015, 137, 10484–10487. [Google Scholar] [CrossRef]
- Yan, H.; Lin, Y.; Wu, H.; Zhang, W.; Sun, Z.; Cheng, H.; Liu, W.; Wang, C.; Li, J.; Huang, X.; et al. Bottom-up precise synthesis of stable platinum dimers on graphene. Nat. Commun. 2017, 8, 1070. [Google Scholar] [CrossRef]
- Yan, H.; Zhao, X.; Guo, N.; Lyu, Z.; Du, Y.; Xi, S.; Guo, R.; Chen, C.; Chen, Z.; Liu, W.; et al. Atomic engineering of high-density isolated Co atoms on graphene with proximal-atom controlled reaction selectivity. Nat. Commun. 2018, 9, 3197. [Google Scholar] [CrossRef] [Green Version]
- Jiang, K.; Siahrostami, S.; Akey, A.J.; Li, Y.; Lu, Z.; Lattimer, J.; Hu, Y.; Stokes, C.; Gangishetty, M.; Chen, G.; et al. Transition-Metal Single Atoms in a Graphene Shell as Active Centers for Highly Efficient Artificial Photosynthesis. Chem 2017, 3, 950–960. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Jia, Y.; Gao, G.; Yan, X.; Chen, N.; Chen, J.; Soo, M.T.; Wood, B.; Yang, D.; Du, A.; et al. Graphene Defects Trap Atomic Ni Species for Hydrogen and Oxygen Evolution Reactions. Chem 2018, 4, 285–297. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Zhao, J.; Chen, Z. Fe-Anchored Graphene Oxide: A Low-Cost and Easily Accessible Catalyst for Low-Temperature CO Oxidation. J. Phys. Chem. C 2012, 116, 2507–2514. [Google Scholar] [CrossRef]
- Yang, M.; Zhou, M.; Zhang, A.; Zhang, C. Graphene Oxide: An Ideal Support for Gold Nanocatalysts. J. Phys. Chem. C 2012, 116, 22336–22340. [Google Scholar] [CrossRef]
- Vilé, G.; Albani, D.; Nachtegaal, M.; Chen, Z.; Dontsova, D.; Antonietti, M.; López, N.; Pérez-Ramírez, J. A Stable Single-Site Palladium Catalyst for Hydrogenations. Angew. Chem. Int. Ed. 2015, 54, 11265–11269. [Google Scholar] [CrossRef]
- Thomas, J.M. Tens of thousands of atoms replaced by one. Nature 2015, 525, 325–326. [Google Scholar] [CrossRef]
- Ong, W.-J.; Tan, L.-L.; Ng, Y.H.; Yong, S.-T.; Chai, S.-P. Graphitic Carbon Nitride (g-C3N4)-Based Photocatalysts for Artificial Photosynthesis and Environmental Remediation: Are We a Step Closer To Achieving Sustainability? Chem. Rev. 2016, 116, 7159–7329. [Google Scholar] [CrossRef]
- Zhou, Z.; Zhang, Y.; Shen, Y.; Liu, S.; Zhang, Y. Molecular engineering of polymeric carbon nitride: Advancing applications from photocatalysis to biosensing and more. Chem. Soc. Rev. 2018, 47, 2298–2321. [Google Scholar] [CrossRef]
- Rivera-Cárcamo, C.; Serp, P. Single Atom Catalysts on Carbon-Based Materials. ChemCatChem 2018, 10, 5058–5091. [Google Scholar] [CrossRef]
- Deng, D.; Chen, X.; Yu, L.; Wu, X.; Liu, Q.; Liu, Y.; Yang, H.; Tian, H.; Hu, Y.; Du, P.; et al. A single iron site confined in a graphene matrix for the catalytic oxidation of benzene at room temperature. Sci. Adv. 2015, 1, e1500462. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Huang, X.; Xi, S.; Miao, S.; Ding, J.; Cai, W.; Liu, S.; Yang, X.; Yang, H.; Gao, J.; et al. Single Cobalt Atoms Anchored on Porous N-Doped Graphene with Dual Reaction Sites for Efficient Fenton-like Catalysis. J. Am. Chem. Soc. 2018, 140, 12469–12475. [Google Scholar] [CrossRef]
- Zhang, H.; Li, J.; Xi, S.; Du, Y.; Hai, X.; Wang, J.; Xu, H.; Wu, G.; Zhang, J.; Lu, J.; et al. A Graphene-Supported Single-Atom FeN5 Catalytic Site for Efficient Electrochemical CO2 Reduction. Angew. Chem. Int. Ed. Engl. 2019, 58, 14871–14876. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Chen, W.; Shen, Z.; Chang, S.; Zhao, M.; Dai, X. Nitrogen coordinated silicon-doped graphene as a potential alternative metal-free catalyst for CO oxidation. Carbon 2017, 111, 448–458. [Google Scholar] [CrossRef]
- Li, X.-F.; Li, Q.-K.; Cheng, J.; Liu, L.; Yan, Q.; Wu, Y.; Zhang, X.-H.; Wang, Z.-Y.; Qiu, Q.; Luo, Y. Conversion of Dinitrogen to Ammonia by FeN3-Embedded Graphene. J. Am. Chem. Soc. 2016, 138, 8706–8709. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Zhang, A.; Dai, Z.; Feng, Y.P.; Zhang, C. Strain-Enhanced Stabilization and Catalytic Activity of Metal Nanoclusters on Graphene. J. Phys. Chem. C 2010, 114, 16541–16546. [Google Scholar] [CrossRef]
- Yin, H.; Tang, H.; Wang, D.; Gao, Y.; Tang, Z. Facile Synthesis of Surfactant-Free Au Cluster/Graphene Hybrids for High-Performance Oxygen Reduction Reaction. ACS Nano 2012, 6, 8288–8297. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Fu, Q.; Zhang, Y.-Y.; Weng, X.; Li, H.; Chen, M.; Jin, L.; Dong, A.; Mu, R.; Jiang, P.; et al. Graphene cover-promoted metal-catalyzed reactions. Proc. Natl. Acad. Sci. USA 2014, 111, 17023–17028. [Google Scholar] [CrossRef] [Green Version]
- Sutter, P.; Sadowski, J.T.; Sutter, E.A. Chemistry under Cover: Tuning Metal–Graphene Interaction by Reactive Intercalation. J. Am. Chem. Soc. 2010, 132, 8175–8179. [Google Scholar] [CrossRef]
- Sutter, P.W.; Flege, J.-I.; Sutter, E.A. Epitaxial graphene on ruthenium. Nat. Mater. 2008, 7, 406–411. [Google Scholar] [CrossRef]
- Stradi, D.; Barja, S.; Diaz, C.; Garnica, M.; Borca, B.; Hinarejos, J.J.; Sánchez-Portal, D.; Alcami, M.; Arnau, A.; De Parga, A.L.V.; et al. Lattice-matched versus lattice-mismatched models to describe epitaxial monolayer graphene on Ru(0001). Phys. Rev. B 2013, 88, 245401. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
Different roles of graphene in graphene-based solid-state catalysts. Graphene can act as a support for catalysts, a cover that protect the underneath catalysts, and a catalytic center of a catalyst.
Figure 1.
Different roles of graphene in graphene-based solid-state catalysts. Graphene can act as a support for catalysts, a cover that protect the underneath catalysts, and a catalytic center of a catalyst.

Figure 2.
Sketch of graphene-supported nanoscale and atomic-scale catalysts. Color Scheme: Graphene in grey; embedded single-atom catalyst in yellow; supported nano catalyst in blue.
Figure 2.
Sketch of graphene-supported nanoscale and atomic-scale catalysts. Color Scheme: Graphene in grey; embedded single-atom catalyst in yellow; supported nano catalyst in blue.

Figure 3.
Au8 cluster adsorbed on defective graphene. Left: Single vacancy in graphene substrate. Right: The chemisorption of Au8 on defective graphene, the iso-surface of charge redistribution is imposed. Color Scheme: C in grey, Au in yellow, charge accumulation (depletion) in green (blue).
Figure 3.
Au8 cluster adsorbed on defective graphene. Left: Single vacancy in graphene substrate. Right: The chemisorption of Au8 on defective graphene, the iso-surface of charge redistribution is imposed. Color Scheme: C in grey, Au in yellow, charge accumulation (depletion) in green (blue).

Figure 4.
Single Au atom adsorbed on defective graphene. (a) Atomic configuration of adsorption of Au atom. (b) DOS projected on Au and C. Please note that the magnetic moment is on the Au atom.
Figure 4.
Single Au atom adsorbed on defective graphene. (a) Atomic configuration of adsorption of Au atom. (b) DOS projected on Au and C. Please note that the magnetic moment is on the Au atom.

Figure 5.
The sketch of experimentally fabricated oxygen stabilized Co single-atom catalyst on graphene. C in grey, Co in orange and O in red.
Figure 5.
The sketch of experimentally fabricated oxygen stabilized Co single-atom catalyst on graphene. C in grey, Co in orange and O in red.

Figure 6.
The sketch of experimentally fabricated M1N4/G catalyst. The generic metal atom M in gold, C in grey, N in blue.
Figure 6.
The sketch of experimentally fabricated M1N4/G catalyst. The generic metal atom M in gold, C in grey, N in blue.

Figure 7.
The adsorption of Au16 on graphene with 5% of tensile strain. The iso-surface of charge redistribution is imposed. Color scheme: C in grey, Au in yellow, Charge depletion (accumulation) in blue (green).
Figure 7.
The adsorption of Au16 on graphene with 5% of tensile strain. The iso-surface of charge redistribution is imposed. Color scheme: C in grey, Au in yellow, Charge depletion (accumulation) in blue (green).

Figure 8.
Sketch of graphene-protected chemical reactions catalyzed by a metal surface.

Figure 9.
Graphene itself as a catalyst: Activating graphene by engineering the underlying substrate.
Figure 9.
Graphene itself as a catalyst: Activating graphene by engineering the underlying substrate.

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Yam, K.M.; Guo, N.; Jiang, Z.; Li, S.; Zhang, C. Graphene-Based Heterogeneous Catalysis: Role of Graphene. Catalysts 2020, 10, 53. https://doi.org/10.3390/catal10010053
AMA Style
Yam KM, Guo N, Jiang Z, Li S, Zhang C. Graphene-Based Heterogeneous Catalysis: Role of Graphene. Catalysts. 2020; 10(1):53. https://doi.org/10.3390/catal10010053
Chicago/Turabian StyleYam, Kah Meng, Na Guo, Zhuoling Jiang, Shulong Li, and Chun Zhang. 2020. "Graphene-Based Heterogeneous Catalysis: Role of Graphene" Catalysts 10, no. 1: 53. https://doi.org/10.3390/catal10010053
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.