Vitamin D3 Metabolites Demonstrate Prognostic Value in EGFR-Mutant Lung Adenocarcinoma and Can be Deployed to Oppose Acquired Therapeutic Resistance


 ,
,
Abstract
:1. Introduction
2. Results
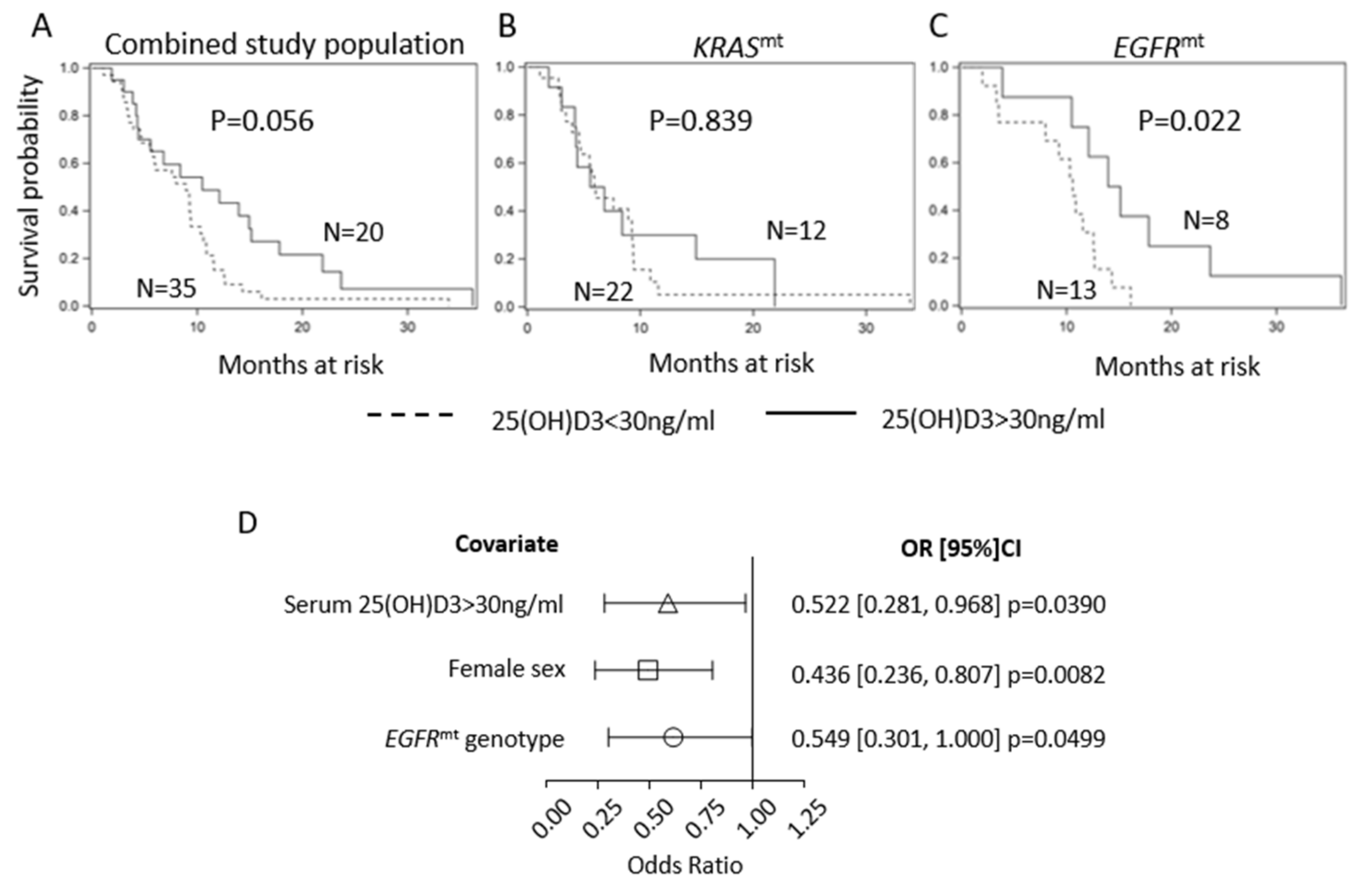
2.1. Serum 25(OH)D3 Predicts PFS in EGFR-Mutant LUAD but Not KRAS-Mutant LUAD
2.2. EGFR TKI Resistance Is Associated with EMT
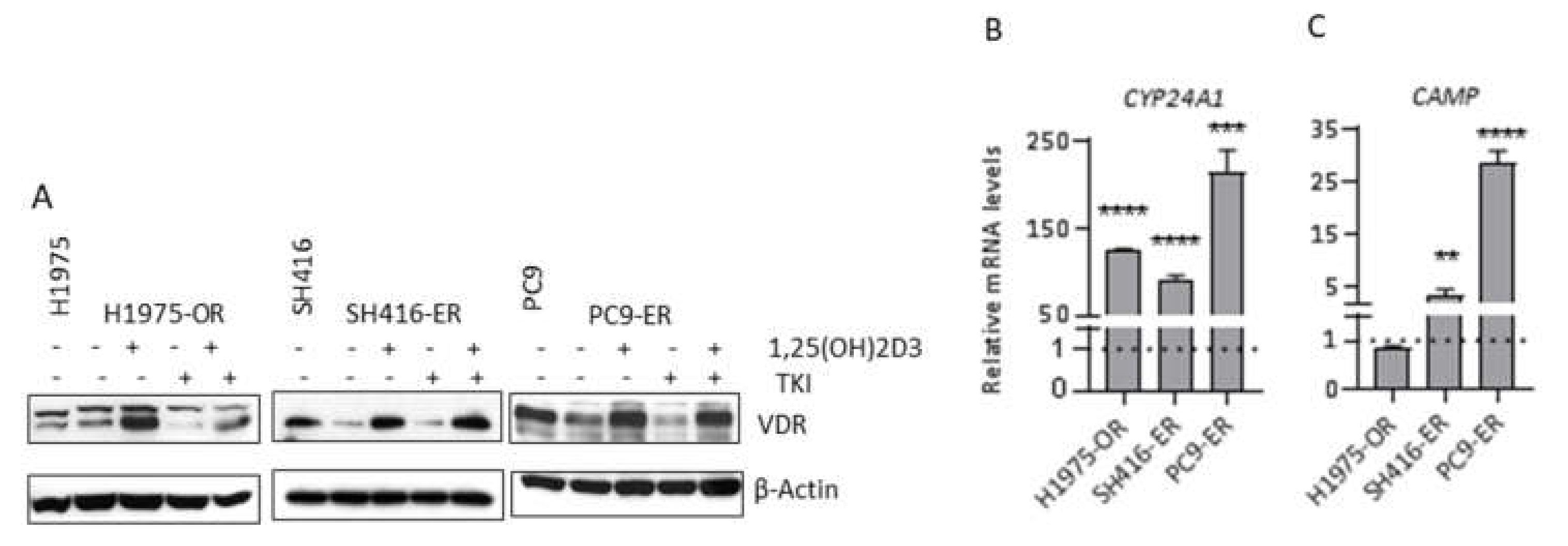
2.3. VDR Expression and Signaling Are Maintained in EGFR TKI-Resistant Cells
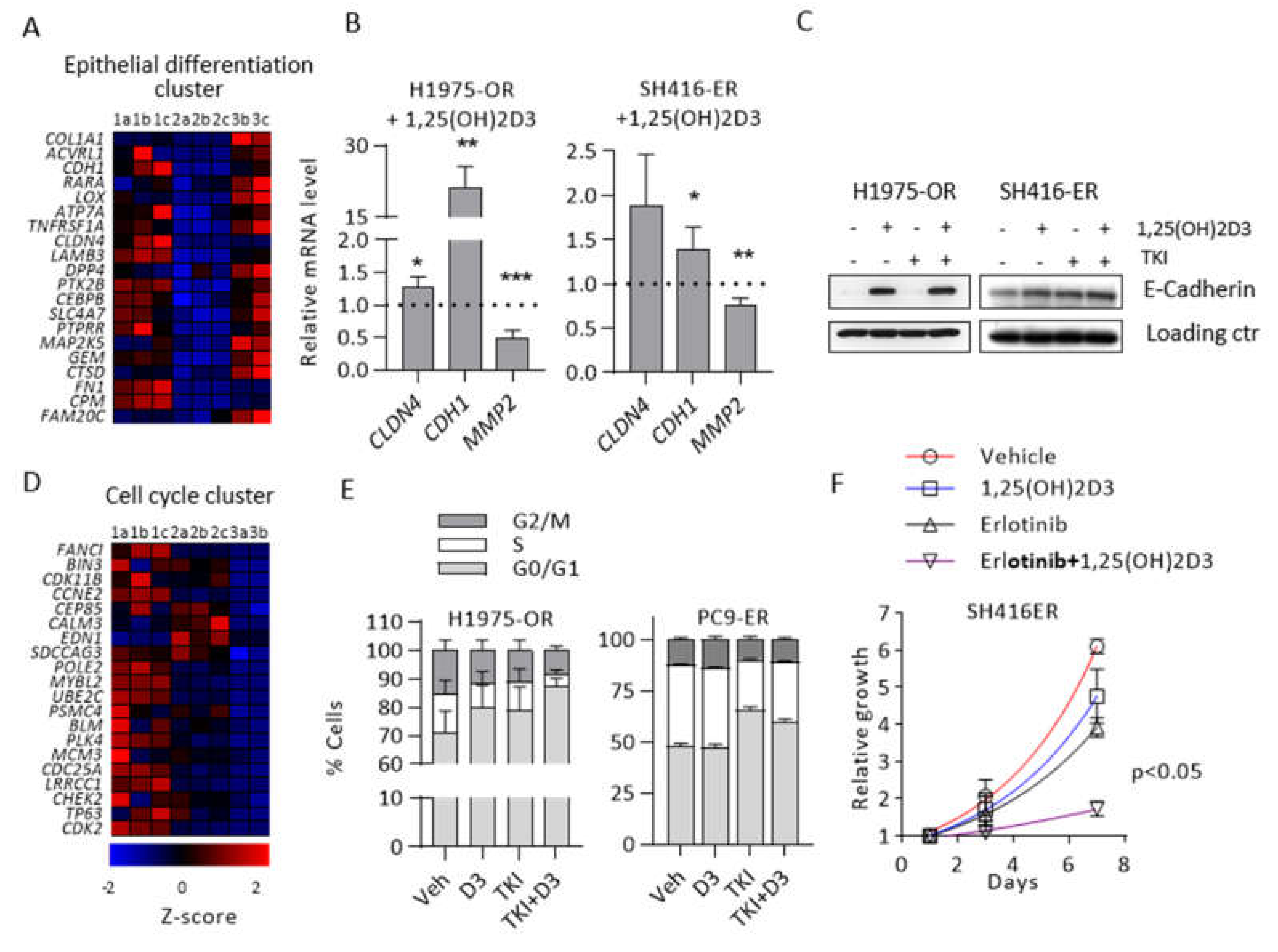
2.4. 1,25(OH)2D3 Promotes Epithelial Differentiation and Cell Cycle Arrest in EMT-Associated Models of EGFR TKI Resistance
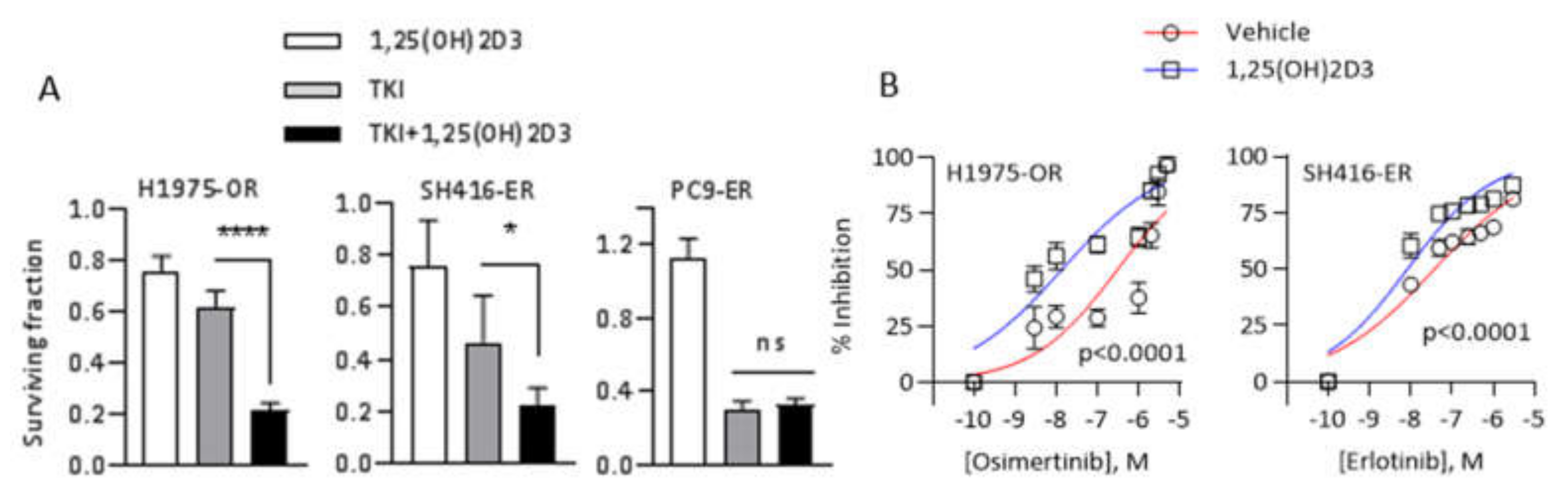
2.5. 1,25(OH)2D3 Promotes Sensitivity to EGFR TKIs in EMT-Associated Models of EGFR TKI Resistance
3. Discussion
4. Materials and Methods
4.1. Retrospective Clinical Study
4.2. Cell Culture
4.3. EGFR TKI and 1,25(OH)2D3 Treatments
4.4. Sulforhodamine B (SRB) Assay
4.5. IC50 and IC80 Determination
4.6. Cell Cycle Analysis
4.7. Cell Doubling Time Determination
4.8. Transwell Invasion Assay
4.9. RNA Isolation, cDNA Synthesis, and qRT–PCR
4.10. Western Blot Antibodies and Detection Reagents
4.11. RNA Sequencing
4.12. Bioinformatics
4.13. Statistical Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Didkowska, J.; Wojciechowska, U.; Manczuk, M.; Lobaszewski, J. Lung cancer epidemiology: Contemporary and future challenges worldwide. Ann. Transl. Med. 2016, 4, 150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shigematsu, H.; Gazdar, A.F. Somatic mutations of epidermal growth factor receptor signaling pathway in lung cancers. Int. J. Cancer 2006, 118, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Shigematsu, H.; Takahashi, T.; Nomura, M.; Majmudar, K.; Suzuki, M.; Lee, H.; Wistuba, II; Fong, K.M.; Toyooka, S.; Shimizu, N.; et al. Somatic mutations of the HER2 kinase domain in lung adenocarcinomas. Cancer Res. 2005, 65, 1642–1646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sequist, L.V.; Waltman, B.A.; Dias-Santagata, D.; Digumarthy, S.; Turke, A.B.; Fidias, P.; Bergethon, K.; Shaw, A.T.; Gettinger, S.; Cosper, A.K.; et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci. Transl. Med. 2011, 3, 75ra26. [Google Scholar] [CrossRef] [Green Version]
- Chung, J.H.; Rho, J.K.; Xu, X.; Lee, J.S.; Yoon, H.I.; Lee, C.T.; Choi, Y.J.; Kim, H.R.; Kim, C.H.; Lee, J.C. Clinical and molecular evidences of epithelial to mesenchymal transition in acquired resistance to EGFR-TKIs. Lung Cancer 2011, 73, 176–182. [Google Scholar] [CrossRef] [PubMed]
- Suda, K.; Tomizawa, K.; Fujii, M.; Murakami, H.; Osada, H.; Maehara, Y.; Yatabe, Y.; Sekido, Y.; Mitsudomi, T. Epithelial to mesenchymal transition in an epidermal growth factor receptor-mutant lung cancer cell line with acquired resistance to erlotinib. J. Thorac. Oncol. 2011, 6, 1152–1161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Lee, J.C.; Lin, L.; Olivas, V.; Au, V.; LaFramboise, T.; Abdel-Rahman, M.; Wang, X.; Levine, A.D.; Rho, J.K.; et al. Activation of the AXL kinase causes resistance to EGFR-targeted therapy in lung cancer. Nat. Genet. 2012, 44, 852–860. [Google Scholar] [CrossRef] [PubMed]
- Campbell, M.J.; Trump, D.L. Vitamin D Receptor Signaling and Cancer. Endocrinol. Metab. Clin. N. Am. 2017, 46, 1009–1038. [Google Scholar] [CrossRef] [PubMed]
- Welsh, J.; Wietzke, J.A.; Zinser, G.M.; Smyczek, S.; Romu, S.; Tribble, E.; Welsh, J.C.; Byrne, B.; Narvaez, C.J. Impact of the Vitamin D3 receptor on growth-regulatory pathways in mammary gland and breast cancer. J. Steroid Biochem. Mol. Biol. 2002, 83, 85–92. [Google Scholar] [CrossRef]
- Palmer, H.G.; Sanchez-Carbayo, M.; Ordonez-Moran, P.; Larriba, M.J.; Cordon-Cardo, C.; Munoz, A. Genetic signatures of differentiation induced by 1alpha,25-dihydroxyvitamin D3 in human colon cancer cells. Cancer Res. 2003, 63, 7799–7806. [Google Scholar]
- Swami, S.; Krishnan, A.V.; Wang, J.Y.; Jensen, K.; Horst, R.; Albertelli, M.A.; Feldman, D. Dietary vitamin D(3) and 1,25-dihydroxyvitamin D(3) (calcitriol) exhibit equivalent anticancer activity in mouse xenograft models of breast and prostate cancer. Endocrinology 2012, 153, 2576–2587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeong, Y.; Swami, S.; Krishnan, A.V.; Williams, J.D.; Martin, S.; Horst, R.L.; Albertelli, M.A.; Feldman, B.J.; Feldman, D.; Diehn, M. Inhibition of Mouse Breast Tumor-Initiating Cells by Calcitriol and Dietary Vitamin D. Mol. Cancer Ther. 2015, 14, 1951–1961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, T.Y.; Neuhouser, M.L. Serum 25-hydroxyvitamin D, vitamin A, and lung cancer mortality in the US population: A potential nutrient-nutrient interaction. Cancer Causes Control 2012, 23, 1557–1565. [Google Scholar] [CrossRef] [PubMed]
- Tretli, S.; Schwartz, G.G.; Torjesen, P.A.; Robsahm, T.E. Serum levels of 25-hydroxyvitamin D and survival in Norwegian patients with cancer of breast, colon, lung, and lymphoma: A population-based study. Cancer Causes Control 2012, 23, 363–370. [Google Scholar] [CrossRef] [Green Version]
- Heist, R.S.; Zhou, W.; Wang, Z.; Liu, G.; Neuberg, D.; Su, L.; Asomaning, K.; Hollis, B.W.; Lynch, T.J.; Wain, J.C.; et al. Circulating 25-hydroxyvitamin D, VDR polymorphisms, and survival in advanced non-small-cell lung cancer. J. Clin. Oncol. 2008, 26, 5596–5602. [Google Scholar] [CrossRef] [Green Version]
- Vashi, P.G.; Edwin, P.; Popiel, B.; Gupta, D. The relationship between circulating 25-hydroxyvitamin D and survival in newly diagnosed advanced non-small-cell lung cancer. BMC Cancer 2015, 15, 1012. [Google Scholar] [CrossRef] [Green Version]
- Anic, G.M.; Weinstein, S.J.; Mondul, A.M.; Mannisto, S.; Albanes, D. Serum vitamin D, vitamin D binding protein, and lung cancer survival. Lung Cancer 2014, 86, 297–303. [Google Scholar] [CrossRef] [Green Version]
- Akiba, T.; Morikawa, T.; Odaka, M.; Nakada, T.; Kamiya, N.; Yamashita, M.; Yabe, M.; Inagaki, T.; Asano, H.; Mori, S.; et al. Vitamin D Supplementation and Survival of Patients with Non-small Cell Lung Cancer: A Randomized, Double-Blind, Placebo-Controlled Trial. Clin. Cancer Res. 2018, 24, 4089–4097. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Kanterewicz, B.; Shoemaker, S.; Hu, Q.; Liu, S.; Atwood, K.; Hershberger, P. Differential response to 1alpha,25-dihydroxyvitamin D3 (1alpha,25(OH)2D3) in non-small cell lung cancer cells with distinct oncogene mutations. J. Steroid Biochem. Mol. Biol. 2013, 136, 264–270. [Google Scholar] [CrossRef] [Green Version]
- Verone-Boyle, A.R.; Shoemaker, S.; Attwood, K.; Morrison, C.D.; Makowski, A.J.; Battaglia, S.; Hershberger, P.A. Diet-derived 25-hydroxyvitamin D3 activates vitamin D receptor target gene expression and suppresses EGFR mutant non-small cell lung cancer growth in vitro and in vivo. Oncotarget 2016, 7, 995–1013. [Google Scholar] [CrossRef] [Green Version]
- Holick, M.F.; Binkley, N.C.; Bischoff-Ferrari, H.A.; Gordon, C.M.; Hanley, D.A.; Heaney, R.P.; Murad, M.H.; Weaver, C.M.; Endocrine, S. Evaluation, treatment, and prevention of vitamin D deficiency: An Endocrine Society clinical practice guideline. J. Clin. Endocrinol. Metab. 2011, 96, 1911–1930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, H.A.; Arcila, M.E.; Rekhtman, N.; Sima, C.S.; Zakowski, M.F.; Pao, W.; Kris, M.G.; Miller, V.A.; Ladanyi, M.; Riely, G.J. Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers. Clin. Cancer Res. 2013, 19, 2240–2247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, Y.; Togashi, Y.; Yatabe, Y.; Mizuuchi, H.; Jangchul, P.; Kondo, C.; Shimoji, M.; Sato, K.; Suda, K.; Tomizawa, K.; et al. EGFR Exon 18 Mutations in Lung Cancer: Molecular Predictors of Augmented Sensitivity to Afatinib or Neratinib as Compared with First- or Third-Generation TKIs. Clin. Cancer Res. 2015, 21, 5305–5313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yano, S.; Wang, W.; Li, Q.; Matsumoto, K.; Sakurama, H.; Nakamura, T.; Ogino, H.; Kakiuchi, S.; Hanibuchi, M.; Nishioka, Y.; et al. Hepatocyte growth factor induces gefitinib resistance of lung adenocarcinoma with epidermal growth factor receptor-activating mutations. Cancer Res. 2008, 68, 9479–9487. [Google Scholar] [CrossRef] [Green Version]
- Cortot, A.B.; Repellin, C.E.; Shimamura, T.; Capelletti, M.; Zejnullahu, K.; Ercan, D.; Christensen, J.G.; Wong, K.K.; Gray, N.S.; Janne, P.A. Resistance to irreversible EGF receptor tyrosine kinase inhibitors through a multistep mechanism involving the IGF1R pathway. Cancer Res. 2013, 73, 834–843. [Google Scholar] [CrossRef] [Green Version]
- Shi, P.; Oh, Y.T.; Zhang, G.; Yao, W.; Yue, P.; Li, Y.; Kanteti, R.; Riehm, J.; Salgia, R.; Owonikoko, T.K.; et al. Met gene amplification and protein hyperactivation is a mechanism of resistance to both first and third generation EGFR inhibitors in lung cancer treatment. Cancer Lett. 2016, 380, 494–504. [Google Scholar] [CrossRef]
- Ichihara, E.; Westover, D.; Meador, C.B.; Yan, Y.; Bauer, J.A.; Lu, P.; Ye, F.; Kulick, A.; de Stanchina, E.; McEwen, R.; et al. SFK/FAK Signaling Attenuates Osimertinib Efficacy in Both Drug-Sensitive and Drug-Resistant Models of EGFR-Mutant Lung Cancer. Cancer Res. 2017, 77, 2990–3000. [Google Scholar] [CrossRef] [Green Version]
- Namba, K.; Shien, K.; Takahashi, Y.; Torigoe, H.; Sato, H.; Yoshioka, T.; Takeda, T.; Kurihara, E.; Ogoshi, Y.; Yamamoto, H.; et al. Activation of AXL as a Preclinical Acquired Resistance Mechanism Against Osimertinib Treatment in EGFR-Mutant Non-Small Cell Lung Cancer Cells. Mol. Cancer Res. 2019, 17, 499–507. [Google Scholar] [CrossRef] [Green Version]
- Tang, Z.H.; Jiang, X.M.; Guo, X.; Fong, C.M.; Chen, X.; Lu, J.J. Characterization of osimertinib (AZD9291)-resistant non-small cell lung cancer NCI-H1975/OSIR cell line. Oncotarget 2016, 7, 81598–81610. [Google Scholar] [CrossRef] [Green Version]
- Nukaga, S.; Yasuda, H.; Tsuchihara, K.; Hamamoto, J.; Masuzawa, K.; Kawada, I.; Naoki, K.; Matsumoto, S.; Mimaki, S.; Ikemura, S.; et al. Amplification of EGFR Wild-Type Alleles in Non-Small Cell Lung Cancer Cells Confers Acquired Resistance to Mutation-Selective EGFR Tyrosine Kinase Inhibitors. Cancer Res. 2017, 77, 2078–2089. [Google Scholar] [CrossRef] [Green Version]
- Fraser, D.R.; Kodicek, E. Unique biosynthesis by kidney of a biological active vitamin D metabolite. Nature 1970, 228, 764–766. [Google Scholar] [CrossRef] [PubMed]
- Upadhyay, S.K.; Verone, A.; Shoemaker, S.; Qin, M.; Liu, S.; Campbell, M.; Hershberger, P.A. 1,25-Dihydroxyvitamin D3 (1,25(OH)2D3) Signaling Capacity and the Epithelial-Mesenchymal Transition in Non-Small Cell Lung Cancer (NSCLC): Implications for Use of 1,25(OH)2D3 in NSCLC Treatment. Cancers (Basel) 2013, 5, 1504–1521. [Google Scholar] [CrossRef] [PubMed]
- Bauml, J.; Mick, R.; Zhang, Y.; Watt, C.D.; Vachani, A.; Aggarwal, C.; Evans, T.; Langer, C. Frequency of EGFR and KRAS mutations in patients with non small cell lung cancer by racial background: Do disparities exist? Lung Cancer 2013, 81, 347–353. [Google Scholar] [CrossRef] [Green Version]
- Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014, 511, 543–550. [Google Scholar] [CrossRef] [PubMed]
- Shaurova, T.G.K.D.; Battaglia, S.; Hutson, A.; Hershberger, P.A. Unpublished Work; Roswell Park Comprehensive Cancer Center: Buffalo, NY, USA, 2019. [Google Scholar]
- Chatkin, J.M.; Abreu, C.M.; Fritscher, C.C.; Wagner, M.B.; Pinto, J.A. Is there a gender difference in non-small cell lung cancer survival? Gend. Med. 2004, 1, 41–47. [Google Scholar] [CrossRef]
- Sakurai, H.; Asamura, H.; Goya, T.; Eguchi, K.; Nakanishi, Y.; Sawabata, N.; Okumura, M.; Miyaoka, E.; Fujii, Y.; Japanese Joint Committee for Lung Cancer Registration. Survival differences by gender for resected non-small cell lung cancer: A retrospective analysis of 12,509 cases in a Japanese Lung Cancer Registry study. J. Thorac Oncol. 2010, 5, 1594–1601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sagerup, C.M.; Smastuen, M.; Johannesen, T.B.; Helland, A.; Brustugun, O.T. Sex-specific trends in lung cancer incidence and survival: A population study of 40,118 cases. Thorax 2011, 66, 301–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wakelee, H.A.; Wang, W.; Schiller, J.H.; Langer, C.J.; Sandler, A.B.; Belani, C.P.; Johnson, D.H.; Eastern Cooperative Oncology Group. Survival differences by sex for patients with advanced non-small cell lung cancer on Eastern Cooperative Oncology Group trial 1594. J. Thorac Oncol. 2006, 1, 441–446. [Google Scholar] [CrossRef]
- Sun, J.M.; Hwang, D.W.; Ahn, J.S.; Ahn, M.J.; Park, K. Prognostic and predictive value of KRAS mutations in advanced non-small cell lung cancer. PLoS ONE 2013, 8, e64816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosell, R.; Carcereny, E.; Gervais, R.; Vergnenegre, A.; Massuti, B.; Felip, E.; Palmero, R.; Garcia-Gomez, R.; Pallares, C.; Sanchez, J.M.; et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): A multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2012, 13, 239–246. [Google Scholar] [CrossRef]
- Palmer, H.G.; Gonzalez-Sancho, J.M.; Espada, J.; Berciano, M.T.; Puig, I.; Baulida, J.; Quintanilla, M.; Cano, A.; de Herreros, A.G.; Lafarga, M.; et al. Vitamin D(3) promotes the differentiation of colon carcinoma cells by the induction of E-cadherin and the inhibition of beta-catenin signaling. J. Cell Biol. 2001, 154, 369–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, J.; Zhang, Z.; Musch, M.W.; Ning, G.; Sun, J.; Hart, J.; Bissonnette, M.; Li, Y.C. Novel role of the vitamin D receptor in maintaining the integrity of the intestinal mucosal barrier. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 294, G208–G216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, W.; Chiu, Y.F.; Kuo, M.H.; Lee, K.L.; Lee, A.C.; Yu, C.C.; Chang, J.L.; Huang, W.C.; Hsiao, S.H.; Lin, S.E.; et al. Expression of Neuroendocrine Factor VGF in Lung Cancer Cells Confers Resistance to EGFR Kinase Inhibitors and Triggers Epithelial-to-Mesenchymal Transition. Cancer Res. 2017, 77, 3013–3026. [Google Scholar] [CrossRef] [Green Version]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dickinson, P.A.; Cantarini, M.V.; Collier, J.; Frewer, P.; Martin, S.; Pickup, K.; Ballard, P. Metabolic Disposition of Osimertinib in Rats, Dogs, and Humans: Insights into a Drug Designed to Bind Covalently to a Cysteine Residue of Epidermal Growth Factor Receptor. Drug Metab. Dispos. 2016, 44, 1201–1212. [Google Scholar] [CrossRef] [Green Version]
- Hidalgo, M.; Siu, L.L.; Nemunaitis, J.; Rizzo, J.; Hammond, L.A.; Takimoto, C.; Eckhardt, S.G.; Tolcher, A.; Britten, C.D.; Denis, L.; et al. Phase I and pharmacologic study of OSI-774, an epidermal growth factor receptor tyrosine kinase inhibitor, in patients with advanced solid malignancies. J. Clin. Oncol. 2001, 19, 3267–3279. [Google Scholar] [CrossRef]
- Molina-Vila, M.A.; Bertran-Alamillo, J.; Reguart, N.; Taron, M.; Castella, E.; Llatjos, M.; Costa, C.; Mayo, C.; Pradas, A.; Queralt, C.; et al. A sensitive method for detecting EGFR mutations in non-small cell lung cancer samples with few tumor cells. J. Thorac Oncol. 2008, 3, 1224–1235. [Google Scholar] [CrossRef] [Green Version]
- Vichai, V.; Kirtikara, K. Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat. Protoc. 2006, 1, 1112–1116. [Google Scholar] [CrossRef]
- Bray, N.L.; Pimentel, H.; Melsted, P.; Pachter, L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 2016, 34, 525–527. [Google Scholar] [CrossRef]
- Soneson, C.; Love, M.I.; Robinson, M.D. Differential analyses for RNA-seq: Transcript-level estimates improve gene-level inferences. F1000Res 2015, 4, 1521. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, X.; Lu, J.; Kulbokas, E.J.; Golub, T.R.; Mootha, V.; Lindblad-Toh, K.; Lander, E.S.; Kellis, M. Systematic discovery of regulatory motifs in human promoters and 3’ UTRs by comparison of several mammals. Nature 2005, 434, 338–345. [Google Scholar] [CrossRef] [PubMed]
- Merico, D.; Isserlin, R.; Stueker, O.; Emili, A.; Bader, G.D. Enrichment map: A network-based method for gene-set enrichment visualization and interpretation. PLoS ONE 2010, 5, e13984. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristic | EGFRmt (n = 21) | KRASmt (n = 34) |
|---|---|---|
| Age (y), mean (SD) | 68 (9) | 65 (9) |
| Female, No. (%) | 14 (64) | 19 (56) |
| African American, No. (%) | 2 (10) | 3 (9) |
| Stage, No. (%) | ||
| IV | 21 (100) | 34 (100) |
| Serum 25(OH)D3 (ng/ml), mean (SD) | 28.78 (11.13) | 24.84 (12.27) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shaurova, T.; Dy, G.K.; Battaglia, S.; Hutson, A.; Zhang, L.; Zhang, Y.; Lovly, C.M.; Seshadri, M.; Goodrich, D.W.; Johnson, C.S.; et al. Vitamin D3 Metabolites Demonstrate Prognostic Value in EGFR-Mutant Lung Adenocarcinoma and Can be Deployed to Oppose Acquired Therapeutic Resistance. Cancers 2020, 12, 675. https://doi.org/10.3390/cancers12030675
Shaurova T, Dy GK, Battaglia S, Hutson A, Zhang L, Zhang Y, Lovly CM, Seshadri M, Goodrich DW, Johnson CS, et al. Vitamin D3 Metabolites Demonstrate Prognostic Value in EGFR-Mutant Lung Adenocarcinoma and Can be Deployed to Oppose Acquired Therapeutic Resistance. Cancers. 2020; 12(3):675. https://doi.org/10.3390/cancers12030675
Chicago/Turabian StyleShaurova, Tatiana, Grace K Dy, Sebastiano Battaglia, Alan Hutson, Letian Zhang, Yunkai Zhang, Christine M Lovly, Mukund Seshadri, David W Goodrich, Candace S Johnson, and et al. 2020. "Vitamin D3 Metabolites Demonstrate Prognostic Value in EGFR-Mutant Lung Adenocarcinoma and Can be Deployed to Oppose Acquired Therapeutic Resistance" Cancers 12, no. 3: 675. https://doi.org/10.3390/cancers12030675