Alcoholic-Hepatitis, Links to Brain and Microbiome: Mechanisms, Clinical and Experimental Research



 ,
,
Abstract
:
1. Introduction
2. Charles Lieber’s Scientific Legacy
3. Interleukin-8 Signaling Pathway in Alcoholic Hepatitis
4. FAT 10 in Alcoholic Hepatitis Pathogenesis
5. The Role of Alcohol-Induced Microtubule Hyperacetylation in ASH Progression
6. Micro-RNA Profiling of Hepatic Macrophages in Alcoholic Liver Disease
7. Signature Pathway of Alcoholic Hepatitis—Integrative Global Analysis
8. Gut-Liver Challenge in Alcoholic Liver Disease
9. Liver Influences on Alcohol Drinking Behavior
10. Phenotyping Human CYP1A1 Polymorphisms Using Model Organisms
11. Alcohol and Non-Infectious Liver Diseases
12. Alcohol and Therapeutics Interaction
13. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ABCA1 | ATP-binding cassette transporter |
| ABCG1 | ATP-binding cassette sub-family G member 1 |
| ACC | Acetyl-CoA carboxylase |
| ADH | alcohol dehydrogenase |
| ADR | Adiponectin |
| AdipoR | Adiponectin receptor |
| AH | acute alcoholic hepatitis |
| ALD | alcoholic liver disease |
| ALDH | acetaldehyde dehydrogenase |
| ALF | alcoholic fatty liver |
| ALT | Alanine aminotransferase |
| AMPK | 5′AMP-activated protein kinase |
| ApoCII | Apolipoprotein C2 |
| ASH | alcoholic steato-hepatitis |
| α-SMA | Alpha-smooth muscle actin |
| AST | aspartate aminotransferase |
| ATM | ataxia-telangiectasis-mutated |
| ATR ataxia and rad | ataxia and rad-3 related |
| AUDIT | Alcohol Use Disorders Identification |
| CCl4 | Carbon tetrachloride |
| CDT | carbohydrate-deficient transferrin |
| Chk | check point kinase |
| ChREBP | Carbohydrate response element-binding protein |
| Col | Collagen type 1 |
| CPT | Carnitine palmitoyl-transferase |
| Ctsb | Capthepsin B |
| CYP | cytochrome P450 |
| DAMPS | damage-associated molecular patterns |
| EMA | European Medicines Agency |
| FABP | Fatty acid-binding protein |
| FAS | Fatty acid synthase |
| FAT 10 | human leukocyte antigen F locus adjacent transcript 10 |
| FDA | Food and drug administration |
| FFA | Free fatty acid |
| Fibroscan | Vibration-Controlled Transient Elastography |
| FMT | Fecal microbial transplantation |
| FXR | farsenoid X receptor |
| G6Pase | Glucose 6-phosphate |
| GAPDH | Glyceraldehyde-3-phosphate dehydrogenase |
| GAA | guanidino-acetate |
| GAMT | guanidino-acetate methyltransferase |
| GGPP | Geranyl-geranyl pyrophosphate |
| γGT | gamma glutamyl transpeptidase, Gamma-glutamyl-transferase |
| GLUT4 | Glucose transporter type 4 |
| GNA15 | canine nucleotide-binding protein Alpha 15 |
| GPx | Glutathione peroxidase |
| GSDMD | Gasdermin-D |
| GSH | Glutathione |
| HBV | hepatitis B virus |
| HCC | hepatocellular carcinoma |
| HCV | hepatitis C virus |
| HDL | high density lipoprotein |
| HFD | high fat diet |
| HMOs | Human milk oligosaccharides |
| 4-HNE | 4-hydroxynonenal |
| IKKβ | Inhibitor of nuclear factor kappa-B kinase subunit beta |
| IARC | International Agency for Research on Cancer |
| IGF | insulin-like growth factor |
| IL | Interleukin |
| IL | Interleukin 8—CXCL8 |
| iNOS | Nitric oxide synthase |
| ITS | internal transcribed spacer |
| LDL | Low-density lipoprotein |
| LA | linoleic acid |
| LCT | long-chain triglycerides |
| LDL | low density lipoprotein |
| LGG | Lactobacillus rhamnosus GG |
| LPS | Lipopolysaccharide |
| MAPK | Mitogen-activated protein kinases |
| MCD | Methionine-choline-deficient diet |
| MCP-1 | Monocyte chemotactic protein 1 |
| MCT | medium-chain triglycerides |
| MCV | mean corpuscular volume of erythrocytes |
| MD2 | Myeloid differentiation 2 |
| MDA | Malondialdehyde |
| MDB | Mallory-Denk body |
| MDF | Maddrey discriminator function |
| MELD | Model for End-Stage Liver Disease |
| MEOS | microsomal ethanol oxidizing system |
| MONCF | monocyte-derived neutrophil chemotactic factor |
| MRI | magnetic resonance imaging |
| NAFLD | Non-alcoholic fatty liver diseases |
| NAS | NALFD activity score |
| NASH | Non-alcoholic steatohepatitis |
| NEFA | Non-esterified fatty acid |
| NFκB | Nuclear factor kappa-B |
| OA | Oleic acid |
| PAMPS | Pathogen-associated molecular patterns |
| PCR | Polymerase chain reaction |
| PMN | Poly-morpho-neutrophyl |
| PPAR | Peroxisome proliferator-activated receptor |
| PPI | proton pump inhibitor |
| ROS | reactive oxygen species |
| SAH | S-adenosyl-homocysteine |
| SAM | S-adenosyl-methionine |
| SCD | Stearoyl-CoA desaturase |
| SF | saturated fat |
| SHP | Small heterodimer partner |
| SIBO | small intestinal bacterial overgrowth |
| SNAP | single nucleotide polymorphisms |
| SOD | Superoxide dismutase |
| SREBP1 | Sterol regulatory element-binding protein 1 |
| TBARS | Thiobarbituric acid reactive substances |
| TC | Total cholesterol |
| TG | Total triglycerides |
| TGF-β | Transforming growth factor beta |
| TIMP | Tissue inhibitor of metalloproteinase |
| TLR | Toll like receptor |
| TNF | Tumor necrosis factor |
| UPN | Upper the normal values |
| USF | unsaturated fat |
| WGCNA | Weighted Gene Coexpression Network Analysis |
References
- Drill, V.A. Hepatotoxic agents; mechanism of action and dietary interrelationship. Pharmacol. Rev. 1952, 4, 1–42. [Google Scholar] [PubMed]
- Lieber, C.S. Metabolic effects of acetaldehyde. Biochem. Soc. Trans. 1988, 16, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Lieber, C.S. Alcoholic fatty liver: Its pathogenesis and mechanism of progression to inflammation and fibrosis. Alcohol 2004, 34, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Lieber, C.S.; Rubin, E.; DeCarli, L.M. Hepatic microsomal ethanol oxidizing system (MEOS): Differentiation from alcohol dehydrogenase and NADPH oxidase. Biochem. Biophys. Res. Commun. 1970, 40, 858–865. [Google Scholar] [CrossRef]
- Lieber, C.S. The microsomal ethanol oxidizing system: Its role in ethanol and xenobiotic metabolism. Biochem. Soc. Trans. 1988, 16, 232–239. [Google Scholar] [CrossRef] [Green Version]
- Teschke, R.; Hasumura, Y.; Lieber, C.S. Hepatic microsomal ethanol oxidizing system: Solubilization, isolation and characterization. Arch. Biochem. Biophys. 1974, 163, 404–415. [Google Scholar] [CrossRef]
- Lieber, C.S.; DeCarli, L.M. Ethanol oxidation by hepatic microsomes: Adaptive increase after ethanol feeding. Science 1968, 162, 917–918. [Google Scholar] [CrossRef]
- Teschke, R. Alcoholic Liver Disease: Alcohol Metabolism, Cascade of Molecular Mechanisms, Cellular Targets, and Clinical Aspects. Biomedicines 2018, 6, 106. [Google Scholar] [CrossRef] [Green Version]
- Neuman, M. Cytokines in Alcoholic Liver. Alcohol Res. Health 2003, 27, 307–316. [Google Scholar]
- Lieber, C.S.; DeCarli, L.M. Hepatotoxicity of ethanol. J. Hepatol. 1991, 12, 394–401. [Google Scholar] [CrossRef]
- Liangpunsakul, S.; Toh, E.; Ross, R.A.; Heathers, L.E.; Chandler, K.; Oshodi, A.; McGee, B.; Modlik, E.; Linton, T.; Mangiacarne, D.; et al. Quantity of alcohol drinking positively correlates with serum levels of endotoxin and markers of monocyte activation. Sci. Rep. 2017, 7, 4462. [Google Scholar] [CrossRef] [PubMed]
- Lieber, C.S. Hepatic, metabolic and toxic effects of ethanol: 1991 update. Alcohol. Clin. Exp. Res. 1991, 15, 573–592. [Google Scholar] [CrossRef] [PubMed]
- Lieber, C.S. Alcohol, liver, and nutrition. J. Am. Coll. Nutr. 1991, 10, 602–632. [Google Scholar] [CrossRef] [PubMed]
- Lieber, C.S. Relationships between nutrition, alcohol use, and liver disease. Alcohol Res. Health 2003, 27, 220–231. [Google Scholar] [PubMed]
- Lieber, C.S.; Cao, Q.; DeCarli, L.M.; Leo, M.A.; Mak, K.M.; Ponomarenko, A.; Ren, C.; Wang, X. Role of medium-chain triglycerides in the alcohol-mediated cytochrome P450 2E1 induction of mitochondria. Alcohol. Clin. Exp. Res. 2007, 31, 1660–1668. [Google Scholar] [CrossRef] [PubMed]
- Seitz, H.K.; Lieber, C.S.; Stickel, F.; Salaspuro, M.; Schlemmer, H.P.; Horie, Y. Alcoholic liver disease: From pathophysiology to therapy. Alcohol. Clin. Exp. Res. 2005, 29, 1276–1281. [Google Scholar] [CrossRef]
- Teschke, R. Alcoholic Liver Disease: Current Mechanistic Aspects with Focus on Their Clinical Relevance. Biomedicines 2019, 7, 68. [Google Scholar] [CrossRef] [Green Version]
- Neuman, M.G.; Schmilowitz-Weiss, H.; Hilzenrat, N.; Bourliere, M.; Marcelin, P.; Trepo, C.; Mazulli, T.; Moussa, G.; Patel, A.; Baig, A.A.; et al. Markers of inflammation and fibrosis in alcoholic hepatitis and viral hepatitis C. Int. J. Hepatol. 2012, 2012, 231210. [Google Scholar] [CrossRef] [Green Version]
- Roerecke, M.; Nanau, R.; Rehm, J.; Neuman, M. Ethnicity matters: A systematic review and Meta-Analysis of the non-linear relationship between alcohol consumption and prevalence and incidence of hepatic steatosis. EBioMedicine 2016, 8, 317–330. [Google Scholar] [CrossRef] [Green Version]
- Neuman, M.G.; French, S.W.; French, B.A.; Seitz, H.K.; Cohen, L.B.; Mueller, S.; Osna, N.A.; Kharbanda, K.K.; Seth, D.; Bautista, A.; et al. Alcoholic and Nonalcoholic Steatohepatitis. Exp. Mol. Pathol. 2014, 97, 492–510. [Google Scholar] [CrossRef] [Green Version]
- Neuman, M.G.; Malnick, S.; Maor, Y.; Nanau, R.M.; Melzer, E.; Ferenci, P.; Seitz, H.K.; Mueller, S.; Mell, H.; Samuel, D.; et al. Alcoholic Liver Disease, Clinical and Translational Research. Exp. Mol. Pathol. 2015, 99, 596–610. [Google Scholar] [CrossRef] [PubMed]
- De Lédinghen, V.; Le Bail, B.; Rebouissoux, L.; Fournier, C.; Foucher, J.; Miette, V.; Castéra, L.; Sandrin, L.; Merrouche, W.; Lavrand, F.; et al. Liver stiffness measurement in children using FibroScan: Feasibility study and comparison with Fibrotest, aspartate transaminase to platelets ratio index, and liver biopsy. J. Pediatr. Gastroenterol. Nutr. 2007, 45, 443–450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Shea, R.S.; Dasarathy, S.; McCullough, A.J. Alcoholic liver disease. Hepatology 2010, 51, 307–328. [Google Scholar] [CrossRef] [PubMed]
- Jürgen Rehm, J.; Shield, K.D. Global Burden of Alcohol Use Disorders and Alcohol Liver Disease. Biomedicines 2019, 7, 99. [Google Scholar] [CrossRef] [Green Version]
- Sandahl, T.D.; Jepsen, P.; Thomsen, K.L.; Vilstrup, H. Incidence and mortality of alcoholic hepatitis in Denmark 1999–2008: A nationwide population based cohort study. J. Hepatol. 2011, 54, 760–776. [Google Scholar] [CrossRef]
- Schwarzinger, M.; Thiebaut, S.P.; Baillot, S.; Mallet, V.; Rehm, J. Alcohol use disorders and associated chronic disease-a national retrospective cohort study from France. BMC Public Health 2017, 18, 43. [Google Scholar] [CrossRef] [Green Version]
- Liangpunsakul, S. Clinical characteristics and mortality of hospitalized alcoholic hepatitis patients in the United States. J. Clin. Gastroenterol. 2011, 45, 714–719. [Google Scholar] [CrossRef] [Green Version]
- Masuhima, K.; Morishita, K.; Yoshimura, T.; Lavu, S.; Kobayashi, Y.; Lew, W.; Appella, E.; Kung, H.F.; Leonard, E.J.; Oppenheim, J.J. Molecular cloning of a human monocyte-derived neutrophil chemotactic factor (MONCF) and the induction of MDNCF mRNA by interleukin 1 and tumor necrosis factor. J. Exp. Med. 1988, 167, 1883–1893. [Google Scholar]
- Lane, H.C.; Anand, A.R.; Ganju, R.K. Cbl and Akt regulate CXCL8-induced and CXCR1-and CXCR2-mediated chemotaxis. Int. Immunol. 2006, 18, 1315–1325. [Google Scholar] [CrossRef] [Green Version]
- Rollins, B.J. Chemokines. Blood 1997, 90, 909–912. [Google Scholar] [CrossRef]
- Feniger-Barish, R.; Belkin, D.; Zaslaver, A.; Gal, S.; Dori, M.; Ran, M.; Ben-Baruch, A. GCP-2-induced internalization of IL-8 receptors: Hierarohical relationships between GCP-2 and other ELR (+)-CXC chemokines and mechanisms regulating CXCR2, internalization and recycling. Blood 2002, 95, 1551. [Google Scholar] [CrossRef]
- Mouneimne, G.; Demarais, V.; Sidani, M.; Scemes, E.; Wang, W.; Song, X.; Eddy, R.; Condeelis, J. Spatial and temporal control of cofilin activity is required for directional sensing during chemotaxin. Curr. Biol. 2006, 16, 2193–2205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, R.; Huang, H.; Zhang, Z.; Wang, F.S. The role of neutrophils in the development of liver disease. Cell. Mol. Immunol. 2014, 11, 224–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mookerjee, R.P.; Stadlbauer, V.; Lidder, S.; Wright, G.A.K.; Hodges, S.J.; Davies, N.A.; Jalan, R. Neutrophil dysfunction in alcoholic hepatitis superimposed on cirrhosis is reversible and predicts the outcome. Hepatology 2007, 46, 831–840. [Google Scholar] [CrossRef]
- Jaeschke, H. Neutrophil-mediated tissue injury in alcoholic hepatitis. Alcohol 2002, 27, 23–27. [Google Scholar] [CrossRef]
- Lazaro, R.; Wu, R.; Lee, S.; Zhu, N.L.; Chen, C.L.; French, S.W.; Xu, J.; Machida, K.; Tsukamoto, H. Osteopontin deficiency does not prevent but promotes alcoholic neutrophilic hepatitis in mice. Hepatology 2015, 61, 129–140. [Google Scholar] [CrossRef] [Green Version]
- Wieser, V.; Adolph, T.E.; Enrich, B.; Kuliopulos, A.; Kaser, A.; Tilg, H. Reversal of murine alcoholic steatohepatitis by pepducin-based functional blockade of interlink-8 receptors. Gut 2017, 66, 930–938. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Gong, M.; French, B.A.; Liao, G.; Li, J.; Tillman, B.; French, S.W. Aberrant modulation of the BRCA1 and G1/S cell cycle pathways in alcoholic hepatitis patients with Mallory-Denk bodies revealed by mRNA sequencing. Oncotarget 2015, 6, 42491–42503. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; French, B.A.; Nelson, T.J.; Li, J.; Tillman, B.; French, S.W. IL-8 signaling is up regulated in alcoholic hepatitis and DDC fed mice with Mallory-Denk bodies (MDBs) present. Exp. Mol. Pathol. 2015, 99, 320–325. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Jin, Y.; Zhang, D.; Wang, J.; Wang, G.; Lee, C.G. Investigating the promoter of FAT10 gene in HCC patients. Genes 2018, 9, 319. [Google Scholar] [CrossRef] [Green Version]
- Aichem, A.; Groettrup, M. The ubiquitin-like modifier FAT10 in cancer development. Int. J. Biochem. Cell Biol. 2016, 79, 451–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, F.; Zhan, B. UBA6 and its bispecific pathways for ubiquitin and FAT10. Int. J. Mol. Sci. 2019, 20, 2250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, C.; Xiong, H.; Chen, L.; Liu, X.; Zou, S.; Guan, J.; Wang, K. GRP78 promoter hepatocellular carcinoma proliferation by increasing FAT10 expression through the NFκB pathway. Exp. Cell Res. 2018, 365, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Ren, J.; Cheong, I.S.Y.; Ban, K.H.K.; Ooi, L.L.P.J.; Yong Tan, S.; Kan, A.; Nuchprayoon, I.; Kang, J.; Lee, R.; et al. Expression of the FAT10 gene is highly upregulated in hepatocellular carcinoma and other gastrointestinal and gynecological cancers. Oncogene 2003, 22, 2592–2603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- French, S.W.; French, B.A.; Oliva, J.; Li, J.; Bardag-Gorce, F.; Tillman, B.; Canaan, A. FAT10 knock out mice livers fail to develop Mallory-Denk bodies in the DDC mouse model. Exp. Mol. Pathol. 2012, 93, 309–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliva, J.; Bardag-Gorce, F.; Lin, A.; French, B.A.; French, S.W. The role of cytokines in UBD promoter regulation and Mallory-Denk body-like aggresomes. Exp. Mol. Pathol. 2010, 89, 1–8. [Google Scholar] [CrossRef] [Green Version]
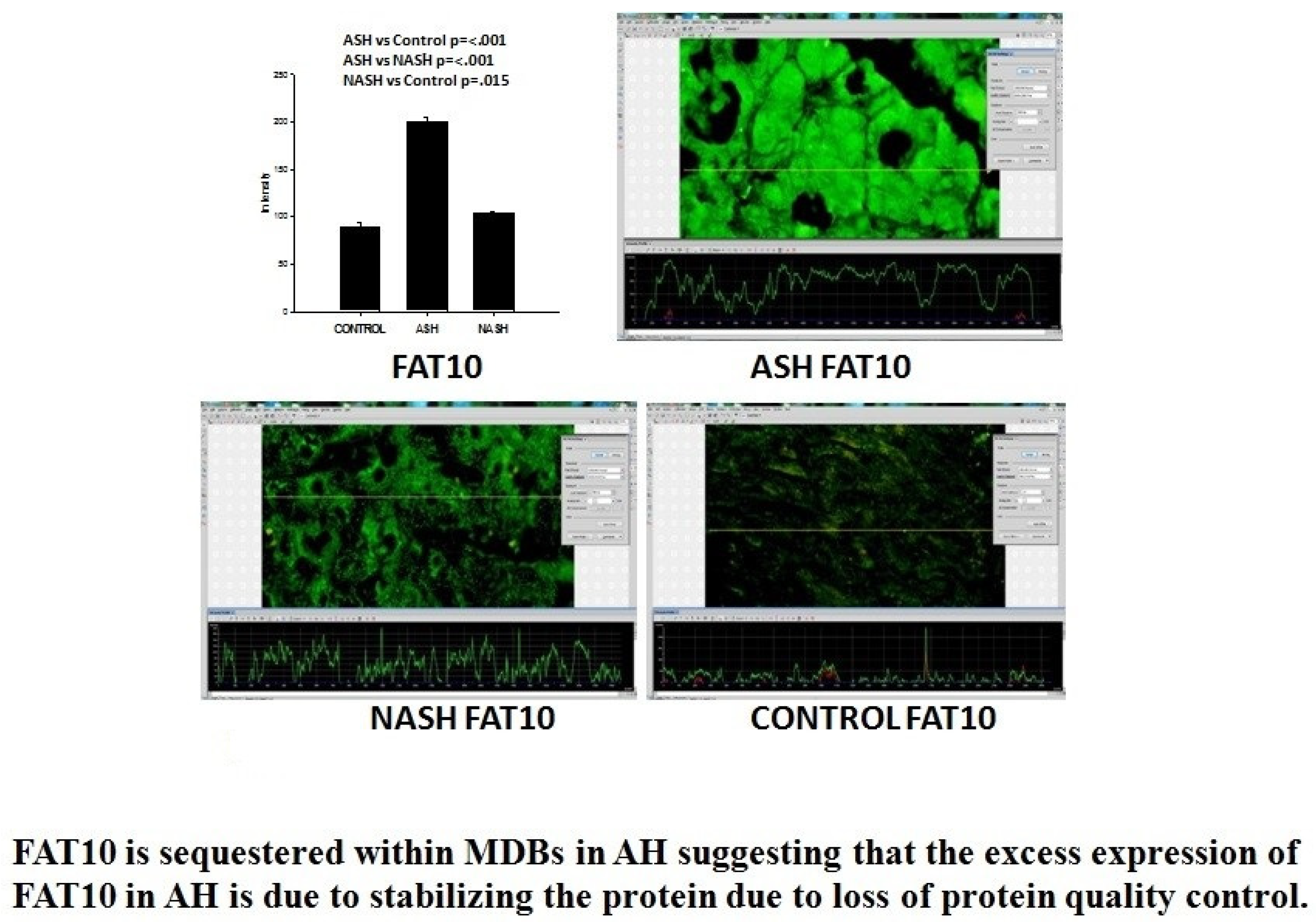
- Ganesan, M.; Hindman, J.; Tillman, B.; Jaramillo, L.; Poluektova, L.I.; French, B.A.; Kharbanda, K.K.; French, S.W.; Osna, N.A. FAT10 suppression stabilizes oxidized proteins in liver cells: Effects of HCV and ethanol. Exp. Mol. Pathol. 2015, 99, 506–516. [Google Scholar] [CrossRef]
- Liu, H.; Gong, M.; French, B.A.; Li, J.; Tillman, B.; French, S.W. Mallory-Denk body (MDB) formation modulates Ufmylation expression epigenetically in alcoholic hepatitis (AH) and non-alcoholic Steatohepatitis. Exp. Mol. Pathol. 2014, 97, 477–483. [Google Scholar] [CrossRef] [Green Version]
- Afifiyan, N.; Tillman, B.; French, B.A.; Sweeny, O.; Masouminia, M.; Samadzadeh, S.; French, S.W. The role of Tec Kinase signalling pathways in the development of Mallory-Denk bodies in balloon cells in alcoholic hepatitis. Exp. Mol. Pathol. 2017, 103, 191–199. [Google Scholar] [CrossRef]
- Qu, C.; Zheng, D.; Li, S.; Liu, Y.; Lidofsky, A.; Holmes, J.A.; Chen, J. Tyrosine kinase SYK is a potential therapeutic target for liver fibrosis. Hepatology 2018, 68, 1125–1139. [Google Scholar] [CrossRef] [Green Version]
- Tuma, D.J.; Casey, C.A. Dangerous byproducts of alcohol breakdown—Focus on adduct. Alcohol Res. Health 2003, 27, 285–290. [Google Scholar] [PubMed]
- Park, P.H.; Miller, R.; Shukla, S.D. Acetylation of histone H3 at lysine 9 by ethanol in rat hepatocytes. Biochem. Biophys. Res. Commun. 2003, 306, 501–504. [Google Scholar] [CrossRef]
- Pal-Bhadra, M.; Bhadra, U.; Jackson, D.E.; Liu, Y.; Lidofsky, A.; Holmes, J.A.; Chen, J.; He, J.; Liao, Y.; Yuan, H.; et al. Distinct methylation patterns in histone H3 at Lys-4 and Lys-9 correlate with up- & down-regulation of genes by ethanol in hepatocytes. Life Sci. 2007, 81, 979–987. [Google Scholar] [PubMed] [Green Version]
- Lee, Y.J.; Shukla, S.D. Histone H3 phosphorylation at serine 10 and serine 28 is mediated by p38 MAPK in rat hepatocytes exposed to ethanol and acetaldehyde. Eur. J. Pharmacol. 2007, 573, 29–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kannarkat, G.T.; Tuma, D.J.; Tuma, P.L. Microtubules are more stable and more highly acetylated in ethanol-treated hepatic cells. J. Hepatol. 2006, 44, 963–970. [Google Scholar] [CrossRef]
- You, M.; Liang, X.; Ajmo, J.M.; Ness, G.C. Involvement of mammalian sirtuin 1 in the action of ethanol in the liver. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 294, G892–G898. [Google Scholar] [CrossRef] [Green Version]
- Picklo, M.J., Sr. Ethanol intoxication increases hepatic N-lysyl protein acetylation. Biochem. Biophys. Res. Commun. 2008, 376, 615–619. [Google Scholar] [CrossRef]
- Lieber, C.S.; Leo, M.A.; Wang, X.; Decarli, L.M. Effect of chronic alcohol consumption on Hepatic SIRT1 and PGC-1 alpha in rats. Biochem. Biophys. Res. Commun. 2008, 370, 44–48. [Google Scholar] [CrossRef]
- Shepard, B.D.; Tuma, P.L. Alcohol-induced protein hyperacetylation: Mechanisms and consequences. World J. Gastroenterol. 2009, 15, 1219–1230. [Google Scholar] [CrossRef] [Green Version]
- Shepard, B.D.; Tuma, D.J.; Tuma, P.L. Chronic ethanol consumption induces global hepatic protein hyperacetylation. Alcohol. Clin. Exp. Res. 2010, 34, 280–291. [Google Scholar] [CrossRef] [Green Version]
- Kouzarides, T. Acetylation: A regulatory modification to rival phosphorylation? EMBO J. 2000, 19, 1176–1179. [Google Scholar] [CrossRef] [PubMed]
- Joseph, R.A.; Shepard, B.D.; Kannarkat, G.T.; Rutledge, T.M.; Tuma, D.J.; Tuma, P.L. Microtubule acetylation and stability may explain alcohol-induced alterations in hepatic protein trafficking. Hepatology 2008, 47, 1745–1753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groebner, J.L.; Fernandez, D.J.; Tuma, D.J.; Tuma, P.L. Alcohol-induced defects in hepatic transcytosis may be explained by impaired dynein function. Mol. Cell. Biochem. 2014, 397, 223–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez, D.J.; Tuma, D.J.; Tuma, P.L. Hepatic microtubule acetylation and stability induced by chronic alcohol exposure impair nuclear translocation of STAT3 and STAT5B, but not Smad2/3. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 303, G1402–G1415. [Google Scholar] [CrossRef] [Green Version]
- Shepard, B.D.; Tuma, D.J.; Tuma, P.L. Lysine acetylation induced by chronic ethanol consumption impairs dynamin-mediated clathrin-coated vesicle release. Hepatology 2012, 55, 1260–1270. [Google Scholar] [CrossRef] [Green Version]
- Goodman, J.M. The gregarious lipid droplet. J. Biol. Chem. 2008, 283, 28005–28009. [Google Scholar] [CrossRef] [Green Version]
- Sahini, N.; Borlak, J. Recent insights into the molecular pathophysiology of lipid droplet formation in hepatocytes. Prog Lipid Res. 2014, 54, 86–112. [Google Scholar] [CrossRef]
- Gross, S.P.; Welte, M.A.; Block, S.M.; Wieschaus, E.F. Dynein-mediated cargo transport in vivo. A switch controls travel distance. J. Cell Biol. 2000, 148, 945–956. [Google Scholar] [CrossRef] [Green Version]
- Shubeita, G.T.; Tran, S.L.; Xu, J.; Vershinin, M.; Cermelli, S.; Cotton, S.L.; Welte, M.A.; Gross, S.P. Consequences of motor copy number on the intracellular transport of kinesin-1-driven lipid droplets. Cell 2008, 135, 1098–1107. [Google Scholar] [CrossRef] [Green Version]
- Crunk, A.E.; Monks, J.; Murakami, A.; Jackman, M.; MacLean, P.S.; Ladinsky, M.; Bales, E.S.; Cain, S.; Orlicky, D.J.; McManaman, J.L. Dynamic regulation of hepatic lipid droplet properties by diet. PLoS ONE 2013, 8, e67631. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Ding, Y.; Chen, Y.; Jackman, M.; Maclean, P.S.; Ladinsky, M.; Bales, E.S.; Cain, S.; Orlicky, D.J.; McManaman, J.L. Dynamic regulation of hepatic lipid droplet properties by diet. J. Lipid Res. 2012, 53, 1245–1253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McVicker, B.L.; Tuma, P.L.; Kharbanda, K.K.; Lee, S.M.; Tuma, D.J. Relationship between oxidative stress and hepatic glutathione levels in ethanol-mediated apoptosis of polarized hepatic cells. World J. Gastroenterol. 2009, 15, 2609–2616. [Google Scholar] [CrossRef] [PubMed]
- Schaffert, C.S.; Todero, S.L.; McVicker, B.L.; Tuma, P.L.; Sorell, M.; Tuma, D.J. WIF-B cells as a model for alcohol-induced hepatocyte injury. Biochem. Pharmacol. 2004, 67, 2167–2174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McVicker, B.L.; Rasineni, K.; Tuma, D.J.; McNiven, M.A.; Casey, C.A. Lipid droplet accumulation and impaired fat efflux in polarized hepatic cells: Consequences of ethanol metabolism. Int. J. Hepatol. 2012, 2012, 978136. [Google Scholar] [CrossRef] [Green Version]
- Groebner, J.L.; Tuma, P.L. The Altered Hepatic Tubulin Code in Alcoholic Liver Disease. Biomolecules 2015, 5, 2140–2159. [Google Scholar] [CrossRef] [Green Version]
- Elliott, P.J.; Jirousek, M. Sirtuins: Novel targets for metabolic disease. Curr. Opin. Investig. Drugs 2008, 9, 371–378. [Google Scholar]
- Dekker, F.J.; van den Bosch, T.; Martin, N.I. Small molecule inhibitors of histone acetyltransferases and deacetylases are potential drugs for inflammatory diseases. Drug Discov. Today 2014, 19, 654–660. [Google Scholar] [CrossRef] [Green Version]
- Manzo, F.; Tambaro, F.P.; Mai, A.; Altucci, L. Histone acetyltransferase inhibitors and preclinical studies. Expert Opin. Ther. Pat. 2009, 19, 761–774. [Google Scholar] [CrossRef]
- Tan, J.; Cang, S.; Ma, Y.; Petrillo, R.L.; Liu, D. Novel histone deacetylase inhibitors in clinical trials as anti-cancer agents. J. Hematol. Oncol. 2010, 3, 5. [Google Scholar] [CrossRef] [Green Version]
- Guirguis, J.; Chhatwal, J.; Dasarathy, J.; Rivas, J.; McMichael, D.; Nagy, L.; McCullough, A.J.; Dasarathy, S. Clinical impact of alcohol-related cirrhosis in the next decade: Estimates based on current epidemiological trends in the United States. Alcohol. Clin. Exp. Res. 2015, 39, 2085–2094. [Google Scholar] [CrossRef] [Green Version]
- Bode, L. Human milk oligosaccharides: Every baby needs a sugar mama. Glycobiology 2012, 22, 1147–1162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ballard, O.; Morrow, A.L. Human milk composition: Nutrients and bioactive factors. Pediatr. Clin. N. Am. 2013, 60, 49–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newburg, D.S.; Ruiz-Palacios, G.M.; Morrow, A.L. Human milk glycans protect infants against enteric pathogens. Ann. Rev. Nutr. 2005, 25, 37–58. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, P.; Seebauer, C.T.; Schnabl, B. Alcoholic liver disease: The gut microbiome and liver cross talk. Alcohol. Clin. Exp. Res. 2015, 39, 763–775. [Google Scholar] [CrossRef] [Green Version]
- Coppa, G.V.; Gabrielli, O.; Buzzega, D.; Zampini, L.; Galeazzi, T.; Maccari, F.; Bertino, E.; Volpi, N. Composition and structure elucidation of human milk glycosaminoglycans. Glycobiology 2011, 21, 295–303. [Google Scholar] [CrossRef] [Green Version]
- Weigel, P.H.; Hascall, V.C.; Tammi, M. Hyaluronan synthases. J. Biol. Chem. 1997, 272, 13997–14000. [Google Scholar] [CrossRef] [Green Version]
- Milinkovic, M.; Antin, J.H.; Hergrueter, C.A.; Underhill, C.B.; Sackstein, R. CD44-hyaluronic acid interactions mediate shear-resistant binding of lymphocytes to dermal endothelium in acute cutaneous GVHD. Blood 2004, 103, 740–742. [Google Scholar] [CrossRef]
- Yang, B.; Zhang, L.; Turley, E.A. Identification of two hyaluronan-binding domains in the hyaluronan receptor RHAMM. J. Biol. Chem. 1993, 268, 8617–8623. [Google Scholar]
- Scheibner, K.A.; Lutz, M.A.; Boodoo, S.; Fenton, M.J.; Powell, J.D.; Horton, M.R. Hyaluronan fragments act as an endogenous danger signal by engaging TLR2. J. Immunol. 2006, 177, 1272–1281. [Google Scholar] [CrossRef] [Green Version]
- Taylor, K.R.; Trowbridge, J.M.; Rudisill, J.A.; Termeer, C.C.; Simon, J.C.; Gallo, R.L. Hyaluronan fragments stimulate endothelial recognition of injury through TLR4. J. Biol. Chem. 2004, 279, 17079–17084. [Google Scholar] [CrossRef] [Green Version]
- Beutler, B. Neo-ligands for innate immune receptors and the etiology of sterile inflammatory disease. Immunol. Rev. 2007, 220, 113–128. [Google Scholar] [CrossRef] [PubMed]
- Petrey, A.C.; de la Motte, C.A. Hyaluronan, a crucial regulator of inflammation. Front. Immunol. 2014, 5, 101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saikia, P.; Roychowdhury, S.; Bellos, D.; Pollard, K.A.; McMullen, M.R.; McCullough, R.L.; McCullough, A.J.; Ghola, P.; de la Motte, C.; Nagy, L.E. Hyaluronic acid 35 normalizes TLR4 signaling in Kupffer cells from ethanol-fed rats via regulation of microRNA291b and its target Tollip. Sci. Rep. 2017, 7, 15671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, X.; Jing, Z.; Cheng, G. 2MicroRNAs: New regulators of Toll-like receptor signalling pathways. Biomed. Res. Int. 2014, 2014, 945169. [Google Scholar] [CrossRef] [Green Version]
- Mandrekar, P.; Bataller, R.; Tsukamoto, H.; Gao, B. Alcoholic hepatitis: Translational approaches to develop targeted therapies. Hepatology 2016, 64, 1343–1355. [Google Scholar] [CrossRef]
- Xu, J.; Chi, F.; Guo, T.; Punj, V.; Lee, V.N.; French, S.W.; Tsukamoto, H. NOTCH reprograms mitochondrial metabolism for proinflammatory macrophage activation. J. Clin. Invest. 2015, 125, 1579–1590. [Google Scholar] [CrossRef] [Green Version]
- Blaya, J.; Marhuenda, F.C.; Pascual, J.A.; Ros, M. Microbiota characterization of compost using omics approaches opens new perspectives for Phytophthora root rot control. PLoS ONE 2016, 11, e0158048. [Google Scholar] [CrossRef] [Green Version]
- Roth, N.C.; Saberi, B.; Macklin, J.; Kanel, G.; French, S.W.; Govindarajan, S.; Buzzanco, A.S.; Stolz, A.A.; Donovan, J.A.; Kaplowitz, N. Prediction of histologic alcoholic hepatitis based on clinical presentation limits the need for liver biopsy. Hepatol. Commun. 2017, 1, 1070–1084. [Google Scholar] [CrossRef]
- Morales-Ibanez, O.; Dominguez, M.; Ki, S.H.; Marcos, M.; Chaves, J.F.; Nguyen-Khac, E.; Houchi, H.; Affò, S.; Sancho-Bru, P.; Altamirano, J.; et al. Human and experimental evidence supporting a role for osteopontin in alcoholic hepatitis. Hepatology 2013, 58, 1742–1756. [Google Scholar] [CrossRef] [Green Version]
- Sancho-Bru, P.; Altamirano, J.; Rodrigo-Torres, D.; Coll, M.; Millán, C. Liver progenitor cell markers correlate with liver damage and predict short-term mortality in patients with alcoholic hepatitis. Hepatology 2012, 55, 141–193. [Google Scholar] [CrossRef]
- Wang, L.; Fouts, D.E.; Starkel, P.; Wang, L.; McCole, D.F.; Brandl, K.; Starkel, P. Intestinal REG3 lectins protect against alcoholic steatohepatitis by reducing mucosa-associated microbiota and preventing bacterial translocation. Cell Host Microbe 2016, 18, 227–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, A.W.; Fouts, D.E.; Brandl, J.; Tärkel, P.; Torralba, M.; Schott, E.; Tsukamoto, H.; Nelson, K.E.; Brenner, D.A.; Schnabl, B. Enteric Dysbiosis Associated with a Mouse Model of Alcoholic Liver Disease. Hepatology 2012, 53, 96–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leclercq, S.; Matamoros, S.; Cani, P.D.; Neyrinck, A.M.; Jamar, F.; Stärkel, P.; Windey, K.; Tremaroli, V.; Bäckhed, F.; Verbeke, K.; et al. Intestinal permeability, gut-bacterial dysbiosis, and behavioral markers of alcohol-dependence severity. Proc. Natl. Acad. Sci. USA 2014, 111, E4485–E4493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, A.; Inamine, T.; Hochrath, K. Intestinal fungi contribute to development of alcoholic liver disease. J. Clin. Invest. 2017, 127, 2829–2841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, P.; Stärkel, P.; Turner, J.R.; Ho, S.B.; Schnabl, B. Dysbiosis-induced intestinal inflammation activates TNFRI and mediates alcoholic liver disease in mice. Hepatology 2015, 61, 883–894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cresci, G.A.; Glueck, B.; McMullen, M.R.; Xin, W.; Allende, D.; Nagy, L.E. Prophylactic tributyrin treatment mitigates chronic-binge alcohol-induced intestinal barrier and liver injury. J. Gastroenterol. Hepatol. 2017, 32, 1587–1597. [Google Scholar] [CrossRef]
- Llorente, C.; Jepsen, P.; Inamine, T.; Wang, L.; Bluemel, S.; Wang, H.J.; Loomba, R.; Bajaj, J.S.; Schubert, M.L.; Sikaroodi, M.; et al. Gastric acid suppression promotes alcoholic liver disease by inducing overgrowth of intestinal. Enterococcus. Nat. Commun. 2017, 8, 837. [Google Scholar] [CrossRef]
- D’Mello, C.; Swain, M.G. Liver-brain interactions in inflammatory liver diseases: Implications for fatigue and mood disorders. Brain Behav. Immun. 2014, 35, 9–20. [Google Scholar] [CrossRef]
- Robinson, G.; Most, D.; Ferguson, L.B.; Mayfield, J.; Harris, R.A.; Blednov, Y.A. Neuroimmune pathways in alcohol consumption: Evidence from behavioral and genetic studies in rodents and humans. Int. Rev. Neurobiol. 2014, 118, 13–39. [Google Scholar]
- Saba, L.M.; Flink, S.C.; Vanderlinden, L.A.; Israel, Y.; Tampier, L.; Colombo, G.; Kianmaa, K.; Bell, R.L.; Printz, M.P.; Flodman, P.; et al. The sequenced rat brain transcriptome--its use in identifying networks predisposing alcohol consumption. FEBS J. 2015, 282, 3556–3578. [Google Scholar] [CrossRef] [Green Version]
- Tabakoff, B.; Saba, L.; Printz, M.; Flodman, P.; Hodgkinson, C.; Goldman, D.; Koob, G.; Richardson, H.N.; Kechris, K.; Bell, R.L.; et al. WHO ISBRA Study on State Trait Markers of Alcoholism. Genetical genomic determinants of alcohol consumption in rats and humans. BMC Biol. 2009, 7, 70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrall, K.K.; Kechris, K.J.; Tabakoff, B.; Hoffman, P.L.; Hines, L.M.; Tsukamoto, H.; Pravenec, M.; Printz, M.; Saba, L.M. Uncovering the liver’s role in immunity through RNA co-expression networks. Mamm. Genome 2016, 27, 469–484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drakoulis, N.; Cascorbi, I.; Brockmöller, J.; Gross, C.R.; Roots, I. Polymorphisms in the human CYP1A1 gene as susceptibility factors for lung cancer: Exon-7 mutation (4889 A to G), and a T to C mutation in the 3’-flanking region. Clin. Investig. 1994, 72, 240–248. [Google Scholar] [CrossRef] [PubMed]
- Guengerich, F.P.; Parikh, A.; Turesky, R.J.; Josephy, P.D. Inter-individual differences in the metabolism of environmental toxicants: Cytochrome P450 1A2 as a prototype. Mutat. Res. 1999, 428, 115–124. [Google Scholar] [CrossRef]
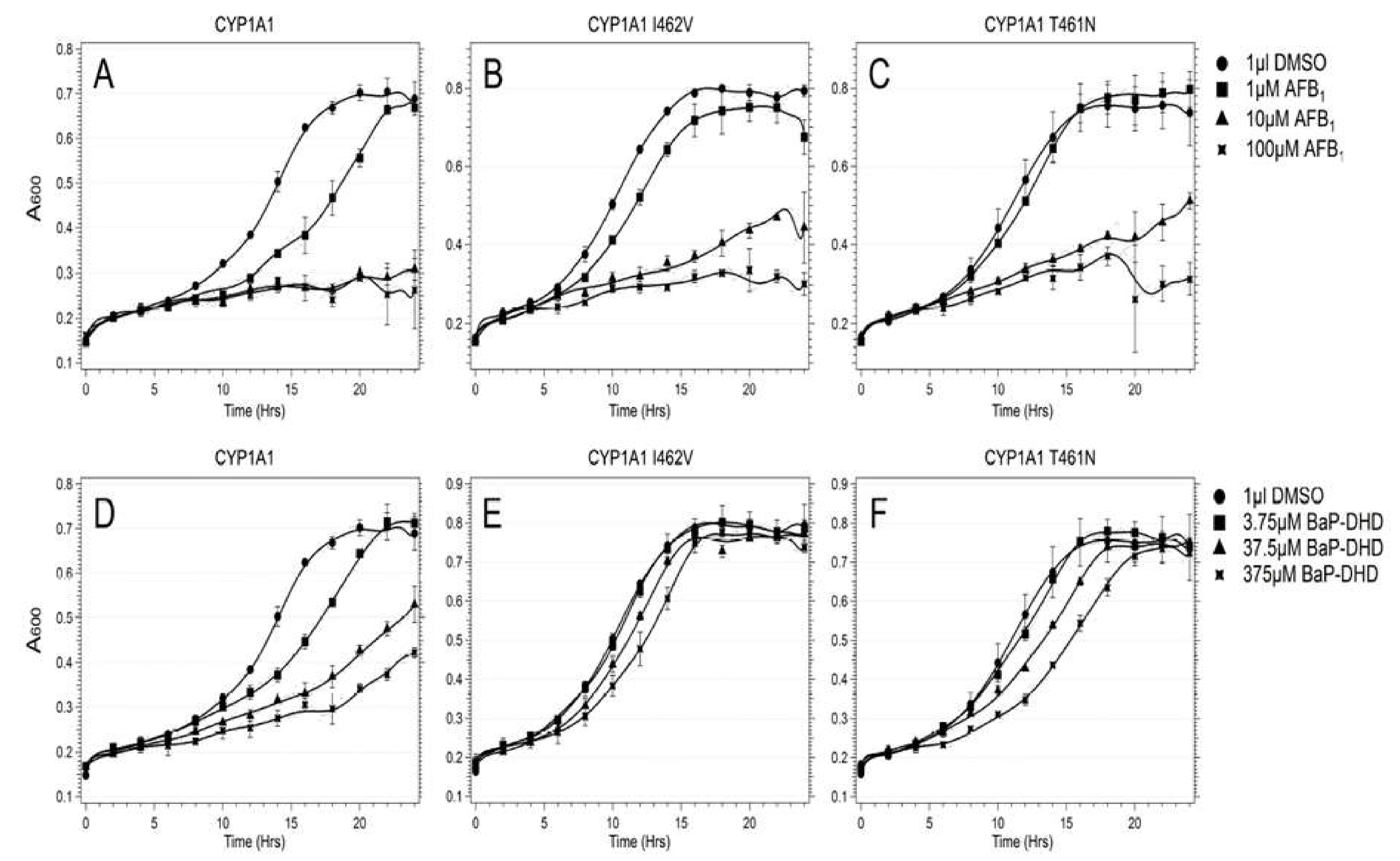
- Freedland, J.; Cera, C.; Fasullo, M. CYP1A1 I462V polymorphism is associated with reduced genotoxicity in yeast despite positive association with increased cancer risk. Mutat. Res. 2017, 815, 35–43. [Google Scholar] [CrossRef] [Green Version]
- Fasullo, M.; Smith, A.; Egner, P.; Cera, C. Activation of Aflatoxin B1 by expression of CYP1A2 polymorphisms in Saccharomyces cerevisiae. Mutat. Res. 2014, 761, 18–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teschke, R. Alcoholic steatohepatitis (ASH) and acute alcoholic hepatitis (AH): Cascade of events, clinical features, and pharmacotherapy options. Exp. Opin. Pharmacother. 2018, 19, 779–793. [Google Scholar] [CrossRef]
- Becket, A.G.; Livingstone, A.V.; Hill, K.R. Acute alcoholic hepatitis. Br. Med. J. 1961, 2, 1113–1119. [Google Scholar] [CrossRef] [Green Version]
- Davidson, C.S. Alcoholic hepatitis. N. Engl. J. Med. 1971, 284, 1378. [Google Scholar] [CrossRef]
- Popper, H.; Uenfriend, S. Hepatic fibrosis. Correlation of biochemical and morphologic investigations. Am. J. Med. 1970, 49, 707–721. [Google Scholar] [CrossRef]
- Edmondson, H.A.; Peters, R.L.; Frankel, H.H.; Borowsky, S. The early stage of liver injury in the alcoholic. Medicine 1967, 46, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Green, J.; Mistilis, S.; Schiff, L. Acute alcoholic hepatitis. Arch. Intern. Med. 1963, 112, 67–70. [Google Scholar] [CrossRef] [PubMed]
- Neuman, M.G.; Maor, Y.; Nanau, R.M.; Melzer, E.; Mell, H.; Opris, M.; Cohen, L.; Malnick, S. Alcoholic Liver Disease: Role of Cytokines. Biomolecules 2015, 5, 2023–2034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neuman, M.G.; Monteiro, M.; Rehm, J. Drug interactions between psychoactive substances and antiretroviral therapy in individuals infected with human immunodeficiency and hepatitis viruses. Subst. Use Misuse 2006, 41, 1395–1463. [Google Scholar] [CrossRef] [PubMed]
- Neary, J.; Goodwin, S.E.; Cohen, L.B.; Neuman, M.G. Alcohol misuse link to POEMS syndrome in a patient. Cancers 2017, 9, 129. [Google Scholar] [CrossRef] [Green Version]
- Rehm, J.; Samokhvalov, A.V.; Neuman, M.G.; Room, R.; Parry, C.; Lönnroth, K.; Patra, J.; Poznyak, V.; Popova, S. The association between alcohol use, alcohol use disorders and tuberculosis (TB). BMC Public Health 2009. [Google Scholar] [CrossRef]
- Roerecke, M.; Vafaei, A.; Hasan, O.S.M.; Chrystoja, B.R.; Cruz, M.; Lee, R.; Neuman, M.G.; Rehm, J. Alcohol consumption and risk of liver cirrhosis: A systematic review and meta-analysis. Am. J. Gastroenterol. 2019, 114, 1574–1586. [Google Scholar] [CrossRef]
- Powell, L.W. The role of alcoholism in hepatic iron storage disease. Ann. N.Y. Acad. Sci. 1975, 252, 124. [Google Scholar] [CrossRef]
- Powell, L.W.; Fletcher, L.M.; Holliday, J.W. Distinction between hemochromatosis and alcoholic siderosis. In Alcoholic Liver Disease. Pathology and Pathogenesis, 2nd ed.; Hall, P., Ed.; Edward Arnold: London, UK, 1995; pp. 199–216. [Google Scholar]
- Simon, M.; Bourel, M.; Genetet, B.; Fauchet, R.; Edan, G.; Brissot, P. Idiopathic hemochromatosis and iron load in alcoholic liver disease. Differentiation in HLA phenotypes. Gastroenterology 1977, 73, 633. [Google Scholar]
- Bothwell, T.H.; Charlton, R.W. Hemochromatosis. In Diseases of the Liver, 6th ed.; Schiff, L., Schiff, E.R., Eds.; Lippincott: Philadelphia, PA, USA, 1987; pp. 1001–1035. [Google Scholar]
- Adhoute, X.; Foucher, J.; Laharie, D.; Terrebonne, E.; Vergniol, J.; Castéra, L.; Lovato, B.; Chanteloup, E.; Merrouche, W.; Couzigou, P.; et al. Diagnosis of liver fibrosis using FibroScan and other noninvasive methods in patients with hemochromatosis: A prospective study. Gastroenterol. Clin. Biol. 2008, 32, 180–187. [Google Scholar] [CrossRef]
- Althausen, T.L.; Doig, R.K.; Weiden, S.; Motteram, R.; Turner, C.N.; Moore, A. Hemochromatosis: Investigation of twenty-three cases, with special reference to etiology, nutrition, iron metabolism, and studies of hepatic and pancreatic function. AMA Arch. Intern. Med. 1951, 88, 553–570. [Google Scholar] [CrossRef] [PubMed]
- Lundvall, O. Alcohol and porphyria cutanea tarda. In Alcoholic Cirrhosis and Other Toxic Hepatopathies; Engel, A., Larsson, T., Eds.; Nordiska Bokhandelns Forlag: Stockholm, Sweden, 1970; pp. 356–372. [Google Scholar]
- Lefkowitch, J.H.; Grossman, M.E. Hepatic pathology in porphyria cutanea tarda. Liver 1983, 3, 19. [Google Scholar] [CrossRef]
- Hift, R.; Kirsch, R. Porphyria cutanea tarda. In Alcoholic Liver Disease. Pathology and Pathogenesis, 2nd ed.; Hall, P., Ed.; Edward Arnold: London, UK, 1995; pp. 219–231. [Google Scholar]
- Joly, P.; Lacan, P.; Garcia, C.; Meley, R.; Pondarré, C.; Francina, A. A novel deletion/insertion caused by a replication error in the β-globin gene locus control region. Hemoglobin 2011, 35, 316–322. [Google Scholar] [CrossRef] [PubMed]
- Neuman, M.G. Cytokines—Central factors in alcoholic liver disease. Alcohol Res. Health. 2003, 27, 307–316. [Google Scholar] [PubMed]
- Lieber, C.S. Interaction of ethanol with drugs, hepatotoxic agents, carcinogens and vitamins. Alcohol Alcohol. 1990, 25, 157. [Google Scholar] [CrossRef]
- Strubelt, O. Interactions between ethanol and other hepatotoxic agents. Biochem. Pharmacol. 1980, 29, 1445–1449. [Google Scholar] [CrossRef]
- Teschke, R.; Vierke, W.; Gellert, J. Effect of ethanol on carbon tetrachloride levels and hepatotoxicity after acute carbon tetrachloride poisoning. Arch. Toxicol. 1984, 56, 78–82. [Google Scholar] [CrossRef]
- Seeff, L.B.; Cuccherini, B.A.; Zimmerman, H.J.; Adler, E.; Benjamin, S.B. Acetaminophen hepatotoxicity in alcoholics. Ann. Intern. Med. 1986, 104, 399–404. [Google Scholar] [CrossRef]
- Zimmerman, H.J.; Maddrey, W.C. Acetaminophen (paracetamol) hepatotoxicity with regular intake of alcohol. Analysis of instances of a therapeutic misadventure. Hepatology 1995, 22, 767. [Google Scholar] [CrossRef]
- Sato, C.; Lieber, C.S. Increased hepatotoxicity of acetaminophen after chronic ethanol consumption in the rat. Gastroenterology 1981, 80, 140. [Google Scholar] [CrossRef]
- Zimmerman, H.J. Effects of alcohol on other hepatotoxins. Alcohol. Clin. Exp. Res. 1986, 10, 3. [Google Scholar] [CrossRef] [PubMed]
- Teschke, R.; Eickhoff, A.; Brown, A.C.; Neuman, M.G.; Schulze, J. Diagnostic Biomarkers in Liver Injury by Drugs, Herbs, and Alcohol: Tricky Dilemma after EMA Correctly and Officially Retracted Letter of Support. Int. J. Mol. Sci. 2019, 21, 212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neuman, M.G. Biomarkers of Drug-Induced Liver Toxicity. Ther. Drug Monit. 2019, 41, 227–234. [Google Scholar] [CrossRef]
- Teschke, R. Microsomal Ethanol-Oxidizing System: Success Over 50 Years and an Encouraging Future. Alcohol. Clin. Exp. Res. 2019, 43, 386–400. [Google Scholar] [CrossRef] [PubMed]
- Neuman, M.G.; Schneider, M.; Nanau, R.M.; Parry, C. HIV-Antiretroviral Therapy Induced Liver, Gastrointestinal, and Pancreatic Injury. Int. J. Hepatol. 2012, 2012, 760706. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| # | Fat Macro | Fat Micro | PMN | Lymph | Other Findings | Fibrosis Stage | MDBs |
|---|---|---|---|---|---|---|---|
| 01-011 | 1+ | 0 | 0 | 0 | 0 | 4+ | 1+ |
| 01-015 | 3+ | 1+ | 1+ | 1+ | 0 | 4+ | 3+ |
| 01-016 | 4+ | 0 | 3+ | 1+ | bile thrombi, EM | 4+ | 4+ |
| 03-001 | 1+ | 0 | 3+ | 4+ | bile thrombi, EM | 4+ | 4+ |
| 03-005 | 2+ | 0 | 0 | 4+ | EM | 4+ | 1+ |
| 03-006 | 1+ | 0 | 4+ | 1+ | satellitosis, EM | 4+ | 4+ |
| 03-007 | 1+ | 0 | 0 | 2+ | 0 | 4+ | 1+ |
| 03-012 | 2+ | 0 | 3+ | 3+ | best satellitosis, EM | 4+ | 4+ |
| 03-014 | 2+ | 0 | 2+ | 0 | most MDBs | 4+ | 4+ |
| 03-015 | 4+ | 0 | 1+ | 0 | 0 | 4+ | 2+ |
| 03-017 | 3+ | 1+ | 2+ | 3+ | EM | 3+ | 1+ |
| 03-018 | 4+ | 0 | 4+ | 2+ | Satellitosis | 4+ | 3+ |
| 03-019 | 3+ | 0 | 3+ | 0 | satellitosis, EM | 4+ | 4+ |
| 03-020 | 1+ | 0 | 3+ | 0 | Satellitosis | 4+ | 1+ |
| 03-022 | 3+ | 1+ | 0 | 2+ | autophagy, EM | 4+ | 3+ |
| 03-023 | 1+ | 0 | 4+ satellitosis | 1+ | autophagy of MDBs, EM | 4+ | 4+ |
| 03-024 | 4+ | 1+ | 3+ | 1+ | PMN lymphocytes, EM | 4+ | 4+ |
| 03-025 | 4+ | 1+ | 1+ | 1+ | autophagy, EM | 3+ | 3+ |
| 03-027 | 1+ | 0 | 4+ | 4+ | PMN satellitosis | 4+ | 4+ |
| 03-039 | 0 | 0 | 1+ | 4+ | duct lymphocytes PMN | 4+ | 0 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Neuman, M.G.; Seitz, H.K.; French, S.W.; Malnick, S.; Tsukamoto, H.; Cohen, L.B.; Hoffman, P.; Tabakoff, B.; Fasullo, M.; Nagy, L.E.; et al. Alcoholic-Hepatitis, Links to Brain and Microbiome: Mechanisms, Clinical and Experimental Research. Biomedicines 2020, 8, 63. https://doi.org/10.3390/biomedicines8030063
Neuman MG, Seitz HK, French SW, Malnick S, Tsukamoto H, Cohen LB, Hoffman P, Tabakoff B, Fasullo M, Nagy LE, et al. Alcoholic-Hepatitis, Links to Brain and Microbiome: Mechanisms, Clinical and Experimental Research. Biomedicines. 2020; 8(3):63. https://doi.org/10.3390/biomedicines8030063
Chicago/Turabian StyleNeuman, Manuela G., Helmut Karl Seitz, Samuel W. French, Stephen Malnick, Heidekazu Tsukamoto, Lawrence B. Cohen, Paula Hoffman, Boris Tabakoff, Michael Fasullo, Laura E. Nagy, and et al. 2020. "Alcoholic-Hepatitis, Links to Brain and Microbiome: Mechanisms, Clinical and Experimental Research" Biomedicines 8, no. 3: 63. https://doi.org/10.3390/biomedicines8030063