Combinatorial Discriminant Analysis Applied to RNAseq Data Reveals a Set of 10 Transcripts as Signatures of Exposure of Cattle to Mycobacterium avium subsp. paratuberculosis

 ,
,  ,
,  and
and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animal Resource
2.2. Sample Preparation, RNA Extraction and Quality Control
2.3. RNA-Seq Library Preparation and Sequencing
2.4. Availability of Data
2.5. RNA-Seq Data Analysis
2.6. Signature Identification
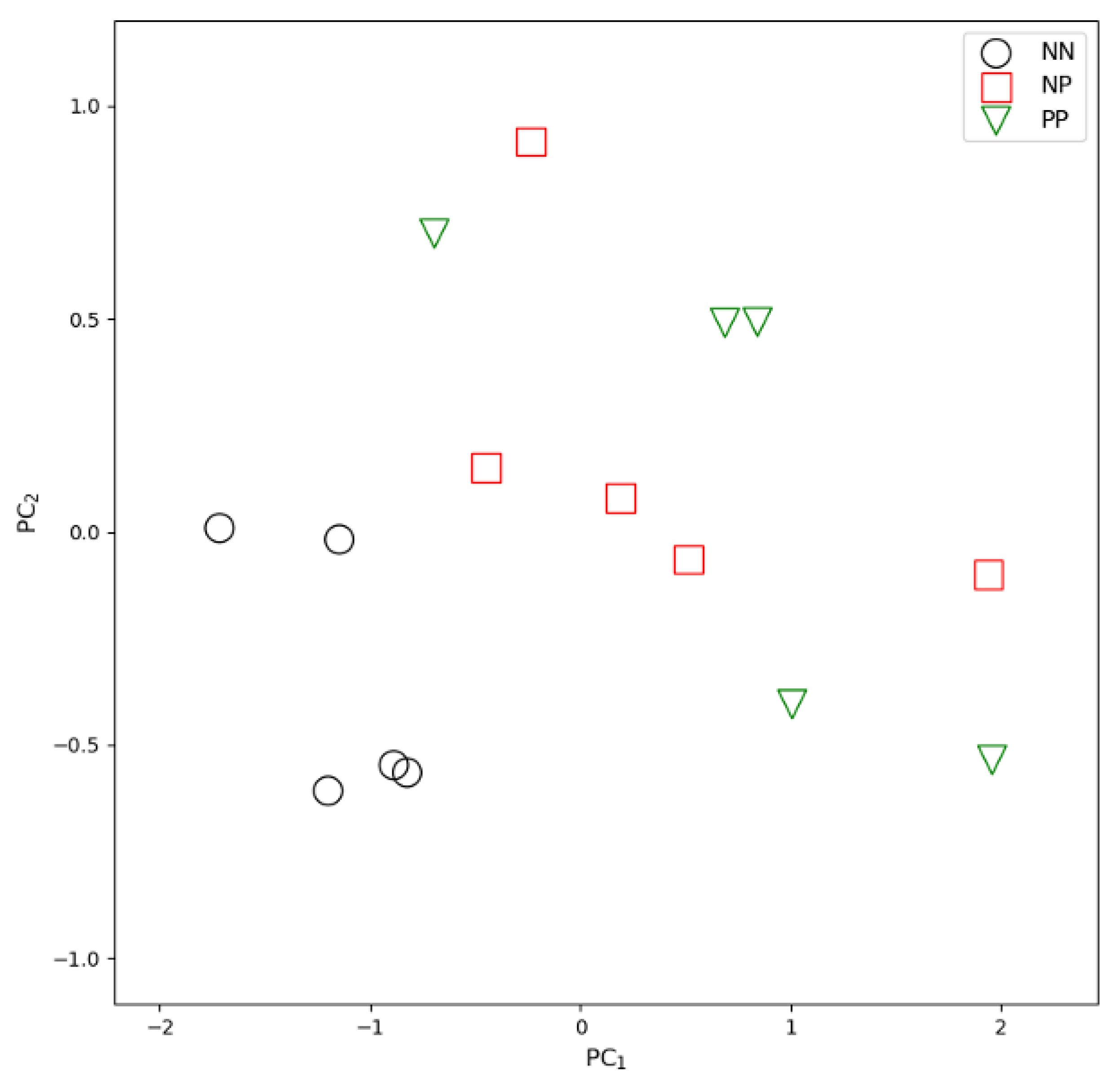
3. Results
3.1. Results of the NGS Pipeline Analysis
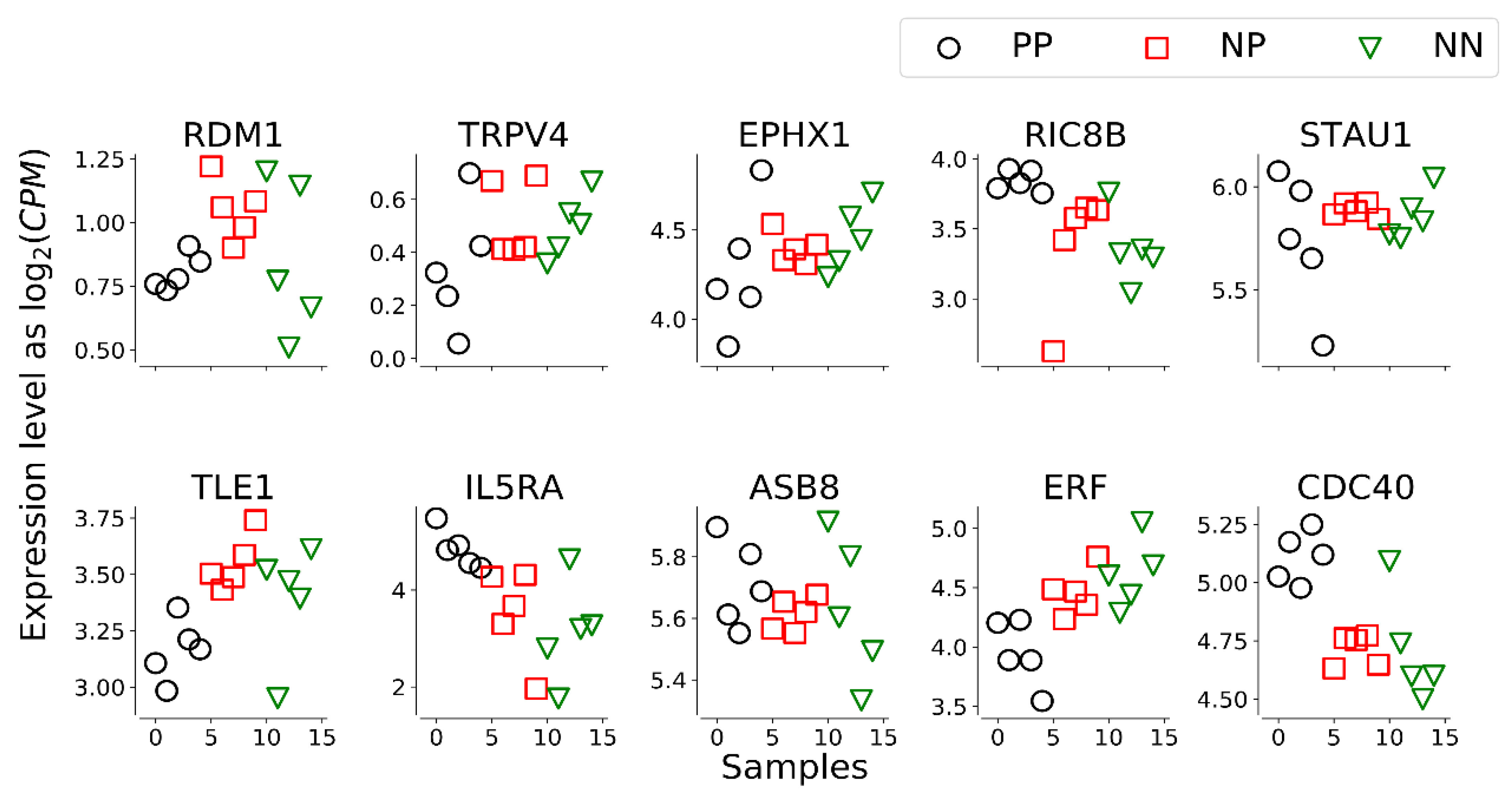
3.2. Results of the Signature Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Olsen, I.; Sigurğardóttir, G.; Djønne, B. Paratuberculosis with special reference to cattle. Vet. Q. 2002, 24, 12–28. [Google Scholar] [CrossRef] [PubMed]
- Whittington, R.J.; Sergeant, E.S.G. Progress towards understanding the spread, detection and control of Mycobacterium avium subsp. paratuberculosis in animal populations. Aust. Vet. J. 2001, 79, 267–278. [Google Scholar] [CrossRef] [PubMed]
- Bach, H. What role does mycobacterium avium subsp. paratuberculosis play in Crohn’s disease? Curr. Infect. Dis. Rep. 2015, 17, 463. [Google Scholar] [CrossRef] [PubMed]
- Liverani, E.; Scaioli, E.; Cardamone, C.; Dal Monte, P.; Belluzzi, A. Mycobacterium avium subspecies paratuberculosis in the etiology of Crohn’s disease, cause or epi- phenomenon? World J. Gastroenterol. 2014, 20, 13060–13070. [Google Scholar] [CrossRef]
- Britton, L.E.; Cassidy, J.P.; O’Donovan, J.; Gordon, S.V.; Markey, B. Potential application of emerging diagnostic techniques to the diagnosis of bovine Johne’s disease (paratuberculosis). Vet. J. 2016, 209, 32–39. [Google Scholar] [CrossRef]
- Casey, M.E.; Meade, K.G.; Nalpas, N.C.; Taraktsoglou, M.; Browne, J.A.; Killick, K.E.; Park, S.D.; Gormley, E.; Hokamp, K.; Magee, D.A.; et al. Analysis of the Bovine Monocyte-Derived Macrophage Response to Mycobacterium avium Subspecies Paratuberculosis Infection Using RNA-seq. Front. Immunol. 2015, 6, 23. [Google Scholar] [CrossRef] [Green Version]
- Farrell, D.; Shaughnessy, R.G.; Britton, L.; MacHugh, D.E.; Markey, B.; Gordon, S.V. The Identification of Circulating MiRNA in Bovine Serum and Their Potential as Novel Biomarkers of Early Mycobacterium avium subsp paratuberculosis Infection. PLoS ONE 2015, 10, e0134310. [Google Scholar] [CrossRef] [Green Version]
- Malvisi, M.; Palazzo, F.; Morandi, N.; Lazzari, B.; Williams, J.L.; Pagnacco, G.; Minozzi, G. Responses of Bovine Innate Immunity to Mycobacterium avium subsp. paratuberculosis Infection Revealed by Changes in Gene Expression and Levels of MicroRNA. PLoS ONE 2016, 11, e0164461. [Google Scholar] [CrossRef] [Green Version]
- Marino, R.; Capoferri, R.; Panelli, S.; Minozzi, G.; Strozzi, F.; Trevisi, E.; Snel, G.G.M.; Ajmone-Marsan, P.; Williams, J.L. Johne’s disease in cattle: An in vitro model to study early response to infection of Mycobacterium avium subsp. paratuberculosis using RNA-seq. Mol. Immunol. 2017, 91, 259–271. [Google Scholar] [CrossRef]
- Shaughnessy, R.G.; Farrell, D.; Riepema, K.; Bakker, D.; Gordon, S.V. Analysis of Biobanked Serum from a Mycobacterium avium subsp paratuberculosis Bovine Infection Model Confirms the Remarkable Stability of Circulating miRNA Profiles and Defines a Bovine Serum miRNA Repertoire. PLoS ONE 2015, 10, e0145089. [Google Scholar] [CrossRef]
- Johnstone, I.M.; Titterington, D.M. Statistical challenges of high-dimensionaldata. Philos. Trans. A Math. Phys. Eng. Sci. 2009, 367, 4237–4253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scotlandi, K.; Remondini, D.; Castellani, G.; Manara, M.C.; Nardi, F.; Cantiani, L.; Francesconi, M.; Mercuri, M.; Caccuri, A.M.; Serra, M.; et al. Overcoming resistance to conventional drugs in Ewing sarcoma and identification of molecular predictors of outcome. J. Clin. Oncol. 2009, 27, 2209–2216. [Google Scholar] [CrossRef] [PubMed]
- Terragna, C.; Renzulli, M.; Remondini, D.; Tagliafico, E.; Di Raimondo, F.; Patriarca, F.; Martinelli, G.; Roncaglia, E.; Masini, L.; Tosi, P.; et al. Correlation between eight-gene expression profiling and response to therapy of newly diagnosed multiple myeloma patients treated with thalidomide-dexamethasone incorporated into double autologous transplantation. Ann. Hematol. 2013, 92, 1271–1280. [Google Scholar] [CrossRef] [PubMed]
- Terragna, C.; Remondini, D.; Martello, M.; Zamagni, E.; Pantani, L.; Patriarca, F.; Pezzi, A.; Levi, G.; Offidani, M.; Proserpio, I.; et al. The genetic and genomic background of multiple myeloma patients achieving complete response after induction therapy with bortezomib, thalidomide and dexamethasone (VTD). Oncotarget 2016, 7, 9666–9679. [Google Scholar] [CrossRef] [PubMed]
- FastQC Software. Babraham Institute—Babraham Bioinformatics. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 3 May 2019).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence alignment/map (SAM) format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef]
- Curti, N.; Giampieri, E.; Levi, G.; Castellani, G.; Remondini, D. DNetPRO: A network approach for low-dimensional signatures from high-throughput data. bioRxiv 2019, 773622. [Google Scholar] [CrossRef] [Green Version]
- Mizzi, C.; Fabbri, A.; Rambaldi, S.; Bertini, F.; Curti, N.; Sinigardi, S.; Luzi, R.; Venturi, G.; Micheli, D.; Muratore, G.; et al. Unraveling pedestrian mobility on a road network using ICTs data during great tourist events. EPJ Data Sci. 2018, 7, 44. [Google Scholar] [CrossRef] [Green Version]
- Curti, N.; Giampieri, E.; Mizzi, C.; Fabbri, A.; Bazzani, A.; Castellani, G.; Remondini, D. A network approach for dimensionality reduction from high-throughput data, SDPS18. 2018; in press. [Google Scholar]
- Brandes, U. A Faster Algorithm for Betweenness Centrality. J. Math. Sociol. 2001, 25, 163–177. [Google Scholar] [CrossRef]
- Fagiolo, G. Clustering in complex directed networks. Phys. Rev. E 2007, 76, 026107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burakoff, R.; Chao, S.; Perencevich, M.; Ying, J.; Friedman, S.; Makrauer, F.; Odze, R.; Khurana, H.; Liew, C.C. Blood-based biomarkers can differentiate ulcerative colitis from Crohn’s disease and noninflammatory diarrhea. Inflamm. Bowel. Dis. 2011, 17, 1719–1725. [Google Scholar] [CrossRef] [PubMed]
- Burakoff, R.; Pabby, V.; Onyewadume, L.; Odze, R.; Adackapara, C.; Wang, W.; Friedman, S.; Hamilton, M.; Korzenik, J.; Levine, J.; et al. Blood-based biomarkers used to predict disease activity in Crohn’s disease and ulcerative colitis. Inflamm. Bowel. Dis. 2015, 21, 1132–1140. [Google Scholar] [CrossRef]
- Marshall, K.W.; Mohr, S.; Khettabi, F.E.; Nossova, N.; Chao, S.; Bao, W.; Ma, J.; Li, X.J.; Liew, C.C. A blood-based biomarker panel for stratifying current risk for colorectal cancer. Int. J. Cancer. 2010, 126, 1177–1186. [Google Scholar] [CrossRef]
- David, J.; Barkema, H.W.; Mortier, R.; Ghosh, S.; Guan, L.L.; De Buck, J. Gene expression profiling and putative biomarkers of calves 3 months after infection with Mycobacterium avium subspecies paratuberculosis. Vet. Immunol. Immunopathol. 2014, 160, 107–117. [Google Scholar] [CrossRef]
- Purdie, A.C.; Plain, K.M.; Begg, D.J.; De Silva, K.; Whittington, R.J. Expression of genes associated with the antigen presentation and processing pathway are consistently regulated in early Mycobacterium avium subsp. paratuberculosis infection. Comp. Immunol. Microbiol. Infect. Dis. 2012, 35, 151–162. [Google Scholar] [CrossRef]
- Kozik, P.; Hodson, N.A.; Sahlender, D.A.; Simecek, N.; Soromani, C.; Wu, J.; Collinson, L.M.; Robinson, M.S. A human genome-wide screen for regulators of clathrin-coated vesicle formation reveals an unexpected role for the V-ATPase. Nat. Cell Biol. 2013, 15, 50–60. [Google Scholar] [CrossRef] [Green Version]
- Pieters, J. Entry and survival of pathogenic mycobacteria in macrophages. Microbes Infect. 2001, 3, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, J.A.; Hart, P.D.A. Response of cultured macrophages to Mycobacterium tuberculosis, with observations on fusion of lysosomes with phagosomes. J. Exp. Med. 1971, 134, 713–740. [Google Scholar] [CrossRef] [Green Version]
- Van Rensburg, I.C.; Wagman, C.; Stanley, K.; Beltran, C.; Ronacher, K.; Walzl, G.; Loxton, A.G. Successful TB treatment induces B-cells expressing FASL and IL5RA mRNA. Oncotarget 2017, 8, 2037–2043. [Google Scholar] [CrossRef] [PubMed]
- Verykokakis, M.; Papadaki, C.; Vorgia, E.; Le Gallic, L.; Mavrothalassitis, G. The RAS-dependent ERF control of cell proliferation and differentiation is mediated by c-Myc repression. J. Biol. Chem. 2007, 282, 30285–30294. [Google Scholar] [CrossRef] [Green Version]
- Pello, O.M.; De Pizzol, M.; Mirolo, M.; Soucek, L.; Zammataro, L.; Amabile, A.; Doni, A.; Nebuloni, M.; Swigart, L.B.; Evan, G.I.; et al. Role of c-MYC inalternative activation of human macrophages and tumor-associated macrophagebiology. Blood 2012, 119, 411–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas-Tikhonenko, A.; Viard-Leveugle, I.; Dews, M.; Wehrli, P.; Sevignani, C.; Yu, D.; Ricci, S.; el-Deiry, W.; Aronow, B.; Kaya, G.; et al. Myc-transformed epithelial cells down-regulate clusterin, which inhibits their growth in vitro and carcinogenesis in vivo. Cancer Res. 2004, 64, 3126–3136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grace, M.S.; Bonvini, S.J.; Belvisi, M.G.; McIntyre, P. Modulation of the TRPV4 ion channel as a therapeutic target for disease. Pharmacol. Ther. 2017, 177, 9–22. [Google Scholar] [CrossRef] [PubMed]
- D’Aldebert, E.; Cenac, N.; Rousset, P.; Martin, L.; Rolland, C.; Chapman, K.; Selves, J.; Alric, L.; Vinel, J.P.; Vergnolle, N. Transient receptor potential vanilloid activated inflammatory signals by intestinal epithelial cells and colitis in mice. Gastroenterology 2011, 140, 275–285. [Google Scholar] [CrossRef] [PubMed]
- Tong, L.; Liu, J.; Yan, W.; Cao, W.; Shen, S.; Li, K.; Li, L.; Niu, G. RDM1 plays an oncogenic role in human lung adenocarcinoma cells. Sci. Rep. 2018, 8, 11525. [Google Scholar] [CrossRef] [Green Version]
- Hamimes, S.; Arakawa, H.; Stasiak, A.Z.; Kierzek, A.M.; Hirano, S.; Yang, Y.G.; Takata, M.; Stasiak, A.; Buerstedde, J.M.; Van Dyck, E. RDM1, a novel RNA recognition motif (RRM)-containing protein involved in the cell response to cisplatin in vertebrates. J. Biol. Chem. 2005, 280, 9225–9235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calder, M.D.; Madan, P.; Watson, A.J. Bovine oocytes and early embryos express Staufen and ELAVL RNA-binding proteins. Zygote 2008, 16, 161–168. [Google Scholar] [CrossRef] [Green Version]
- Sivonova, M.; Dobrota, D.; Matakova, T.; Grobarcikova, S.; Habala, V.; Salagovic, J.; Tajtakova, M.; Pidanicova, A.; Valansky, L.; Lachvacs, L.; et al. Microsomal epoxide hydrolase polymorphisms, cigarette smoking and prostate cancer risk in the Slovak population. Neoplasma 2012, 59, 79–84. [Google Scholar] [CrossRef]
- Zhu, Q.; Xing, W.; Qian, B.; Von Dippe, P.; Shneider, B.L.; Fox, V.L.; Levy, D. Inhibition of human m-epoxide hydrolase gene expression in a case of hypercholanemia. Biochim. Biophys. 2003, 1638, 208–216. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.Z.; Li, J.J.; Zhang, F.R.; Shen, Y.; Yao, M.; Wan, D.F.; Gu, J.R. Exogenous expression of SOCS box-deficient mutant ASB-8 suppresses the growth of lung adenocarcinoma SPC-A1 cells. Sheng Wu Hua Xue Yu Sheng Wu Wu Li Xue Bao (Shanghai) 2003, 35, 548–553. [Google Scholar] [PubMed]
- Ali, Z.; Haroon Khan, A.; Rehman, U.; Faisal, M.; Ahmad, I.N.; Mamoon, N.; Nasir, H.; Hameed, Z. Is TLE1 Expression Limited to Synovial Sarcoma? Our Experience at Shifa International Hospital, Pakistan. Cureus 2019, 11, e6259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jagdis, A.; Rubin, B.P.; Tubbs, R.R.; Pacheco, M.; Nielsen, T.O. Prospective evaluation of TLE1 as a diagnostic immunohistochemical marker in synovial sarcoma. Am. J. Surg. Pathol. 2009, 33, 1743–1751. [Google Scholar] [CrossRef]
- Von Dannecker, L.E.; Mercadante, A.F.; Malnic, B. Ric-8B, an olfactory putative GTP exchange factor, amplifies signal transduction through the olfactory-specific G-protein Gαolf. J. Neurosci. 2005, 25, 3793–3800. [Google Scholar] [CrossRef]
- Von Dannecker, L.E.; Mercadante, A.F.; Malnic, B. Ric-8B promotes functional expression of odorant receptors. Proc. Natl. Acad. Sci. USA 2006, 103, 9310–9314. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Gent Name | PP vs. NN | NP vs. NN | PP vs. NP |
|---|---|---|---|
| RDM1 | 0.153 | 0.492 | −0.338 |
| TRPV4 | 1.535 * | 1.457 * | 0.948 * |
| EPHX1 | 0.132 | 0.104 | 0.028 |
| RIC8B | −0.538 * | −0.415 | −0.122 |
| STAU1 | 0.081 | 0.155 | −0.074 |
| TLE1 | 0.238 | 0.445 | −0.207 |
| IL5RA | −1.556 * | −1.191 | −0.366 |
| ASB8 | −0.095 | −0.078 | −0.016 |
| ERF | 0.681 * | 0.554 | 0.126 |
| CDC40 | −0.429 * | −0.387 | −0.042 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Malvisi, M.; Curti, N.; Remondini, D.; De Iorio, M.G.; Palazzo, F.; Gandini, G.; Vitali, S.; Polli, M.; Williams, J.L.; Minozzi, G. Combinatorial Discriminant Analysis Applied to RNAseq Data Reveals a Set of 10 Transcripts as Signatures of Exposure of Cattle to Mycobacterium avium subsp. paratuberculosis. Animals 2020, 10, 253. https://doi.org/10.3390/ani10020253
Malvisi M, Curti N, Remondini D, De Iorio MG, Palazzo F, Gandini G, Vitali S, Polli M, Williams JL, Minozzi G. Combinatorial Discriminant Analysis Applied to RNAseq Data Reveals a Set of 10 Transcripts as Signatures of Exposure of Cattle to Mycobacterium avium subsp. paratuberculosis. Animals. 2020; 10(2):253. https://doi.org/10.3390/ani10020253
Chicago/Turabian StyleMalvisi, Michela, Nico Curti, Daniel Remondini, Maria Grazia De Iorio, Fiorentina Palazzo, Gustavo Gandini, Silvia Vitali, Michele Polli, John L. Williams, and Giulietta Minozzi. 2020. "Combinatorial Discriminant Analysis Applied to RNAseq Data Reveals a Set of 10 Transcripts as Signatures of Exposure of Cattle to Mycobacterium avium subsp. paratuberculosis" Animals 10, no. 2: 253. https://doi.org/10.3390/ani10020253