TWIST1 Heterodimerization with E12 Requires Coordinated Protein Phosphorylation to Regulate Periostin Expression

 , , and
, , and 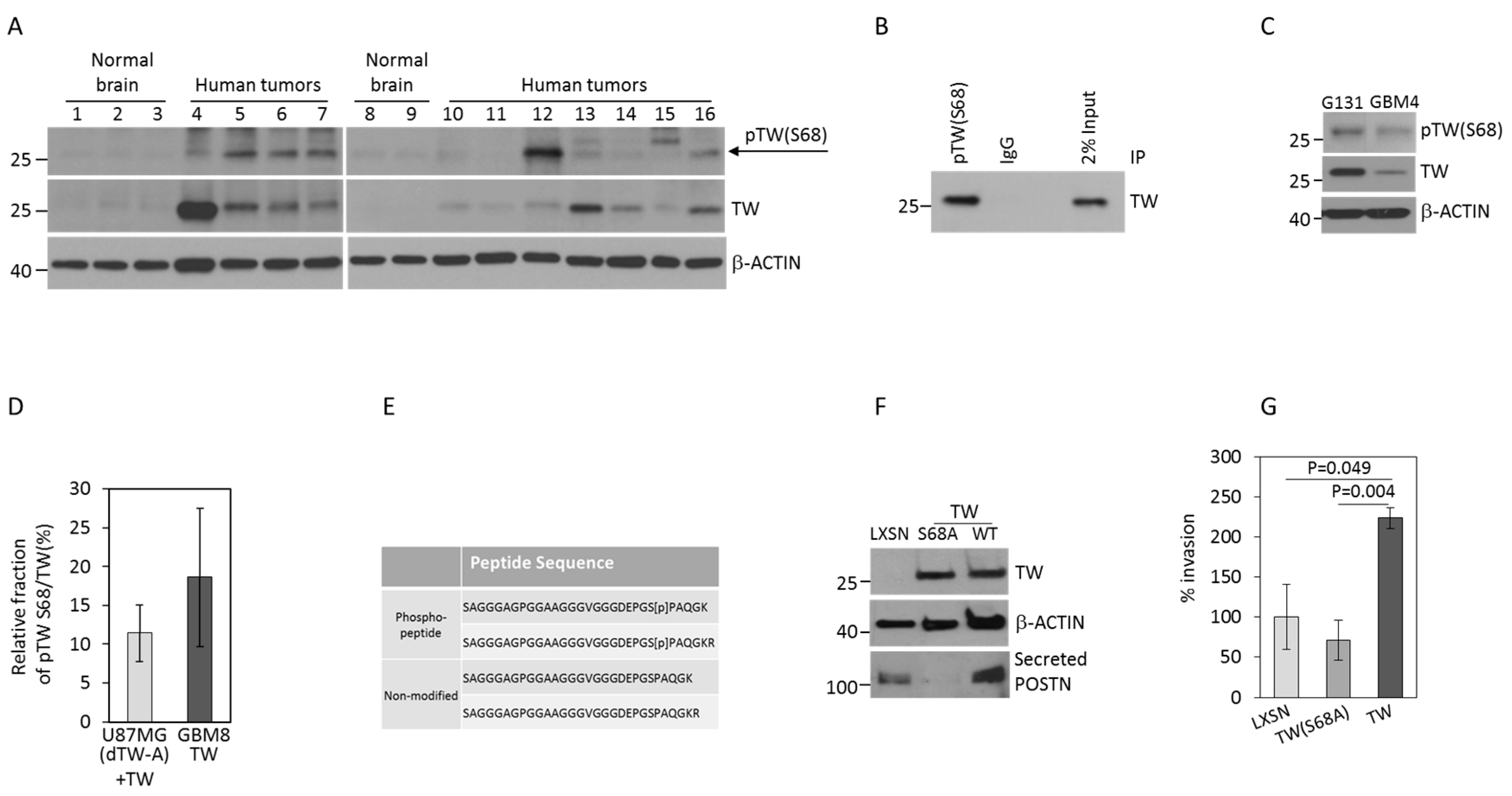{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. TW S68 Phosphorylation Detected in Human GBM and GBM Cells Promotes Invasion
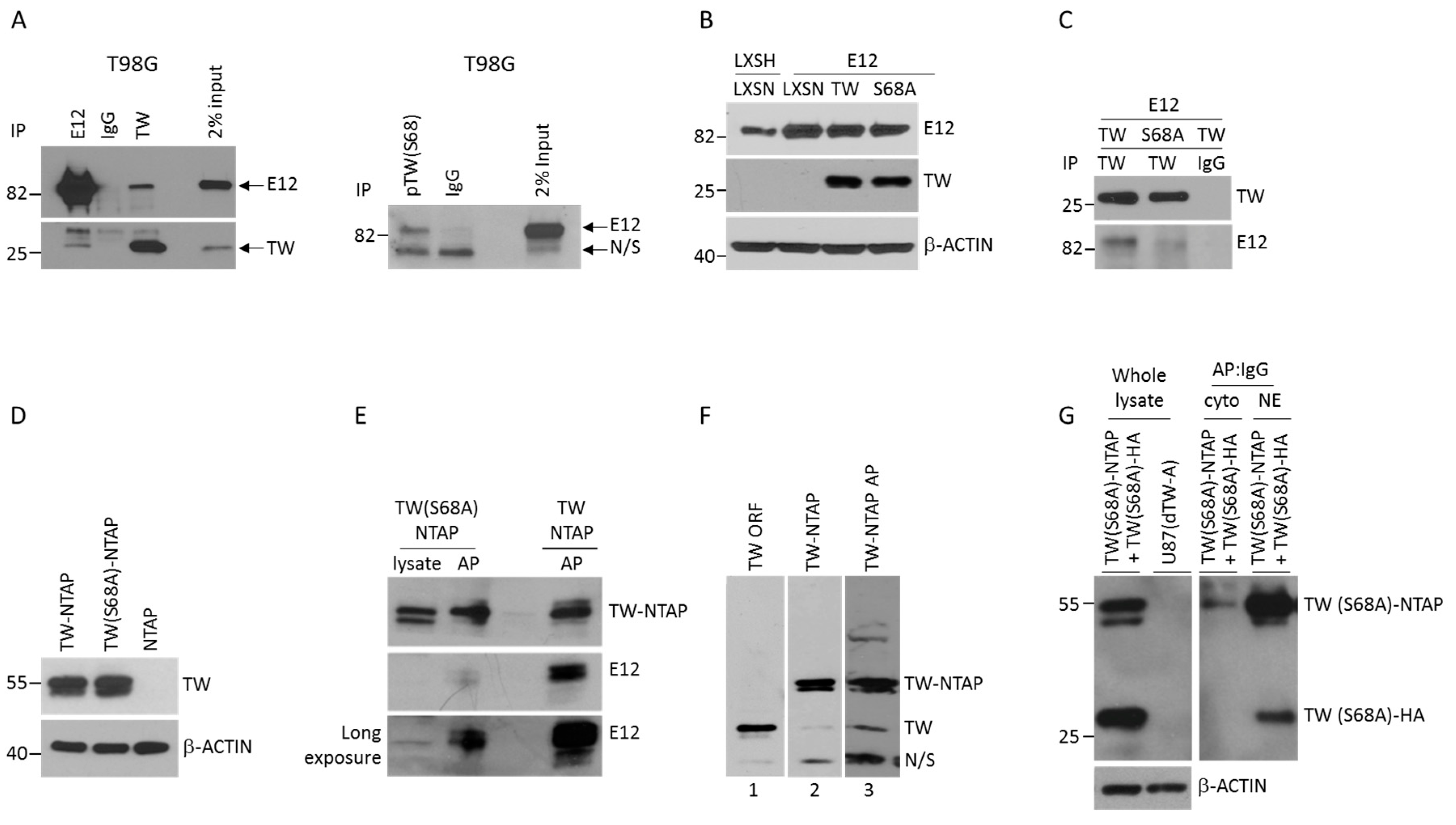
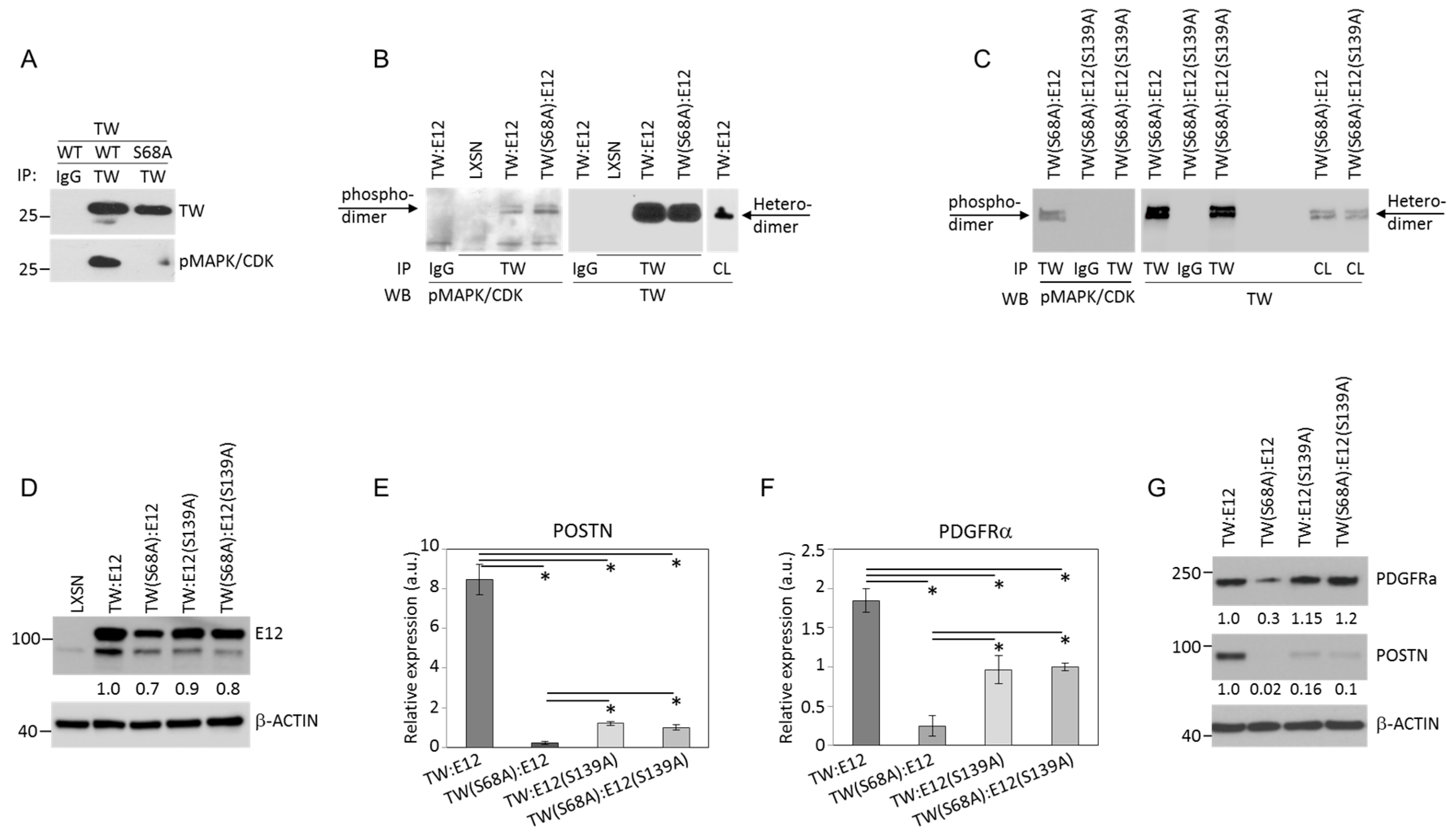
2.2. Serine 68 Phosphorylation in TW Promotes Interaction with the E12 Binding Partner
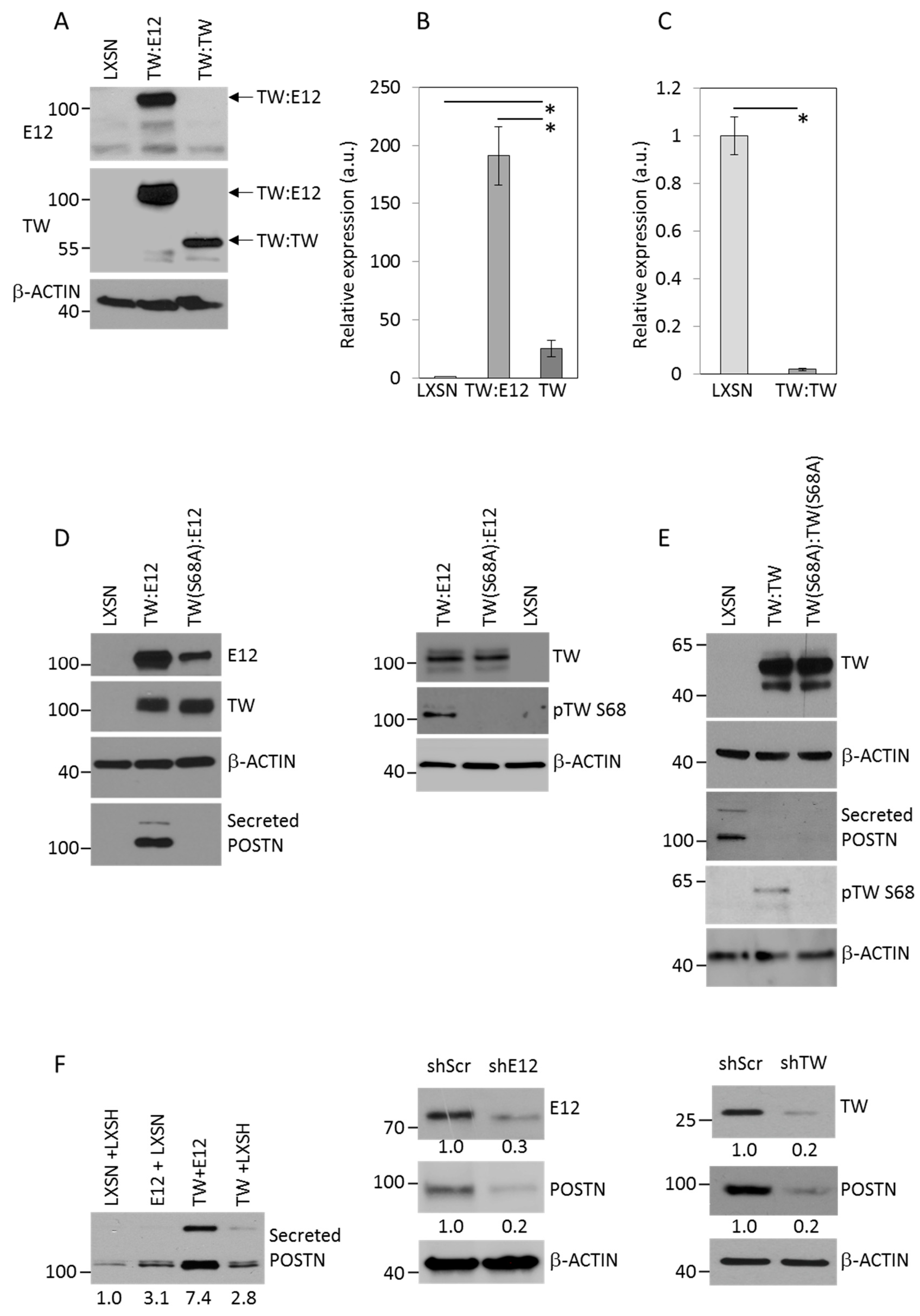
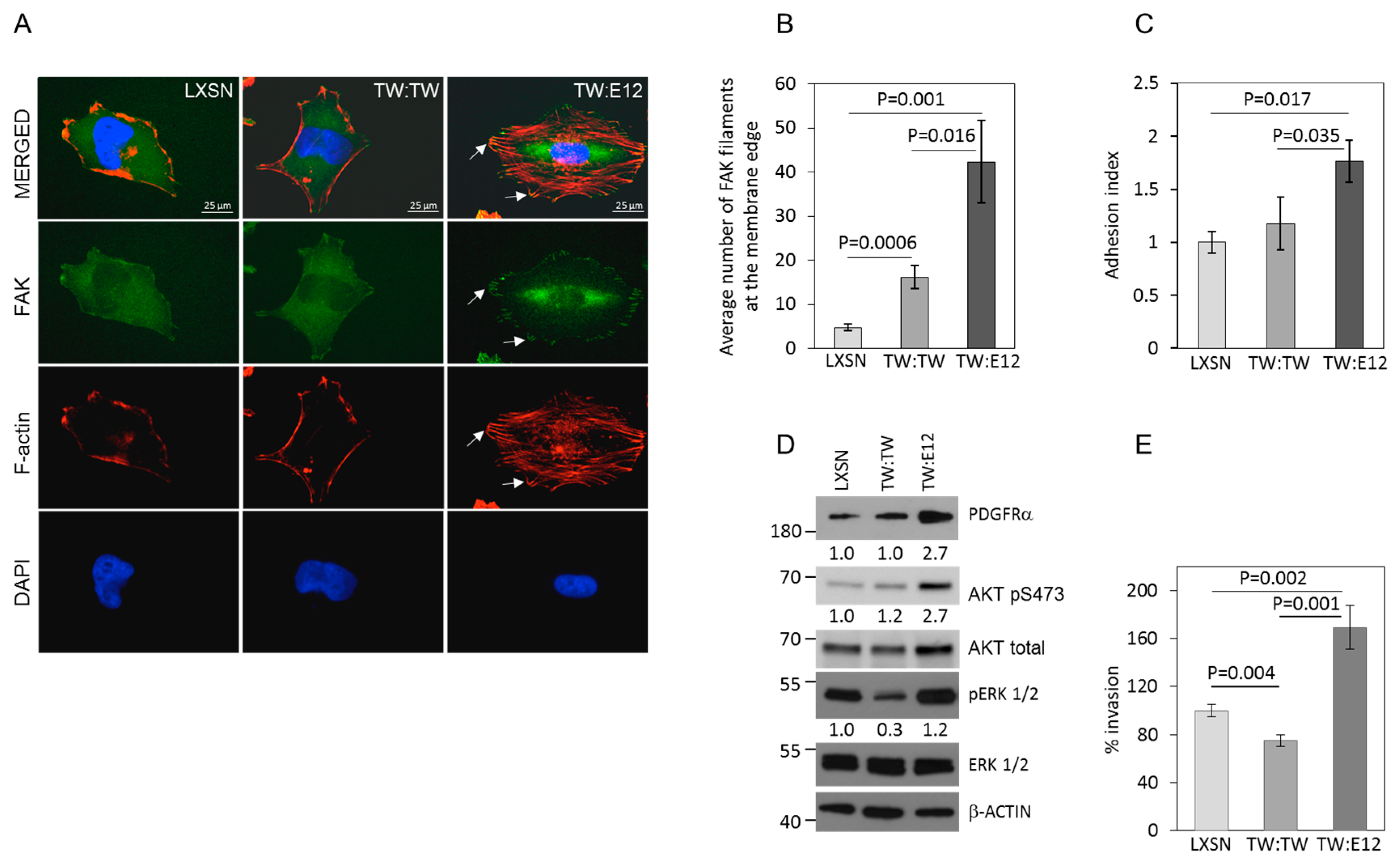
2.3. TW:E12 Heterodimers and TW:TW Homodimers Differentially Regulate Expression of Periostin (POSTN), Glioma Cell Adhesion, and Invasion
2.4. E12 S139 and TW S68 Phosphorylation Regulate POSTN mRNA Expression
3. Discussion
4. Materials and Methods
4.1. Cells and Tissue
4.2. Expression Constructs and Virus Production
4.3. Antibody Validation
4.4. Immuno-Precipitation and Affinity Purification
4.5. Invasion, Adhesion Assays, and Immunofluorescence
4.6. Quantitative Real Time PCR
4.7. Mass Spectrometry
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Iwadate, Y. Epithelial-mesenchymal transition in glioblastoma progression. Oncol. Lett. 2016, 11, 1615–1620. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phillips, H.S.; Kharbanda, S.; Chen, R.; Forrest, W.F.; Soriano, R.H.; Wu, T.D.; Misra, A.; Nigro, J.M.; Colman, H.; Soroceanu, L.; et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell 2006, 9, 157–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tso, C.L.; Shintaku, P.; Chen, J.; Liu, Q.; Liu, J.; Chen, Z.; Yoshimoto, K.; Mischel, P.S.; Cloughesy, T.F.; Liau, L.M.; et al. Primary glioblastomas express mesenchymal stem-like properties. Mol. Cancer Res. 2006, 4, 607–619. [Google Scholar] [CrossRef] [PubMed]
- Vehlow, A.; Cordes, N. Invasion as target for therapy of glioblastoma multiforme. Biochim. Biophys. Acta 2013, 1836, 236–244. [Google Scholar] [CrossRef] [PubMed]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Gonzalez, L.; Alonso, J. Periostin: A Matricellular Protein With Multiple Functions in Cancer Development and Progression. Front. Oncol. 2018, 8, 225. [Google Scholar] [CrossRef]
- Morra, L.; Moch, H. Periostin expression and epithelial-mesenchymal transition in cancer: A review and an update. Virchows Arch 2011, 459, 465–475. [Google Scholar] [CrossRef]
- Elias, M.C.; Tozer, K.R.; Silber, J.R.; Mikheeva, S.; Deng, M.; Morrison, R.S.; Manning, T.C.; Silbergeld, D.L.; Glackin, C.A.; Reh, T.A.; et al. TWIST is expressed in human gliomas and promotes invasion. Neoplasia 2005, 7, 824–837. [Google Scholar] [CrossRef]
- Mikheeva, S.A.; Mikheev, A.M.; Petit, A.; Beyer, R.; Oxford, R.G.; Khorasani, L.; Maxwell, J.P.; Glackin, C.A.; Wakimoto, H.; Gonzalez-Herrero, I.; et al. TWIST1 promotes invasion through mesenchymal change in human glioblastoma. Mol. Cancer 2010, 9, 194. [Google Scholar] [CrossRef]
- Mikheev, A.M.; Mikheeva, S.A.; Trister, A.D.; Tokita, M.J.; Emerson, S.N.; Parada, C.A.; Born, D.E.; Carnemolla, B.; Frankel, S.; Kim, D.H.; et al. Periostin is a novel therapeutic target that predicts and regulates glioma malignancy. Neuro. Oncol. 2015, 17, 372–382. [Google Scholar] [CrossRef] [PubMed]
- Dobrian, A.D. A tale with a Twist: A developmental gene with potential relevance for metabolic dysfunction and inflammation in adipose tissue. Front. Endocrinol. (Lausanne) 2012, 3, 108. [Google Scholar] [CrossRef] [PubMed]
- Firulli, A.B.; Conway, S.J. Phosphoregulation of Twist1 provides a mechanism of cell fate control. Curr. Med. Chem. 2008, 15, 2641–2647. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.; Zhou, J.; Fu, J.; He, T.; Qin, J.; Wang, L.; Liao, L.; Xu, J. Phosphorylation of serine 68 of Twist1 by MAPKs stabilizes Twist1 protein and promotes breast cancer cell invasiveness. Cancer Res. 2011, 71, 3980–3990. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.; Fu, J.; Shen, T.; Lin, X.; Liao, L.; Feng, X.H.; Xu, J. The Small C-terminal Domain Phosphatase 1 Inhibits Cancer Cell Migration and Invasion by Dephosphorylating Ser(P)68-Twist1 to Accelerate Twist1 Protein Degradation. J. Biol. Chem. 2016, 291, 11518–11528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Firulli, B.A.; Redick, B.A.; Conway, S.J.; Firulli, A.B. Mutations within helix I of Twist1 result in distinct limb defects and variation of DNA binding affinities. J. Biol. Chem. 2007, 282, 27536–27546. [Google Scholar] [CrossRef]
- Connerney, J.; Andreeva, V.; Leshem, Y.; Muentener, C.; Mercado, M.A.; Spicer, D.B. Twist1 dimer selection regulates cranial suture patterning and fusion. Dev. Dyn. 2006, 235, 1345–1357. [Google Scholar] [CrossRef]
- Sharma, V.P.; Fenwick, A.L.; Brockop, M.S.; McGowan, S.J.; Goos, J.A.; Hoogeboom, A.J.; Brady, A.F.; Jeelani, N.O.; Lynch, S.A.; Mulliken, J.B.; et al. Mutations in TCF12, encoding a basic helix-loop-helix partner of TWIST1, are a frequent cause of coronal craniosynostosis. Nat. Genet. 2013, 45, 304–307. [Google Scholar] [CrossRef]
- Firulli, B.A.; Krawchuk, D.; Centonze, V.E.; Vargesson, N.; Virshup, D.M.; Conway, S.J.; Cserjesi, P.; Laufer, E.; Firulli, A.B. Altered Twist1 and Hand2 dimerization is associated with Saethre-Chotzen syndrome and limb abnormalities. Nat. Genet. 2005, 37, 373–381. [Google Scholar] [CrossRef] [Green Version]
- Castanon, I.; Von Stetina, S.; Kass, J.; Baylies, M.K. Dimerization partners determine the activity of the Twist bHLH protein during Drosophila mesoderm development. Development 2001, 128, 3145–3159. [Google Scholar]
- Gajula, R.P.; Chettiar, S.T.; Williams, R.D.; Nugent, K.; Kato, Y.; Wang, H.; Malek, R.; Taparra, K.; Cades, J.; Annadanam, A.; et al. Structure-function studies of the bHLH phosphorylation domain of TWIST1 in prostate cancer cells. Neoplasia 2015, 17, 16–31. [Google Scholar] [CrossRef] [PubMed]
- Jacqueroud, L.; Bouard, C.; Richard, G.; Payen, L.; Devouassoux-Shisheboran, M.; Spicer, D.B.; Caramel, J.; Collin, G.; Puisieux, A.; Tissier, A.; et al. The Heterodimeric TWIST1-E12 Complex Drives the Oncogenic Potential of TWIST1 in Human Mammary Epithelial Cells. Neoplasia 2016, 18, 317–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Nikhil, K.; Viccaro, K.; Chang, L.; Jacobsen, M.; Sandusky, G.; Shah, K. The Aurora-A-Twist1 axis promotes highly aggressive phenotypes in pancreatic carcinoma. J. Cell. Sci. 2017, 130, 1078–1093. [Google Scholar] [CrossRef] [PubMed]
- Mikheev, A.M.; Mikheeva, S.A.; Severs, L.J.; Funk, C.C.; Huang, L.; McFaline-Figueroa, J.L.; Schwensen, J.; Trapnell, C.; Price, N.D.; Wong, S.; et al. Targeting TWIST1 through loss of function inhibits tumorigenicity of human glioblastoma. Mol. Oncol. 2018, 12, 1188–1202. [Google Scholar] [CrossRef] [PubMed]
- Goossens, S.; Vandamme, N.; Van Vlierberghe, P.; Berx, G. EMT transcription factors in cancer development re-evaluated: Beyond EMT and MET. Biochim. Biophys. Acta Rev. Cancer 2017, 1868, 584–591. [Google Scholar] [CrossRef] [PubMed]
- Oshima, A.; Tanabe, H.; Yan, T.; Lowe, G.N.; Glackin, C.A.; Kudo, A. A novel mechanism for the regulation of osteoblast differentiation: transcription of periostin, a member of the fasciclin I family, is regulated by the bHLH transcription factor, twist. J. Cell Biochem. 2002, 86, 792–804. [Google Scholar] [CrossRef] [PubMed]
- Tian, B.; Zhang, Y.; Zhang, J. Periostin is a new potential prognostic biomarker for glioma. Tumour Biol. 2014, 35, 5877–5883. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wang, Y.; Jiang, C. Stromal protein periostin identified as a progression associated and prognostic biomarker in glioma via inducing an invasive and proliferative phenotype. Int. J. Oncol. 2013, 42, 1716–1724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, G.; Hemmings, B.A. Phosphorylation of basic helix-loop-helix transcription factor Twist in development and disease. Biochem. Soc. Trans. 2012, 40, 90–93. [Google Scholar] [CrossRef] [PubMed]
- Gajula, R.P.; Chettiar, S.T.; Williams, R.D.; Thiyagarajan, S.; Kato, Y.; Aziz, K.; Wang, R.; Gandhi, N.; Wild, A.T.; Vesuna, F.; et al. The twist box domain is required for Twist1-induced prostate cancer metastasis. Mol. Cancer Res. 2013, 11, 1387–1400. [Google Scholar] [CrossRef]
- Yochum, Z.A.; Cades, J.; Mazzacurati, L.; Neumann, N.M.; Khetarpal, S.K.; Chatterjee, S.; Wang, H.; Attar, M.A.; Huang, E.H.; Chatley, S.N.; et al. A First-in-Class TWIST1 Inhibitor with Activity in Oncogene-Driven Lung Cancer. Mol. Cancer Res. 2017, 15, 1764–1776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, A.T.; Liu, Y.; Ayyanathan, K.; Benner, C.; Jiang, Y.; Prokop, J.W.; Paz, H.; Wang, D.; Li, H.R.; Fu, X.D.; et al. An evolutionarily conserved DNA architecture determines target specificity of the TWIST family bHLH transcription factors. Genes Dev. 2015, 29, 603–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wakimoto, H.; Kesari, S.; Farrell, C.J.; Curry, W.T., Jr.; Zaupa, C.; Aghi, M.; Kuroda, T.; Stemmer-Rachamimov, A.; Shah, K.; Liu, T.C.; et al. Human glioblastoma-derived cancer stem cells: establishment of invasive glioma models and treatment with oncolytic herpes simplex virus vectors. Cancer Res. 2009, 69, 3472–3481. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; MacQuarrie, K.L.; Analau, E.; Tyler, A.E.; Dilworth, F.J.; Cao, Y.; Diede, S.J.; Tapscott, S.J. MyoD and E-protein heterodimers switch rhabdomyosarcoma cells from an arrested myoblast phase to a differentiated state. Genes Dev. 2009, 23, 694–707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mikheev, A.M.; Mikheeva, S.A.; Liu, B.; Cohen, P.; Zarbl, H. A functional genomics approach for the identification of putative tumor suppressor genes: Dickkopf-1 as suppressor of HeLa cell transformation. Carcinogenesis 2004, 25, 47–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, G.I.; Gingras, A.C. Affinity-purification mass spectrometry (AP-MS) of serine/threonine phosphatases. Methods 2007, 42, 298–305. [Google Scholar] [CrossRef] [PubMed]
- Neuhold, L.A.; Wold, B. HLH forced dimers: tethering MyoD to E47 generates a dominant positive myogenic factor insulated from negative regulation by Id. Cell 1993, 74, 1033–1042. [Google Scholar] [CrossRef]
- Mikheev, A.M.; Mikheev, S.A.; Zhang, Y.; Aebersold, R.; Zarbl, H. CArG binding factor A (CBF-A) is involved in transcriptional regulation of the rat Ha-ras promoter. Nucleic. Acids Res. 2000, 28, 3762–3770. [Google Scholar] [CrossRef] [Green Version]
- Rappsilber, J.; Mann, M.; Ishihama, Y. Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat. Protoc. 2007, 2, 1896–1906. [Google Scholar] [CrossRef]
- Nesvizhskii, A.I.; Keller, A.; Kolker, E.; Aebersold, R. A statistical model for identifying proteins by tandem mass spectrometry. Anal. Chem. 2003, 75, 4646–4658. [Google Scholar] [CrossRef]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mikheeva, S.A.; Camp, N.D.; Huang, L.; Jain, A.; Jung, S.Y.; Avci, N.G.; Tokita, M.; Wolf-Yadlin, A.; Zhang, J.; Tapscott, S.J.; et al. TWIST1 Heterodimerization with E12 Requires Coordinated Protein Phosphorylation to Regulate Periostin Expression. Cancers 2019, 11, 1392. https://doi.org/10.3390/cancers11091392
Mikheeva SA, Camp ND, Huang L, Jain A, Jung SY, Avci NG, Tokita M, Wolf-Yadlin A, Zhang J, Tapscott SJ, et al. TWIST1 Heterodimerization with E12 Requires Coordinated Protein Phosphorylation to Regulate Periostin Expression. Cancers. 2019; 11(9):1392. https://doi.org/10.3390/cancers11091392
Chicago/Turabian StyleMikheeva, Svetlana A., Nathan D. Camp, Lei Huang, Antrix Jain, Sung Yun Jung, Naze G. Avci, Mari Tokita, Alejandro Wolf-Yadlin, Jing Zhang, Stephen J. Tapscott, and et al. 2019. "TWIST1 Heterodimerization with E12 Requires Coordinated Protein Phosphorylation to Regulate Periostin Expression" Cancers 11, no. 9: 1392. https://doi.org/10.3390/cancers11091392