Glutathione S-Transferase pi-1 Knockdown Reduces Pancreatic Ductal Adenocarcinoma Growth by Activating Oxidative Stress Response Pathways


Abstract
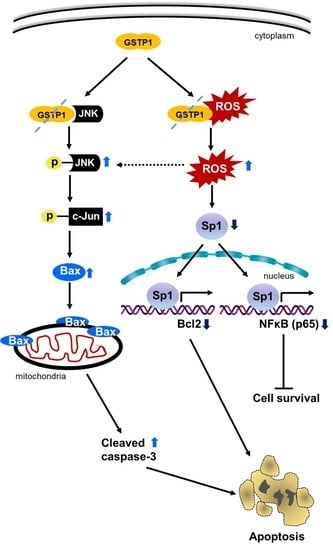
:
1. Introduction
2. Results
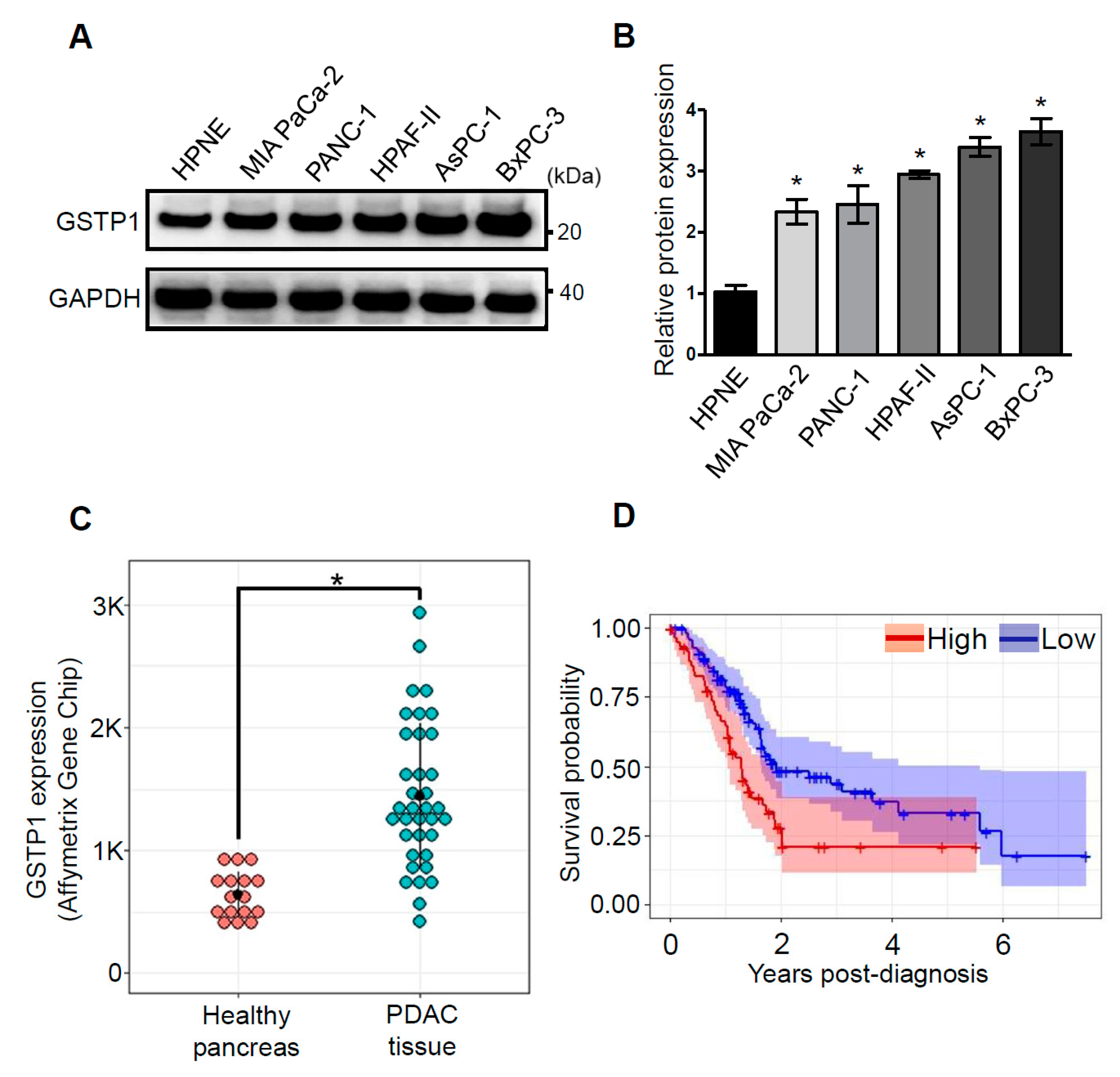
2.1. GSTP1 Is Overexpressed in Human PDAC Cells
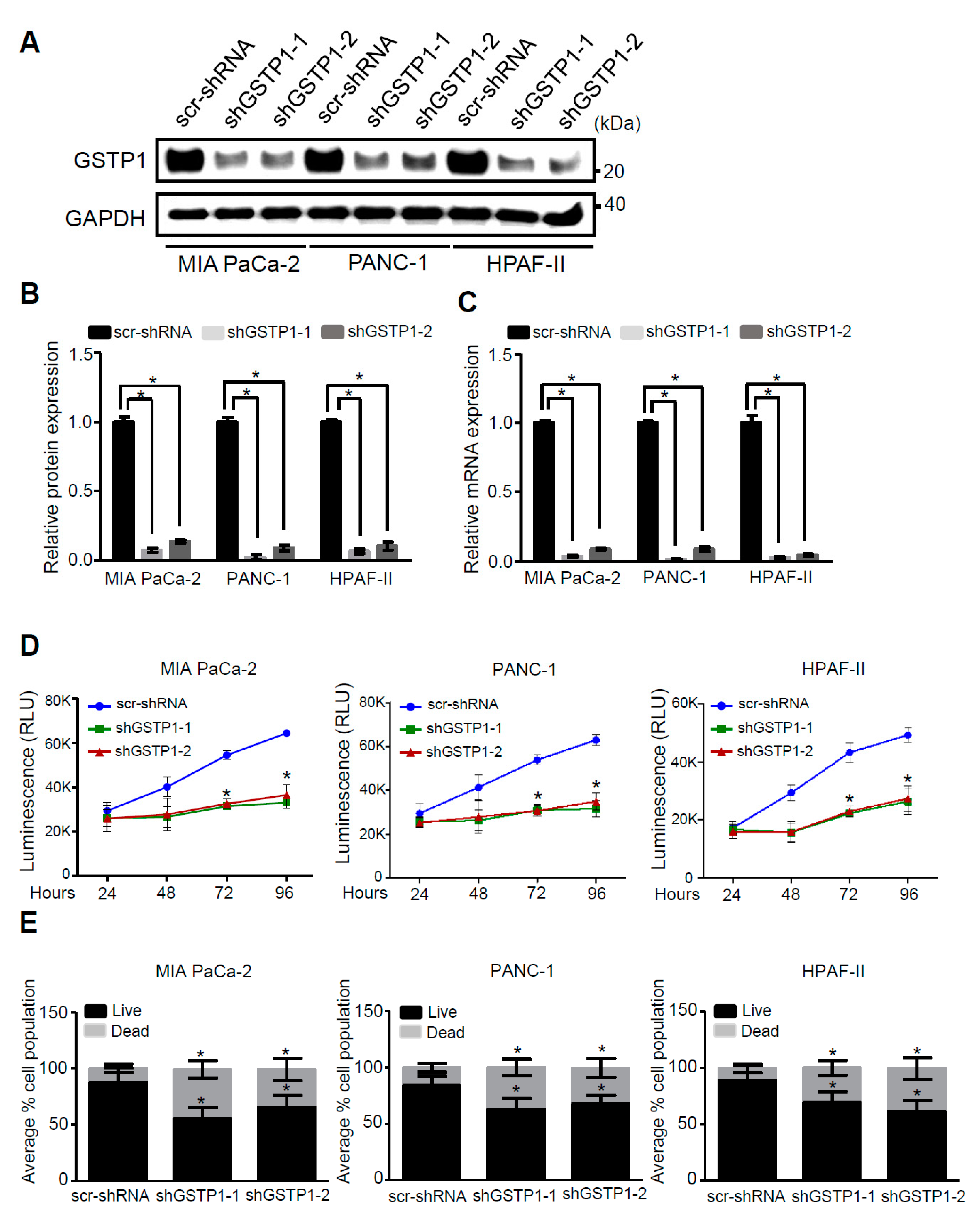
2.2. GSTP1 Knockdown Impairs PDAC Cell Growth
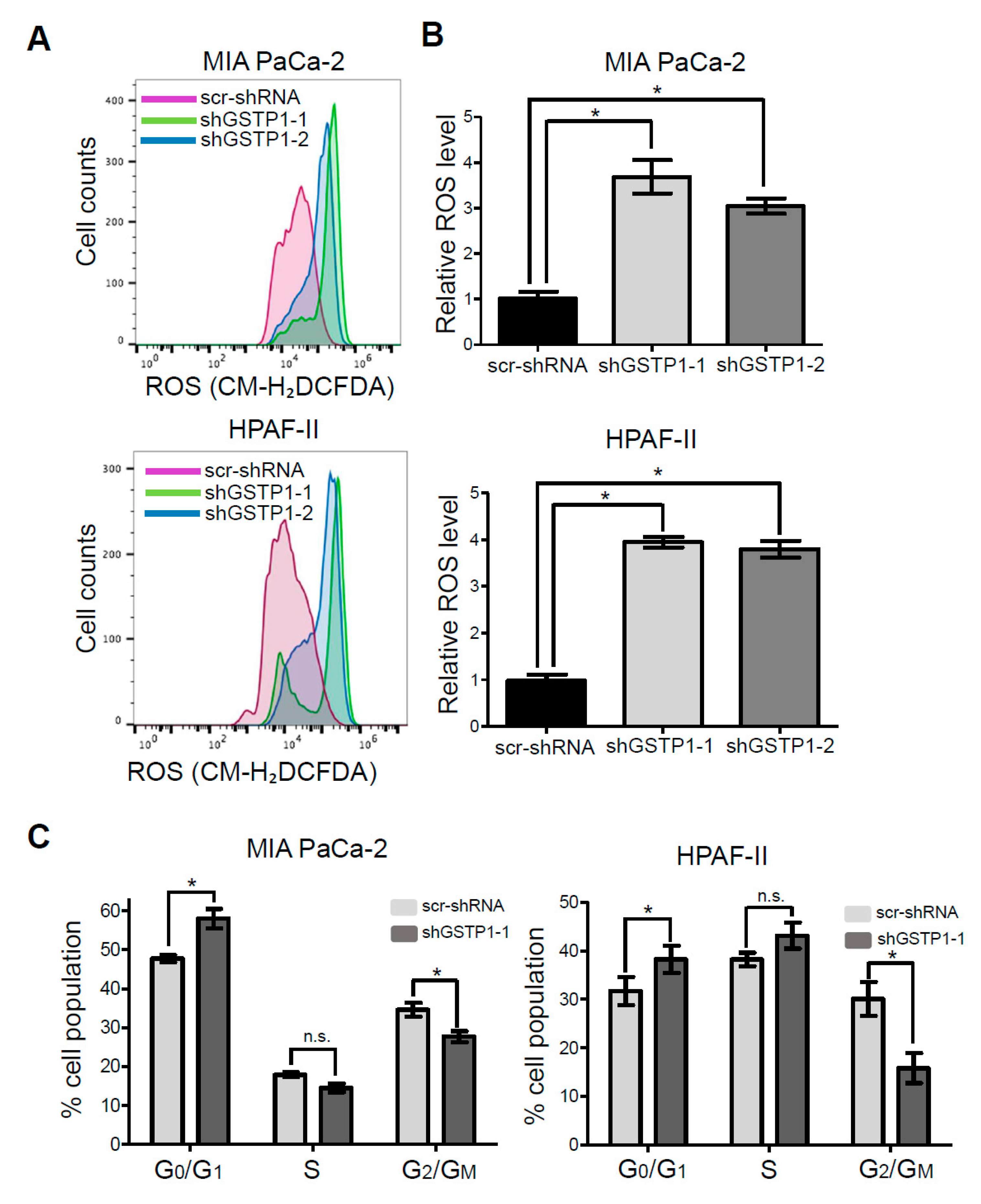
2.3. GSTP1 Knockdown Elevates ROS Levels in PDAC Cells
2.4. GSTP1 Knockdown Prolongs the G0/G1 Phase of the Cell Cycle
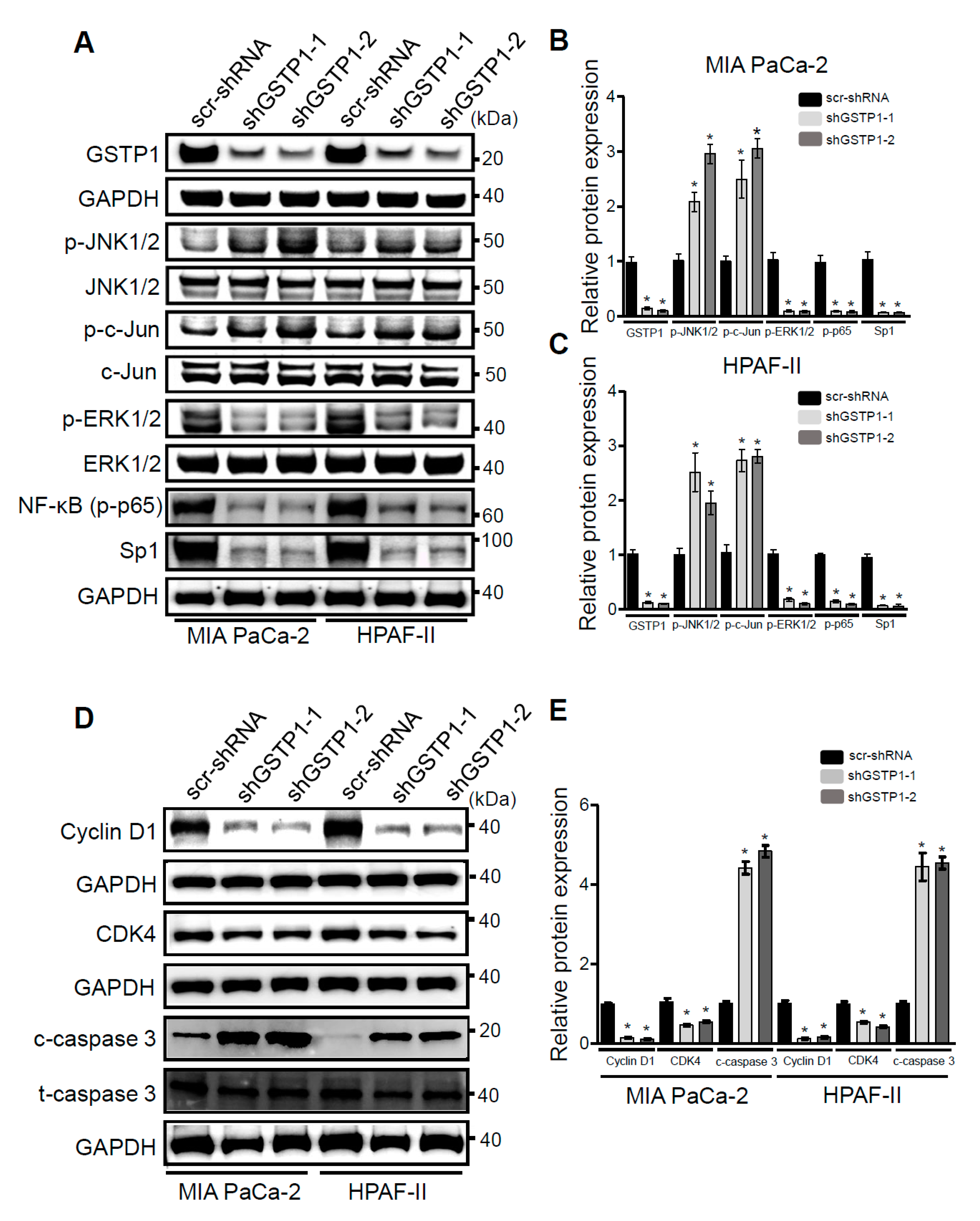
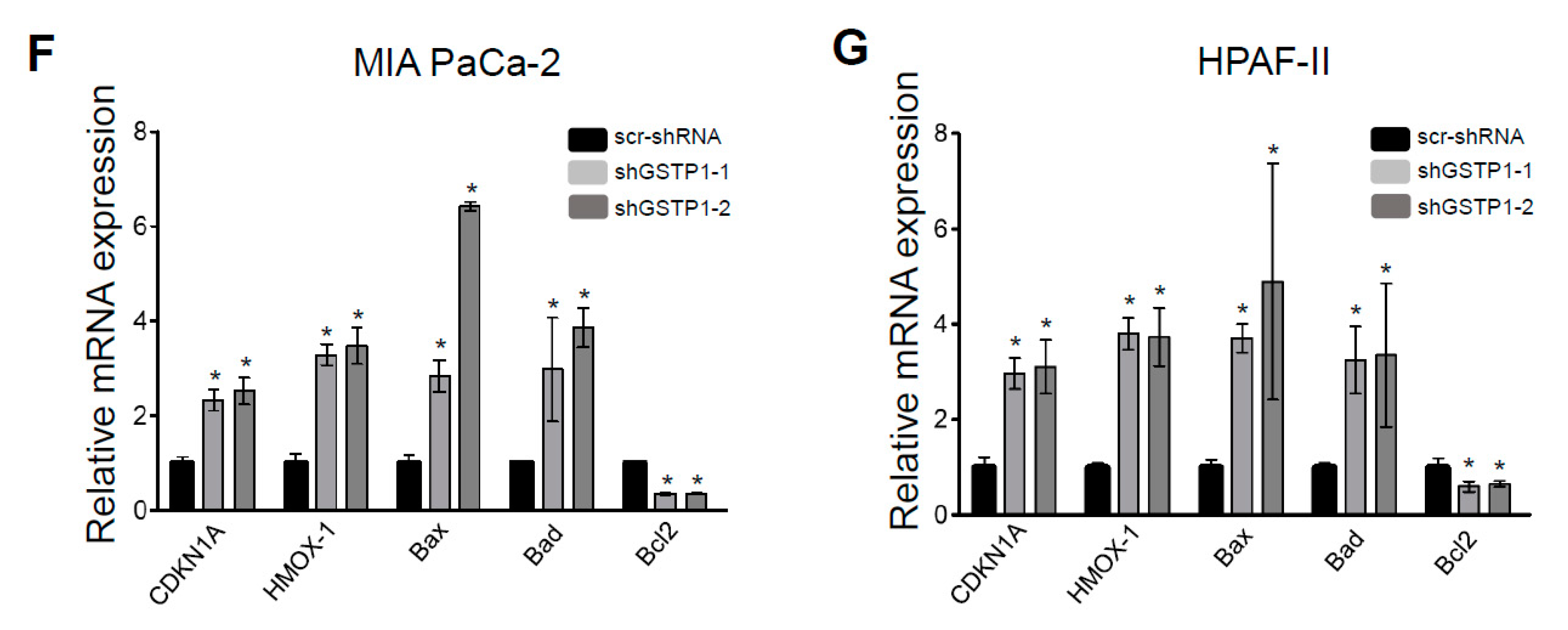
2.5. GSTP1 Knockdown Activates Oxidative Stress-Mediated Apoptotic Signaling in PDAC Cells
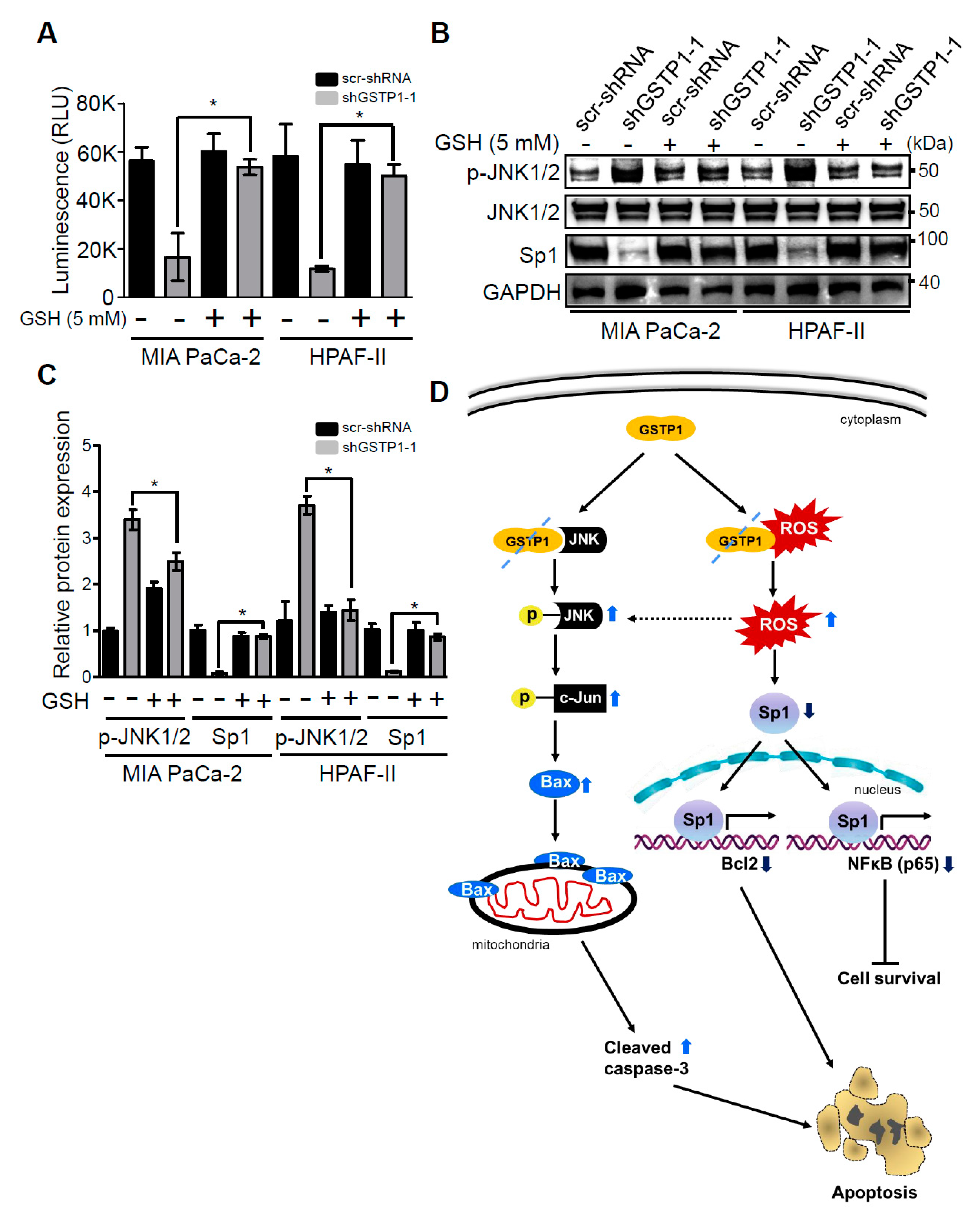
2.6. Addition of Glutathione Reverses the Effects of GSTP1 Knockdown on Cell Viability and Oxidative Stress-Response Signaling
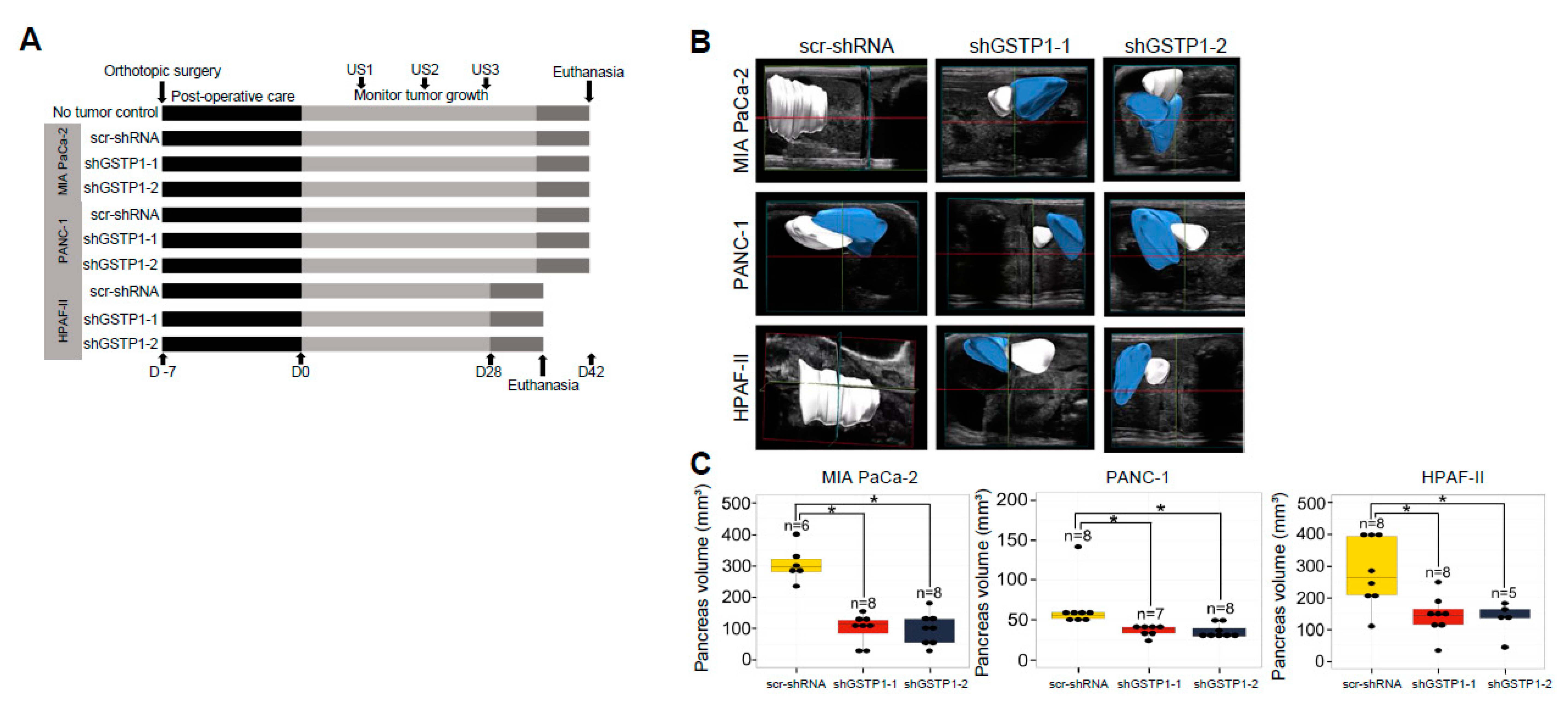
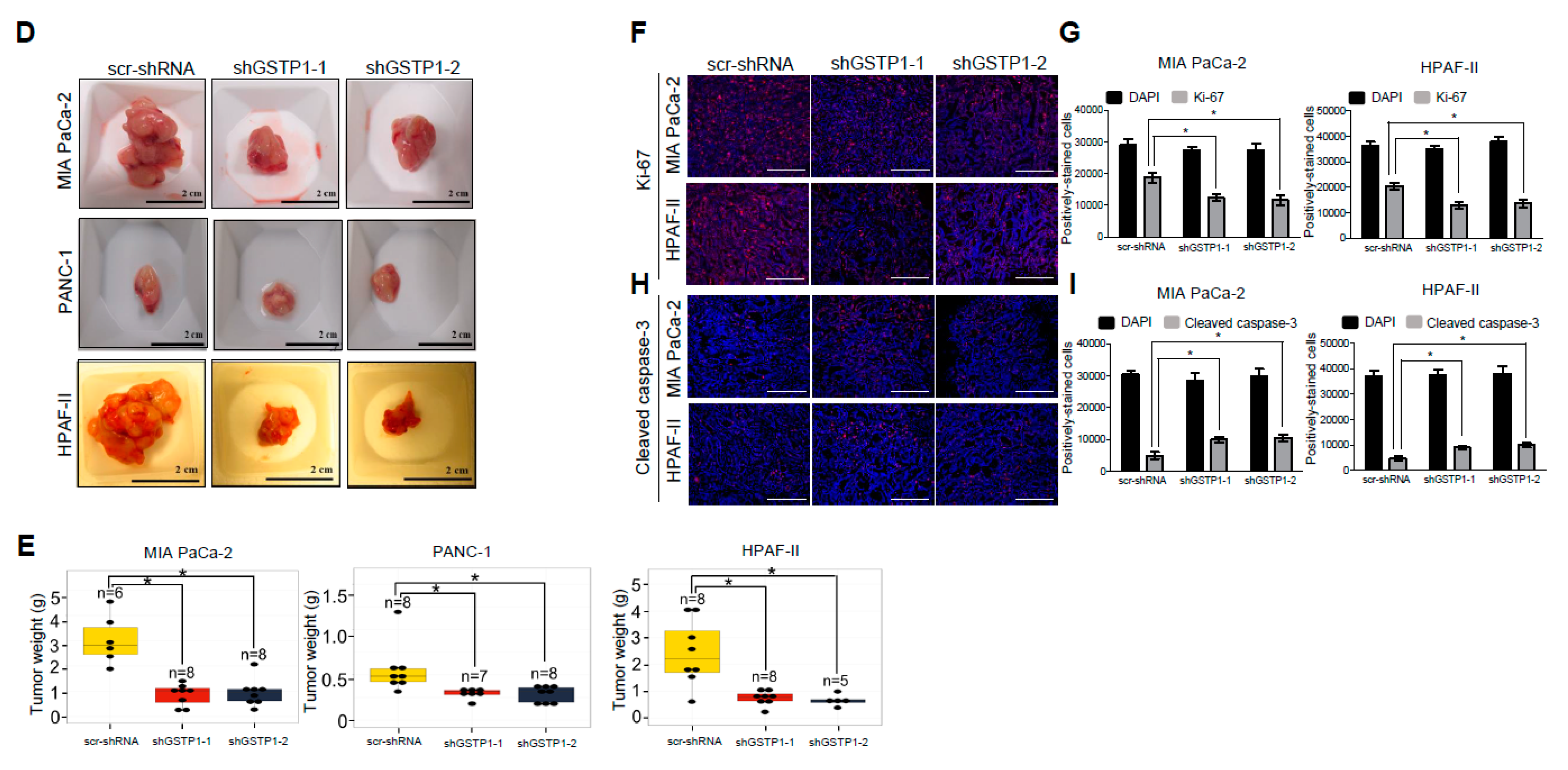
2.7. GSTP1 Knockdown Impairs the Growth of Orthotopic PDAC Tumors In Vivo
2.8. Tumor Cell Proliferation Is Reduced and Apoptosis Is Increased by GSTP1 Knockdown in Pancreatic Tumors
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Cell Culture
4.3. Constructing Knockdown Cell Lines
4.4. Western Blotting
4.5. RNA Extraction and Gene Expression by qRT-PCR
4.6. Cell Viability Assay
4.7. Cell Cycle Arrest Assay
4.8. Detection of ROS Levels by the 2,7-Dichlorodihydrofluorescein Diacetate (CM-H2DCFDA) Assay
4.9. Orthotopic Tumor Studies
4.10. Murine Abdominal Ultrasound Imaging
4.11. Immunohistochemistry
4.12. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Availability of Data and Materials
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Drouillard, A.; Manfredi, S.; Lepage, C.; Bouvier, A.M. Epidemiology of pancreatic cancer. Bull. Cancer 2018, 105, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Rueff, J.; Rodrigues, A.S. Cancer Drug Resistance: A Brief Overview from a Genetic Viewpoint. Methods Mol. Biol. 2016, 1395, 1–18. [Google Scholar]
- Oettle, H.; Post, S.; Neuhaus, P.; Gellert, K.; Langrehr, J.; Ridwelski, K.; Schramm, H.; Fahlke, J.; Zuelke, C.; Burkart, C.; et al. Adjuvant chemotherapy with gemcitabine vs observation in patients undergoing curative-intent resection of pancreatic cancer: A randomized controlled trial. JAMA 2007, 297, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Oettle, H.; Neuhaus, P. Adjuvant therapy in pancreatic cancer: A critical appraisal. Drugs 2007, 67, 2293–2310. [Google Scholar] [CrossRef]
- Ghaneh, P.; Costello, E.; Neoptolemos, J.P. Biology and management of pancreatic cancer. Gut 2007, 56, 1134–1152. [Google Scholar] [CrossRef]
- Chan, A.; Diamandis, E.P.; Blasutig, I.M. Strategies for discovering novel pancreatic cancer biomarkers. J. Proteom. 2013, 81, 126–134. [Google Scholar] [CrossRef] [Green Version]
- Ma, W.W.; Xie, H.; Fetterly, G.; Pitzonka, L.; Whitworth, A.; LeVea, C.; Wilton, J.; Mantione, K.; Schihl, S.; Dy, G.K.; et al. A Phase Ib Study of the FGFR/VEGFR Inhibitor Dovitinib With Gemcitabine and Capecitabine in Advanced Solid Tumor and Pancreatic Cancer Patients. Am. J. Clin. Oncol. 2018, 42, 184–189. [Google Scholar] [CrossRef]
- Zhen, D.B.; Coveler, A.; Zanon, S.; Reni, M.; Chiorean, E.G. Biomarker-driven and molecularly targeted therapies for pancreatic adenocarcinoma. Semin. Oncol. 2018, 45, 3. [Google Scholar] [CrossRef]
- Deberardinis, R.J.; Sayed, N.; Ditsworth, D.; Thompson, C.B. Brick by brick: Metabolism and tumor cell growth. Curr. Opin. Genet. Dev. 2008, 18, 54–61. [Google Scholar] [CrossRef] [Green Version]
- Ramsey, M.R.; Sharpless, N.E. ROS as a tumour suppressor? Nat. Cell Biol. 2006, 8, 1213–1215. [Google Scholar] [CrossRef]
- Takahashi, A.; Ohtani, N.; Yamakoshi, K.; Iida, S.I.; Tahara, H.; Nakayama, K.; Nakayama, K.I.; Ide, T.; Saya, H.; Hara, E. Mitogenic signalling and the p16INK4a-Rb pathway cooperate to enforce irreversible cellular senescence. Nat. Cell Biol. 2006, 8, 1291–1297. [Google Scholar] [CrossRef]
- Williamson, J.M.; Boettcher, B.; Meister, A. Intracellular cysteine delivery system that protects against toxicity by promoting glutathione synthesis. Proc. Natl. Acad. Sci. USA 1982, 79, 6246–6249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Ye, Z.W.; Gao, P.; Reyes, L.; Jones, E.E.; Branham-O’Connor, M.; Blumer, J.B.; Drake, R.R.; Manevich, Y.; Townsend, D.M.; et al. Glutathione S-transferase P influences redox and migration pathways in bone marrow. PLoS ONE 2014, 9, e107478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keen, J.H.; Jakoby, W.B. Glutathione transferases. Catalysis of nucleophilic reactions of glutathione. J. Biol. Chem. 1978, 253, 5654–5657. [Google Scholar] [PubMed]
- Medeiros, R.; Vasconcelos, A.; Costa, S.; Pinto, D.; Ferreira, P.; Lobo, F.; Morais, A.; Oliveira, J.; Lopes, C. Metabolic susceptibility genes and prostate cancer risk in a southern European population: The role of glutathione S-transferases GSTM1, GSTM3, and GSTT1 genetic polymorphisms. Prostate 2004, 58, 414–420. [Google Scholar] [CrossRef]
- Ye, Z.; Song, H.; Higgins, J.; Pharoah, P.; Danesh, J. Five glutathione s-transferase gene variants in 23,452 cases of lung cancer and 30,397 controls: Meta-analysis of 130 studies. PLoS Med. 2006, 3, e91. [Google Scholar] [CrossRef]
- Tew, K.D.; Townsend, D.M. Glutathione-s-transferases as determinants of cell survival and death. Antioxid. Redox Signal. 2012, 17, 1728–1737. [Google Scholar] [CrossRef] [Green Version]
- Adler, V.; Yin, Z.; Fuchs, S.Y.; Benezra, M.; Rosario, L.; Tew, K.D.; Pincus, M.R.; Sardana, M.; Henderson, C.J.; Wolf, C.R.; et al. Regulation of JNK signaling by GSTp. EMBO J. 1999, 18, 1321–1334. [Google Scholar] [CrossRef]
- Louie, S.M.; Grossman, E.A.; Crawford, L.A.; Ding, L.; Camarda, R.; Huffman, T.R.; Miyamoto, D.K.; Goga, A.; Weerapana, E.; Nomura, D.K. GSTP1 Is a Driver of Triple-Negative Breast Cancer Cell Metabolism and Pathogenicity. Cell Chem. Biol. 2016, 23, 567–578. [Google Scholar] [CrossRef] [Green Version]
- Ye, Z.W.; Zhang, J.; Ancrum, T.; Manevich, Y.; Townsend, D.M.; Tew, K.D. Glutathione S-Transferase P-Mediated Protein S-Glutathionylation of Resident Endoplasmic Reticulum Proteins Influences Sensitivity to Drug-Induced Unfolded Protein Response. Antioxid. Redox Signal. 2017, 26, 247–261. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Yin, F.; Hang, Q.; Dong, X.; Chen, J.; Li, L.; Cao, P.; Yin, Z.; Luo, L. Regulation of Endothelial Permeability by Glutathione S-Transferase Pi Against Actin Polymerization. Cell. Physiol. Biochem. 2018, 45, 406–418. [Google Scholar] [CrossRef]
- Findlay, V.J.; Townsend, D.M.; Saavedra, J.E.; Buzard, G.S.; Citro, M.L.; Keefer, L.K.; Ji, X.; Tew, K.D. Tumor cell responses to a novel glutathione S-transferase-activated nitric oxide-releasing prodrug. Mol. Pharmacol. 2004, 65, 1070–1079. [Google Scholar] [CrossRef] [Green Version]
- Checa-Rojas, A.; Delgadillo-Silva, L.F.; Velasco-Herrera, M.D.C.; Andrade-Domínguez, A.; Gil, J.; Santillán, O.; Lozano, L.; Toledo-Leyva, A.; Ramírez-Torres, A.; Talamas-Rohana, P.; et al. GSTM3 and GSTP1: Novel players driving tumor progression in cervical cancer. Oncotarget 2018, 9, 21696–21714. [Google Scholar] [CrossRef] [Green Version]
- Tew, K.D.; Manevich, Y.; Grek, C.; Xiong, Y.; Uys, J.; Townsend, D.M. The role of glutathione S-transferase P in signaling pathways and S-glutathionylation in cancer. Free Radic. Biol. Med. 2011, 51, 299–313. [Google Scholar] [CrossRef] [Green Version]
- Uhlen, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, A.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Proteomics. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Daemen, A.; Peterson, D.; Sahu, N.; McCord, R.; Du, X.; Liu, B.; Kowanetz, K.; Hong, R.; Moffat, J.; Gao, M.; et al. Metabolite profiling stratifies pancreatic ductal adenocarcinomas into subtypes with distinct sensitivities to metabolic inhibitors. Proc. Natl. Acad. Sci. USA 2015, 112, E4410–E4417. [Google Scholar] [CrossRef] [Green Version]
- Deer, E.L.; González-Hernández, J.; Coursen, J.D.; Shea, J.E.; Ngatia, J.; Scaife, C.L.; Firpo, M.A.; Mulvihill, S.J. Phenotype and genotype of pancreatic cancer cell lines. Pancreas 2010, 39, 425–435. [Google Scholar] [CrossRef] [Green Version]
- Kanwal, R.; Pandey, M.; Bhaskaran, N.; Maclennan, G.T.; Fu, P.; Ponsky, L.E.; Gupta, S. Protection against oxidative DNA damage and stress in human prostate by glutathione S-transferase P1. Mol. Carcinog. 2014, 53, 8–18. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Fan, Y.; Xue, B.; Luo, L.; Shen, J.; Zhang, S.; Jiang, Y.; Yin, Z. Human glutathione S-transferase P1-1 interacts with TRAF2 and regulates TRAF2-ASK1 signals. Oncogene 2006, 25, 5787–5800. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Arifoglu, P.; Ronai, Z.; Tew, K.D. Glutathione S-transferase P1-1 (GSTP1-1) inhibits c-Jun N-terminal kinase (JNK1) signaling through interaction with the C terminus. J. Biol. Chem. 2001, 276, 20999–21003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tew, K.D.; Townsend, D.M. Regulatory functions of glutathione S-transferase P1-1 unrelated to detoxification. Drug Metab. Rev. 2011, 43, 179–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tew, K.D. Glutathione-associated enzymes in anticancer drug resistance. Cancer Res. 1994, 54, 4313–4320. [Google Scholar] [CrossRef] [Green Version]
- Fujitani, N.; Yoneda, A.; Takahashi, M.; Takasawa, A.; Aoyama, T.; Miyazaki, T. Silencing of Glutathione S-Transferase Pi Inhibits Cancer Cell Growth via Oxidative Stress Induced by Mitochondria Dysfunction. Sci. Rep. 2019, 9, 14764. [Google Scholar] [CrossRef]
- Arrick, B.A.; Nathan, C.F.; Griffith, O.W.; Cohn, Z.A. Glutathione depletion sensitizes tumor cells to oxidative cytolysis. J. Biol. Chem. 1982, 257, 1231–1237. [Google Scholar]
- Arrick, B.A.; Nathan, C.F. Glutathione metabolism as a determinant of therapeutic efficacy: A review. Cancer Res. 1984, 44, 4224–4232. [Google Scholar]
- Karki, K.; Hedrick, E.; Kasiappan, R.; Jin, U.; Safe, S. Piperlongumine Induces Reactive Oxygen Species (ROS)-Dependent Downregulation of Specificity Protein Transcription Factors. Cancer Prev. Res. 2017, 10, 467–477. [Google Scholar] [CrossRef] [Green Version]
- Elsby, R.; Kitteringham, N.R.; Goldring, C.E.; Lovatt, C.A.; Chamberlain, M.; Henderson, C.J.; Wolf, C.R.; Park, B.K. Increased constitutive c-Jun N-terminal kinase signaling in mice lacking glutathione S-transferase Pi. J. Biol. Chem. 2003, 278, 22243–22249. [Google Scholar] [CrossRef] [Green Version]
- Mohammad, J.; Singh, R.R.; Riggle, C.; Haugrud, B.; Abdalla, M.Y.; Reindl, K.M. JNK inhibition blocks piperlongumine-induced cell death and transcriptional activation of heme oxygenase-1 in pancreatic cancer cells. Apoptosis 2019, 24, 730–744. [Google Scholar] [CrossRef]
- Pathi, S.S.; Lei, P.; Sreevalsan, S.; Chadalapaka, G.; Jutooru, I.; Safe, S. Pharmacologic doses of ascorbic acid repress specificity protein (Sp) transcription factors and Sp-regulated genes in colon cancer cells. Nutr. Cancer 2011, 63, 1133–1142. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Wei, D.; Huang, S.; Peng, Z.; Le, X.; Wu, T.; Yao, J.; Ajani, J.; Xie, K. Transcription factor Sp1 expression is a significant predictor of survival in human gastric cancer. Clin. Cancer Res. 2003, 9, 6371–6380. [Google Scholar]
- Jutooru, I.; Guthrie, A.S.; Chadalapaka, G.; Pathi, S.; Kim, K.; Burghardt, R.; Jin, U.; Safe, S. Mechanism of action of phenethylisothiocyanate and other reactive oxygen species-inducing anticancer agents. Mol. Cell. Biol. 2014, 34, 2382–2395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Safe, S.; Abbruzzese, J.; Abdelrahim, M.; Hedrick, E. Specificity Protein Transcription Factors and Cancer: Opportunities for Drug Development. Cancer Prev. Res. 2018, 11, 371–382. [Google Scholar] [CrossRef] [PubMed]
- O’Hagan, H.M.; Wang, W.; Sen, S.; Shields, C.D.; Lee, S.S.; Zhang, Y.W.; Clements, E.G.; Cai, Y.; Neste, L.V.; Easwaran, H.; et al. Oxidative damage targets complexes containing DNA methyltransferases, SIRT1, and polycomb members to promoter CpG Islands. Cancer Cell 2011, 20, 606–619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pathi, S.S.; Jutooru, I.; Chadalapaka, G.; Sreevalsan, S.; Anand, S.; Thatcher, G.R.; Safe, S. GT-094, a NO-NSAID, inhibits colon cancer cell growth by activation of a reactive oxygen species-microRNA-27a: ZBTB10-specificity protein pathway. Mol. Cancer Res. 2011, 9, 195–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, G.Z.; Tao, H.L.; Zhou, C.; Wang, D.D.; Peng, C.B. Midazolam prevents motor neuronal death from oxidative stress attack mediated by JNK-ERK pathway. Hum. Cell 2018, 31, 64–71. [Google Scholar] [CrossRef]
- Chintharlapalli, S.; Papineni, S.; Lei, P.; Pathi, S.; Safe, S. Betulinic acid inhibits colon cancer cell and tumor growth and induces proteasome-dependent and -independent downregulation of specificity proteins (Sp) transcription factors. BMC Cancer 2011, 11, 371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hedrick, E.; Cheng, Y.; Jin, U.; Kim, K.; Safe, S. Specificity protein (Sp) transcription factors Sp1, Sp3 and Sp4 are non-oncogene addiction genes in cancer cells. Oncotarget 2016, 7, 22245–22256. [Google Scholar] [CrossRef]
- Qie, S.; Diehl, J.A. Cyclin D1, cancer progression, and opportunities in cancer treatment. J. Mol. Med. 2016, 94, 1313–1326. [Google Scholar] [CrossRef] [Green Version]
- Laphanuwat, P.; Likasitwatanakul, P.; Sittithumcharee, G.; Thaphaengphan, A.; Chomanee, N.; Suppramote, O.; Ketaroonrut, N.; Charngkaew, K.; Lam, E.; Okada, S.; et al. Cyclin D1 depletion interferes with oxidative balance and promotes cancer cell senescence. J. Cell Sci. 2018, 131. [Google Scholar] [CrossRef] [Green Version]
- Dong, Y.; Sui, L.; Sugimoto, K.; Tai, Y.; Tokuda, M. Cyclin D1-CDK4 complex, a possible critical factor for cell proliferation and prognosis in laryngeal squamous cell carcinomas. Int. J. Cancer 2001, 95, 209–215. [Google Scholar] [CrossRef]
- Jutooru, I.; Chadalapaka, G.; Lei, P.; Safe, S. Inhibition of NFkappaB and pancreatic cancer cell and tumor growth by curcumin is dependent on specificity protein down-regulation. J. Biol. Chem. 2010, 285, 25332–25344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheng, L.; Wu, J.; Gong, X.; Dong, D.; Sun, X. SP1-induced upregulation of lncRNA PANDAR predicts adverse phenotypes in retinoblastoma and regulates cell growth and apoptosis in vitro and in vivo. Gene 2018, 668, 140–145. [Google Scholar] [CrossRef] [PubMed]
- Hedrick, E.; Crose, L.; Linardic, C.M.; Safe, S. Histone Deacetylase Inhibitors Inhibit Rhabdomyosarcoma by Reactive Oxygen Species-Dependent Targeting of Specificity Protein Transcription Factors. Mol. Cancer Ther. 2015, 14, 2143–2153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ascione, A.; Cianfriglia, M.; Dupuis, M.L.; Mallano, A.; Sau, A.; Tregno, F.P.; Pezzola, S.; Caccuri, A.M. The glutathione S-transferase inhibitor 6-(7-nitro-2,1,3-benzoxadiazol-4-ylthio)hexanol overcomes the MDR1-P-glycoprotein and MRP1-mediated multidrug resistance in acute myeloid leukemia cells. Cancer Chemother. Pharmacol. 2009, 64, 419–424. [Google Scholar] [CrossRef]
- Tregno, F.P.; Sau, A.; Pezzola, S.; Geroni, C.; Lapenta, C.; Spada, M.; Filomeni, G.; Bonanno, E.; Federici, G.; Caccuri, A.M. In vitro and in vivo efficacy of 6-(7-nitro-2,1,3-benzoxadiazol-4-ylthio)hexanol (NBDHEX) on human melanoma. Eur. J. Cancer 2009, 45, 2606–2617. [Google Scholar] [CrossRef]
- Zhuo, R.; Kosak, K.M.; Sankar, S.; Wiles, E.T.; Sun, Y.; Zhang, J.; Ayello, J.; Prestwich, G.D.; Shami, P.J.; Cairo, M.S.; et al. Targeting Glutathione S-transferase M4 in Ewing sarcoma. Front. Pediatr. 2014, 2, 83. [Google Scholar] [CrossRef] [Green Version]
- Ruscoe, J.E.; Rosario, L.A.; Wang, T.; Gaté, L.; Arifoglu, P.; Wolf, C.R.; Henderson, C.J.; Ronai, Z.; Tew, K.D. Pharmacologic or genetic manipulation of glutathione S-transferase P1-1 (GSTpi) influences cell proliferation pathways. J. Pharmacol. Exp. Ther. 2001, 298, 339–345. [Google Scholar] [PubMed]
- Mahadevan, D.; Sutton, G.R. Ezatiostat hydrochloride for the treatment of myelodysplastic syndromes. Expert Opin. Investig. Drugs 2015, 24, 725–733. [Google Scholar] [CrossRef]
- Dhillon, H.; Mamidi, S.; McClean, P.; Reindl, K.M. Transcriptome Analysis of Piperlongumine-Treated Human Pancreatic Cancer Cells Reveals Involvement of Oxidative Stress and Endoplasmic Reticulum Stress Pathways. J. Med. Food 2016, 19, 578–585. [Google Scholar] [CrossRef] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Garber, J.C. On the care and use of US lab animals. Nature 2011, 476, 152. [Google Scholar] [CrossRef] [PubMed] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Sequence | Reverse Sequence |
|---|---|---|
| HPRT | 5′-GAA CGT CTT GCT CGA GAT GTG-3′ | 5′TCC AGC AGG TCA GCA AAG AAT-3′ |
| β-Actin | 5′-TTG CCG ACA GGA TGC AGA-3′ | 5′-GCC GAT CCA CAC GGA GTA CTT-3′ |
| β-Tubulin | 5′-GTT CGC TCA GGT CCT TTT GG-3′ | 5′-CCC TCT GTG TAG TGG CCT TTG-3′ |
| GSTP1 | 5′-CAG GAG GGC TCA CTC AAA GC-3′ | 5′-AGG TGA CGC AGG ATG GTA TTG-3′ |
| CDKN1A HMOX-1 | 5′-GGA CAG CAG AGG AAG ACC ATG T-3′ 5′-AAT TCT CTT GGC TGG CTT CCT-3′ | 5′-GCC GTT TTC GAC CCT GAG A-3′ 5′-CAT AGG CTC CTT CCT CCT TTC C-3′ |
| Bax | 5′-TTG CTT CAG GGT TTC ATC CA-3′ | 5′-ACA CTC GCT CAG CTT CTT G-3′ |
| Bak | 5′-ACA TCA ACC GAC GCT ATG AC-3′ | 5′-TGG TGG CAA TCT TGG TGA A-3′ |
| Bcl2 | 5′-CGC CCT GTG GAT GAC TGA GTA-3′ | 5′-CCT CAG CCC AGA CTC ATC A-3′ |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Singh, R.R.; Mohammad, J.; Orr, M.; Reindl, K.M. Glutathione S-Transferase pi-1 Knockdown Reduces Pancreatic Ductal Adenocarcinoma Growth by Activating Oxidative Stress Response Pathways. Cancers 2020, 12, 1501. https://doi.org/10.3390/cancers12061501
Singh RR, Mohammad J, Orr M, Reindl KM. Glutathione S-Transferase pi-1 Knockdown Reduces Pancreatic Ductal Adenocarcinoma Growth by Activating Oxidative Stress Response Pathways. Cancers. 2020; 12(6):1501. https://doi.org/10.3390/cancers12061501
Chicago/Turabian StyleSingh, Rahul R., Jiyan Mohammad, Megan Orr, and Katie M. Reindl. 2020. "Glutathione S-Transferase pi-1 Knockdown Reduces Pancreatic Ductal Adenocarcinoma Growth by Activating Oxidative Stress Response Pathways" Cancers 12, no. 6: 1501. https://doi.org/10.3390/cancers12061501