Epigenetic Regulation of Skin Cells in Natural Aging and Premature Aging Diseases



1
Istituto di Genetica Molecolare CNR, Pavia; via Abbiategrasso 207, 27100 Pavia, Italy
2
Molecular and Cell Biology Laboratory, Istituto Dermopatico dell’Immacolata, IDI-IRCCS, Via dei Monti di Creta 104, 00167 Rome, Italy
*
Authors to whom correspondence should be addressed.
Cells 2018, 7(12), 268; https://doi.org/10.3390/cells7120268
Submission received: 15 November 2018
/
Revised: 7 December 2018
/
Accepted: 11 December 2018
/
Published: 12 December 2018
(This article belongs to the Special Issue Epigenetic Regulation of Stem Cells Ageing in Health and Disease)
{kind=link}
{kind=link}
Abstract
:Skin undergoes continuous renewal throughout an individual’s lifetime relying on stem cell functionality. However, a decline of the skin regenerative potential occurs with age. The accumulation of senescent cells over time probably reduces tissue regeneration and contributes to skin aging. Keratinocytes and dermal fibroblasts undergo senescence in response to several intrinsic or extrinsic stresses, including telomere shortening, overproduction of reactive oxygen species, diet, and sunlight exposure. Epigenetic mechanisms directly regulate skin homeostasis and regeneration, but they also mark cell senescence and the natural and pathological aging processes. Progeroid syndromes represent a group of clinical and genetically heterogeneous pathologies characterized by the accelerated aging of various tissues and organs, including skin. Skin cells from progeroid patients display molecular hallmarks that mimic those associated with naturally occurring aging. Thus, investigations on progeroid syndromes strongly contribute to disclose the causal mechanisms that underlie the aging process. In the present review, we discuss the role of epigenetic pathways in skin cell regulation during physiologic and premature aging.
1. Introduction
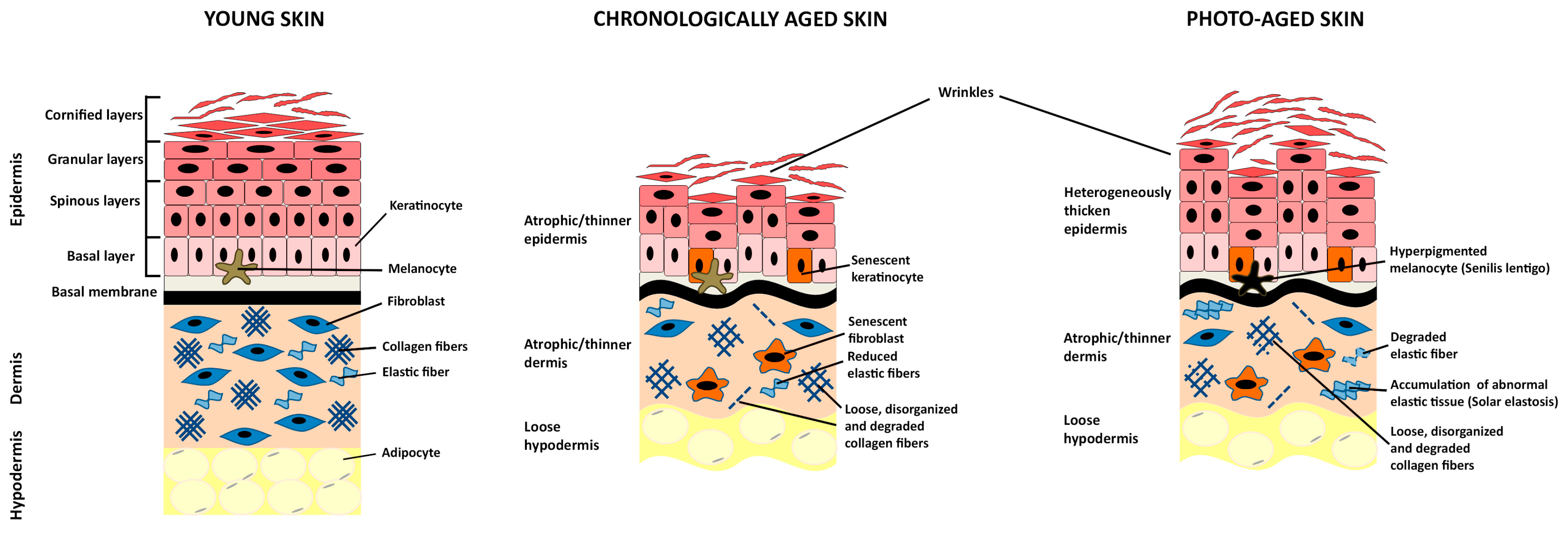
The skin protects the body from environmental stresses, such as water loss and microorganism infection, and is made up of three tissue layers: the epidermis, the dermis, and the hypodermis (Figure 1) [1].
The human epidermis consists of four major cell layers composed of keratinocytes in stages of progressive differentiation [2]. Stem cells (SCs) and transient amplifying (TA)-cells are located in the basal layer and are instrumental for proper human epidermal regeneration. SCs are endowed with self-renewal capacity and persist during a lifetime. They contribute to both epidermal renewal and repair by continuously generating pools of TA-progenitors that persist for limited periods (3–4 months). Indeed, long-lived, slow cycling SCs generate activated SCs that enter into a transient state of rapid proliferation giving rise to TA-cells, which constitute the largest group of proliferating cells endowed with different proliferative capacities [2,3,4,5]. Following several divisions, TA-cells withdraw from the cell-cycle and generate post-mitotic keratinocytes that migrate upwards to compose suprabasal and upper layers by executing their terminal differentiation program. This process is tightly regulated by temporal and spatial gene expression modulation. The suprabasal layers are characterized by early differentiation markers (e.g., keratins 1 and 10), whereas the upper layers express late differentiation genes (e.g., loricrin, involucrin). The outermost layers are characterized by terminally differentiated and enucleated corneocytes that are continuously removed and replaced by cells from the strata below [2]. Thus, the clonal conversion from SC to TA- and thereafter, to post-mitotic cells, is a continuous process that maintains the integrity of the epidermal structure [3]. Epidermal homeostasis relies on a finely tuned processes that dictate the choice between SC self-renewal and differentiation [2]. Keratinocyte SC de-regulations may result in skin aging and/or tumorigenesis.
The dermis is a connective tissue populated by fibroblasts that are responsible for the synthesis and secretion of collagens and other matrix proteins (fibronectin, elastin, and glycans) into the extracellular environment. Matrix components maintain the skin architecture and confer elasticity as well as resistance and strength to tissue [1,6]. Human dermal fibroblasts are composed of heterogeneous subpopulations with distinct gene expression profiles according to the anatomical site of origin and different physiological and pathological conditions [7]. Notably, fibroblasts are implicated in almost every skin process by interacting with both the epidermal and the other resident dermal cells, such as endothelial, neural, adipocytes, and inflammatory cells. The signaling from the dermal compartment is fundamental for the maintenance and homeostasis of epidermal SCs. Moreover, dermal fibroblasts are involved in various physiopathological conditions including wound healing, fibrosis, aging, and skin cancer [1,7].
At last, the hypodermis is the subcutaneous adipose layer underneath the dermis that surrounds hair follicles and connects the skin to the below muscles and bones. Even though the activity of hypodermal adipocytes is relevant for skin homeostasis and the separation between the dermal and hypodermal layers is not always well defined [8], the following review will focus on the epigenetic changes and transcriptional regulatory networks associated to an age-dependent functional decline of epidermal SCs and dermal fibroblasts.
2. Key Regulators of Epidermal Homeostasis
Various signaling and transcriptional pathways regulate in a stage-specific manner the expression of genes implicated in epidermal SC homeostasis, proliferation, differentiation and aging. The clonal evolution of SCs is, in part, regulated by the tumor suppressor p16INK4a and the transcription factor p63 [3,9]. They are modulated during clonal conversion accomplishment, and their over-expression or down-regulation impairs this process [10,11,12]. p16INK4a is an inhibitor of Cdk4/cyclin D complex and maintains the retinoblastoma protein (pRb) in its hypophosphorylated active state, preventing the E2F-mediated transcription and blocking the entry of proliferating cells into S phase [13]. Instead of playing a primary role in the continuous epidermal regeneration [14], p16INK4a is rather a master sensor of aberrant chromatin status that rapidly drives cell cycle exit and senescence [15]. Therefore, a key requirement for the maintenance and survival of the skin SC population throughout life is the repression of p16INK4a, thus, explaining the tight regulation in skin cells of p16INK4a encoding gene (CDKN2A), which belongs to the INK4a/ARF locus [16].
Differently, p63 is a master regulator of epidermal morphogenesis. It acts as a transcription factor and is implicated in the maintenance of keratinocyte self-renewal and/or cell fate decision [17,18]. The TP63 gene encodes several isoforms of p63 due to the presence of alternative promoters, different translation initiation sites, and alternative splicing events [19]. In human epidermis, ∆Np63 is the predominant isoform and plays a key role in keratinocyte proliferation and differentiation process through a Myc-regulated gene network and the interaction with several other transcription factors (AP-1, Klf4, Arnt, PPAR-alpha) [20,21]. Specifically, ∆Np63 and the protein encoded by its transcriptional target REDD1 gene are essential for the proliferative capacity and differentiation of progenitor cells [22,23]. Furthermore, ΔNp63α promotes keratinocyte proliferation by suppressing the expression of senescence-inducing miRNAs [12]. Thus, the regulation of p63 expression is fundamental to skin regeneration.
Transcription factor-dependent and epigenetic regulatory mechanisms tightly collaborate to ensure proper epidermal homeostasis. Indeed, several epigenetic networks work in concert to preserve keratinocyte stemness and promote proliferation by repressing the transcription of the p16INK4a-encoding gene and other cell-cycle inhibitors as well as by inhibiting unscheduled activation of non-lineage- or terminal differentiation-associated genes. The unbalancing of opposite epigenetic enzymatic activities drives the transition from epidermal SC quiescence to activation. On the contrary, specific epigenetic networks may promote keratinocyte terminal differentiation by acting through the p63-regulated networks on epidermal differentiation complex (EDC) genes. In dermal fibroblasts, the epigenetic networks are involved in the repression of INK4a/ARF locus as well as inflammatory genes to fight against senescence and paracrine pro-inflammatory processes [9,24,25,26,27,28].
Finally, the deregulation of epigenetic pathways directing epidermal homeostasis can induce epigenomic instability and, in turn, skin aging.
3. Skin Aging
Aging is characterized by the accumulation of macromolecular damages, impaired tissue renewal, and progressive loss of physiological integrity. One of the hallmarks of aging is cellular senescence that is triggered by several intrinsic (e.g., telomere shortening, ROS overproduction) and extrinsic (e.g., UV radiations, nutrient deprivation, inflammation) stimuli leading to growth arrest and specific phenotypic alterations, such as chromatin and secretome changes. Cellular senescence prevents the uncontrolled proliferation of damaged cells and induces the clearance and the regeneration of the tissue. However, in old organisms, the accumulation of several damages and the deficiency of immunological surveillance result in senescent cell accumulation and impaired tissue homeostasis [29,30,31]. Studies in mouse models indicate a causative role of cellular senescence in driving in vivo aging. Indeed, the mediators of senescence may limit the long-term growth of self-renewing compartments, thus, prompting aging. p16INK4a expression increases significantly with aging and the enhanced clearance of p16INK4a-positive senescent cells delays the onset of aging signs in progeroid mouse models [32,33]. Moreover, the deficiency of p63 in adult mice causes a cell growth arrest that impairs tissue regeneration and induces the appearance of aging features [34].
Skin aging can be distinguished in intrinsic or chronological aging and extrinsic or photo-aging, which are superimposed in the sun-exposed area of the body [35,36].
3.1. Chronological Skin Aging
Chronological skin aging results from the passage of time and is mainly influenced by genetic or metabolic factors. Aged skin exhibits epidermal thinning, fragility, wrinkle formation, and loss of elasticity [35,37]. Histological features are epidermal atrophy, reduced amounts of dermal fibroblasts and collagen fibers, which are loose, thin, and disorganized (Figure 1) [35,37].
The thinning of the epidermis depends on progressive keratinocyte SC dysfunctions and lower epidermal turnover, which are associated with the decline of skin barrier function and wound healing capacity [38]. Studies in mice and humans suggest that the reduced tissue regenerative capacity is not necessarily due to a decline in SC number or self-renewal but rather to a minor ability to produce progenitor, TA- and differentiated cells [39]. Nevertheless, the number of TA-cells increases in aged epidermis likely because they slow down the cell cycle compared to young TA-cells [16]. Moreover, during each replication cycle, telomeres become shorter and trigger a persistent activation of DNA damage response pathways thereby leading to cellular senescence [40]. p16INK4a and p63 are mediators of keratinocyte senescence. Specifically, p16INK4a is undetectable in keratinocytes from young donors and gets up-regulated during both replicative and premature senescence [10,11]. In contrast, p63 expression decreases during keratinocyte replicative senescence and its silencing are sufficient to induce cellular senescence [10,12]. Notably, p16INK4a expression directly correlates with chronological aging of human skin in vivo. The number of p16INK4a-positive cells increases with age in both epidermal and dermal compartments [11,41]. Moreover, aged keratinocytes are characterized by reduced expression of p63 [11,12].
The age-dependent remodeling of the dermis is mainly due to the dysfunction of long-lasting resident fibroblast populations that undergo continuous damage accumulation [42]. Aged fibroblasts lose the ability to remodel and organize the extracellular matrix (ECM) reducing the overall synthesis and secretion of collagens and elastins. Moreover, aged fibroblasts may alter the epidermal homeostasis by paracrine mechanisms [43]. Senescent fibroblasts display the accumulation of distinct heterochromatin structures designated senescence-associated heterochromatin foci (SAHF) that are induced by p16INK4a/pRb pathway activation. SAHF are silent domains that co-localize with the epigenetic mark H3K9me3 and the heterochromatin protein HP1, which may induce a senescent cellular state by transcriptionally repressing the E2F target genes involved in cell proliferation [44]. Aged fibroblasts are characterized by the presence of nuclear γH2AX-positive foci representative of DNA double-strand breaks that increase exponentially with age. These DNA damage foci co-localize with either telomeric DNA, thus, indicating telomere dysfunction [45], or with PML nuclear bodies that sustain damage-induced senescence growth arrest and inflammatory cytokine secretion [46]. In vivo aged dermal fibroblasts display enhanced secretion of the cysteine-rich, angiogenic inducer protein 61 (CYR61 or CCN1) that stimulates the secretion of pro-inflammatory cytokines and matrix metalloproteinases (MMPs). In turn, CCN1 and MMPs contribute to the acquisition and maintenance of the senescent cell state by negatively regulating collagen homeostasis and increasing the degradation of collagen, respectively [47].
3.2. Photo-Aging
Skin is continuously exposed to environmental insults that may lead to extrinsic aging. Specifically, chronic exposure to sunlight results in phenotypic changes globally termed photo-aging. Photo-aged skin is characterized by epidermal thickness heterogeneity, accumulation of immune cells and pigmentation alterations associated to the formation of “senilis lentigines”. However, major alterations occur primarily within the dermis: Collagen fibers disorganization and partial degradation, which cause wrinkling of the skin, the disintegration of elastic fibers, and accumulation of abnormal elastic tissue that characterize “solar elastosis” (Figure 1) [35,48].
Sun exposed skin is affected by both UV-A (320–400 nm) and UV-B (290–320 nm) radiations. UV-A rays are less energetic but penetrate deeper into the dermal layer. They induce DNA, protein, and lipid damages as well as dermal remodeling alterations through the generation of reactive oxygen species (ROS). UV exposure at a sub-erythemal dose is a potent stimulator for producing and releasing skin–fibroblast-derived elastase that leads to drastic alterations of dermal structure and “solar elastosis”. Moreover, UV-A exposure increases the production of MMPs, in particular of MMP-1 that hydrolyzes the interstitial collagen leading to the disorganization and progressive degeneration of dermal ECM [49,50,51]. Chronic UV-A irradiation also affects the synthesis of other ECM molecules of the dermis. It inhibits hyaluronan synthesis by down-regulating the hyaluronic acid synthases (HAS)-1, -2 and -3, thus, altering the dermal proteoglycan composition [42]. Furthermore, aged fibroblasts increase melanogenic gene transcription leading to hyperpigmentation and appearance of “senilis lentigines” [52].
Differently from UV-A, UV-B radiation is mainly absorbed by the epidermis and directly induces DNA lesions, such as cyclobutane-pyrimidine dimers (CPDs) and 6-4 photoproducts (6-4PPs), in keratinocytes. DNA photolesions may result in the generation of DNA mutations leading to cell senescence, apoptosis or carcinogenesis. Moreover, UV-B radiation stimulates keratinocytes to release soluble factors such as cytokines, interleukin (IL)-1α and IL-6 that give rise to an inflammatory response, known as “sunburn”. Through a paracrine mechanism, keratinocytes also induce the secretion of MMP-1 from dermal fibroblasts [53,54]. It may be relevant to note that in vitro, human dermal fibroblasts are more susceptible to UV exposure than epidermal keratinocytes [55].
4. Epigenetic Changes in Skin Aging
Epigenetic regulatory networks consist of three major events: DNA modifications (mainly DNA methylation), histone modifications (mostly histone methylation or acetylation), and recruitment of higher-order chromatin remodelers [28]. DNA and histone modifications affect gene transcription by altering histone–DNA and histone–histone interactions and, thus, regulating the accessibility of transcription factors and components of the transcriptional machinery to the chromatin. Histone modifications, which occur mainly at the amino-terminal portion of histone tails, often act cooperatively and synergistically to either repress or activate transcription [24,26,28].
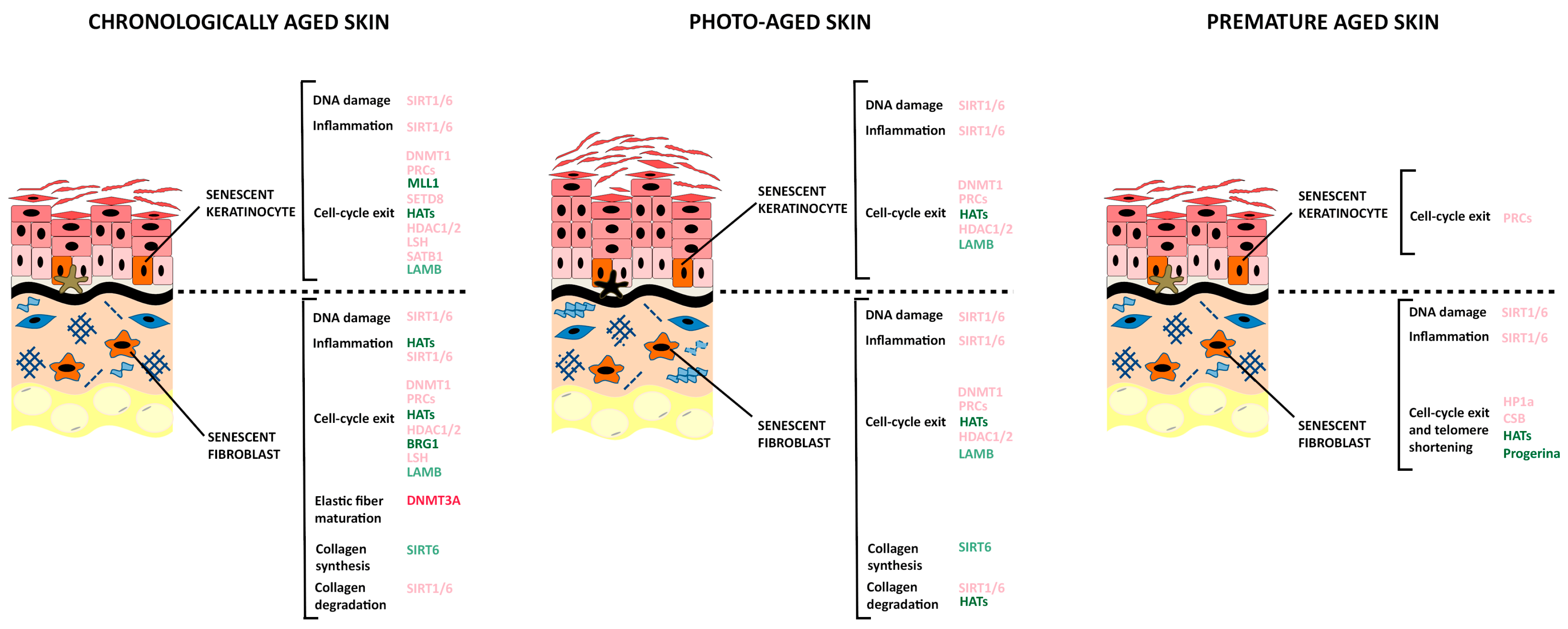
Epigenetic changes are relevant for cellular senescence and aging (Figure 2). They can be modified by endogenous (e.g., intracellular signaling pathways) as well as exogenous stimuli deriving from lifestyle, diet, and environmental exposure (e.g., UV radiations, smoking, physical activity, antioxidant intake, caloric restriction) [56].
The chromatin of elderly subjects is characterized by histone loss, incorporation of different histone variants, altered DNA methylation and histone modification patterns, recruitment of different chromatin modifiers, and altered transcription profiles. Many studies indicate that epigenome modification contributes to the aging process [57]. Indeed, chromatin remodeling and histone post-translational modifications are critical for the recruitment and activation of DNA repair pathways and, therefore, for the maintenance of genomic integrity. The phosphorylation of histone H2A variant (γ-H2AX), the methylation on histone H3 lysine 79 (H3K79me), and the acetylation on histone H3 lysine 9 (H3K9) and lysine 14 (H3K14) have a key role in damage sensing and chromatin opening. On the contrary, the dephosphorylation of H2AX, acetylation or deacetylation of histone H3 and H4 are important for chromatin restoration after DNA-break repair [58]. The histone variant H2A.J, whose up-regulation promotes the expression of inflammatory genes, accumulates in an age-dependent manner in mouse and human skin as well as in irradiated mouse skin [59].
4.1. DNA Methylation
DNA methylation is an epigenetic mechanism that is mostly associated with transcriptional repression and is capable of regulating several aspects of gene expression (e.g., long-term gene silencing and genomic stability maintenance). The methyl group is placed by one of the three DNA methyltransferases (DNMT1, DNMT3A, and DNMT3B) on the fifth position of cytosine, predominantly at the CpG dinucleotide islands, to generate the 5-methyl-cytosine (5mC) base. DNMT1 is known as “maintenance DNMT” since it preserves the original methylation patterns during cell divisions. On the contrary, DNMT3A and DNMT3B are “de novo” DNMTs that catalyze new methylation marks. The presence of 5mC on the DNA mainly inhibits transcription by preventing the binding of specific transcription factors or by recruiting Methyl-CpG-binding proteins (MBPs) and histone deacetylases that, in turn, induce chromatin condensation. However, in some sporadic cases, DNA methylation contributes to gene activation: CpG methylation of the CRE sequence (TGACGTCA) enhances the DNA binding of the C/EBPα transcription factor, which regulates the expression of differentiation genes in several cell types [24,26,60,61].
A passive loss of 5mC marks can occur through several mechanisms, such as down-regulation of DNMT enzymes, cytosolic localization of DNMT, impairment of DNMT recruitment on DNA, decreased levels of the methyl donor S-adenosyl methionine, and inhibition of DNMT enzymatic activity [62]. The active loss of 5mC can be due to the Ten–Eleven Translocation (TET) family enzymes, which convert 5mC into 5-hydroxymethylcytosine (5hmC), and, in turn, into 5-formylcytosines and 5-carboxylcytosines. These modifications are substrates of the base excision repair pathway (BER) and are reverted to cytosine actively reducing the 5mC marks on DNA [63]. However, 5hmC is not only an intermediate of DNA demethylation, but rather a stable epigenetic mark associated with euchromatin [62].
DNA methylation is instrumental for suppressing the expression of genes involved in cell cycle exit and keratinocyte terminal differentiation [64,65,66]. Thus, the maintenance of the DNA methylation patterns is important to preserve skin progenitor cell identity and self-renewal whereas their remodeling after internal or environmental stimuli is associated with skin aging.
Young individuals display similar DNA methylation patterns (methylomes) whereas they become divergent in the elderly. These age-related changes in DNA methylation involve both hypermethylation and hypomethylation events and are associated with the progressive accumulation of epigenetic damage, known as “epigenetic drift” [67]. Studies carried out on monozygotic twins have shown that aging-associated DNA methylation changes are evident in monozygotic twins who lived apart for long periods, suggesting an environmental component of the “epigenetic drift” hypothesis [68,69]. Some of the age-related methylation changes that involve specific genomic regions are directional and not stochastic. Indeed, several studies indicate the presence of aging-associated differentially methylated regions (a-DMRs) that change over time in the same direction [62].
Comparing the methylomes between epidermal and dermal skin samples, a high degree of tissue specificity and inter-individual similarities within tissues have been found [70]. Methylation changes in the epidermis from young and old individuals are highly localized and display only minor quantitative differences demonstrating a limited destabilization of the epigenome during aging [71,72]. However, methylomes from aged skin samples are more heterogeneous than those from young ones, in support of the “epigenetic drift” hypothesis [70,72]. Discontinuous methylation changes can be, therefore, considered as features of aging methylome [72]. Interestingly, increased methylation heterogeneity has also been associated with fibroblast and epithelial cell senescence in culture [73,74,75]. Of note, there was a significant correlation between DNA methylation changes in in vitro long-term fibroblast cultures and in vivo aged fibroblasts, indicating that both processes might be regulated by similar epigenetic events [73]. Methylome data obtained from skin samples can be used to predict the chronological age of donors with elevated accuracy [72]. Intrinsic skin aging is mainly associated with widespread hypermethylation of CpG islands at gene regulatory elements [70,71,72]. Notably, hypermethylated DMRs have been associated with “bivalent” chromatin structures, which characterize genomic regions of transcription regulation in SCs. Aberrant DNA methylation at “bivalent” chromatin domain promoters is associated with a decreased capacity to differentiate in cell culture. Therefore, it is tempting to speculate that DMRs hypermethylation may induce a decline in the ability to produce progenitors, TA-, and differentiated cells [39,71]. Hypomethylated DNA sequences in aged skin are mainly co-associated with transcription enhancer elements in H3K9me3-marked regions [76], whereas, the indication of age-related global loss of DNA methylation as a major feature of intrinsic skin aging is still controversial [72,77]. The knowledge about DNA methylation changes might need a revaluation after the discovery of 5hmC because the conventional bisulfite sequencing method cannot discriminate between 5mC and 5hmC [78].
Age-related methylation changes have a minimal impact on in-cis gene expression, acting primarily to stabilize pre-existing baseline expression levels [71,72,79]. Analysis of gene co-expression networks allowed the identification of gene subsets whose expression changes in correlation with age and, in turn, with the loss of co-regulation by specific transcription factors. Skin samples from elderly display reduced connectivity of gene expression, suggesting that the age-related erosion of methylation patterns is paralleled by a reduced fine-tuning in transcriptional networks, probably mediated by methylation-dependent changes in transcription factor binding. Thus, the loss of epigenetic regulatory fidelity may be defined as a key feature of skin aging epigenome [72].
DNMTs can be involved in the age-related erosion of DNA methylation patterns. By repressing the INK4a/ARF locus, DNMT1 plays a key role in SC maintenance and tissue renewal, therefore, its dysfunction in keratinocytes and fibroblasts is instrumental for skin aging [64]. DNMT1 dysfunction is associated with epidermal cell senescence and impairment of skin homeostasis. The epidermal loss of DNMT1 in mice decreases hair follicle SC activation during aging and leads to progressive alopecia [64,80]. DMNT1 expression inversely correlates with p16INK4a expression and chronological age in human skin samples as well as fibroblasts in culture [81]. Notably, DNMT1 knockdown significantly induces p21 up-regulation and premature senescence in human dermal fibroblasts [81,82]. UHRF1, a component of DNMT1 methylation machinery, is necessary for maintaining cell proliferation and it gets down-regulated upon stress-induced or replicative senescence of human keratinocytes as well as fibroblasts. Notably, decreased UHRF1 expression is a key initial event in the suppression of DNMT1-mediated DNA methylation [26,83]. DNMT1 expression is also regulated by miR-377. A negative correlation has been identified between miR-377 and DNMT1 expression in human skin tissues and dermal fibroblasts collected from UV-unexposed areas of differently aged individuals. By directly targeting DNMT1, miR-377 regulates the methylation of TP53 promoter, and, therefore, induces the senescence of human skin fibroblasts [81]. Furthermore, aged fibroblasts display higher binding of DNMT3A on the Lysyl oxidase-like 1 (LOXL1) promoter. LOXL1 is an amino-oxidase enzyme involved in maturation of elastic dermal fibers, and its down-regulation is associated with the loss of skin elasticity [84].
The remodeling of DNA methylation patterns is also a feature of skin photo-aging. Besides DNA photolesions, UV irradiations induce oxidative stress, inflammatory responses, and immune suppression, which all can influence the epigenetic pattern. Specifically, sun exposure induces an epigenetic shift towards DNA hypomethylation in human skin and the degree of hypomethylation correlates with clinical photo-aging measures [70,77]. Among other events, a reduced expression of DNMT1, associated with the up-regulation of miR-377, is observed in photo-aged skin and UV-A-induced senescent human fibroblasts [81]. Indeed, it has been observed that in human dermal fibroblasts UV-A radiations reduce DNMT1 expression through a ROS-mediated ZEB1 down-regulation [85]. In contrast, UV-B irradiations did not directly induce detectable changes of DNA methylation in human primary and immortalized HaCat keratinocytes [86,87]. However, the expression of TET enzymes (mainly TET2) and 5hmC levels significantly increase after UV-B irradiation [87].
4.2. Histone Methylation
Histone methyltransferases (HMTs) and demethylases (HDMs) covalently modify histones on their amino-terminal tails, altering the local chromatin environment and, in turn, gene transcription. Depending upon which residue of the histone tail is modified, histone methylation may promote transcription repression or activation. The trimethylation of histone 3 at lysine 27 (H3K27me3) and at lysine 9 (H3K9me3) are associated to facultative and constitutive heterochromatin, respectively, and, therefore, to gene silencing. In contrast, the trimethylation of histone 3 at lysine 4 (H3K4me3) and at lysine 36 (H3K36me3) are linked to euchromatin and transcriptional activation [26,88,89,90].
Among the methylated histone modifications, H3K27me3 is the most well-studied at the level of skin homeostasis. The presence of this transcriptional repressor epigenetic mark depends on the opposite activities of two families of chromatin modifiers: the Polycomb Repressor Complex (PRC) enzymes, which own methyltransferase activity and use it to catalyze histone methylation, and the histone demethylase Jumanji proteins that remove the methyl group [26,88,89].
The PRC complex activity involves two sub-complexes: PRC1 and PRC2. PRC2 is the initiator of transcription repression whereas PRC1 is a repressor maintenance complex. The PRC2 complex consists of the EED, Suz12, RBAp46/48, and methyltransferase EZH1/2 subunits that catalyze H3K27me3. This mark is necessary for the binding of PRC1 to the chromatin [88]. Canonical PRC1 is made of four different ortholog proteins, CBX, PCGF, HPH, and the E3-ligase protein (RING) that acts by catalyzing the monoubiquitylation of histone H2A (H2AK119ub1). This last histone modification prevents the RNA polymerase II transcription elongation by inducing chromatin condensation and gene silencing [89]. Through its chromodomain, the CBX subunit recognizes H3K27me3 and allows the recruitment of PRC1 to the target genes. Notably, CBX confers specificity to the complex as the different CBX proteins have a distinct pattern of chromatin binding [26,88,89]. The non-canonical PRC1 (ncPRC1) complexes are characterized by the presence of Rybp/Yaf2 and the lack of CBX components, and, therefore, their recruitment to DNA is independent of H3K27me3 mark [91]. Although the potential mechanism by which the ncPRCs mediate gene repression is largely unknown, these complexes are involved in embryonic development and stem cell maintenance [92,93,94].
PRC proteins play a key role in controlling epidermal SC identity and self-renewal by preventing the unscheduled induction of non-lineage, differentiation, and senescence genes [26]. On the contrary, the histone demethylase JMJD3 regulates the epidermal differentiation [95]. Reduced expression of PRC subunits is associated with skin aging as well as keratinocyte and fibroblast senescence both in vivo and in vitro models [11,41]. In particular, the levels of PRC2 in basal epidermal progenitors and fibroblasts decrease with age. EZH2 reduction leads to decreased levels of H3K27me3, disassociation of PRC1 complex from the chromatin and increased transcription of INK4a/ARF genes [25,96]. One of the most studied PRC1 subunits in human skin is the PCGF4 subunit/Bmi-1. Human keratinocytes and fibroblasts from aged donors are characterized by Bmi-1 down-regulation and early p16INK4a expression, which are also correlated with SC depletion [11,96]. In agreement, Bmi-1 deficiency in mice leads to tissue atrophy and accelerated aging [97,98]. Notably, Bmi-1 is able to revert the aged phenotype modulating both p16INK4a levels and clonal conversion in primary human keratinocytes from elderly donors [11]. In human dermal fibroblasts, miR-141-mediated Bmi-1 down-regulation induces cell senescence via p16INK4a up-regulation [96]. The PRC-mediated transcription repression is counteracted by the action of Trithorax (TrxG) H3K4 histone methyltransferase complexes, which activate gene transcription through the deposition of H3K4me3 marks at H3K27me3 labelled promoters [99,100]. Specifically, the enzyme Histone-lysine N-methyltransferase 2A (KMT2A) or myeloid/lymphoid or mixed-lineage leukemia 1 (MLL1) activate the transcription of the p16INK4a-encoding gene during replicative and oncogene-induced senescence [101]. Therefore, the decline of PRC complexes may induce p16INK4a expression by favoring the accessibility of p16INK4a-encoding gene to MLL1.
PRC complexes seem to have an active role also in photo-aging. Indeed, UV-A radiations modulate the expression of EZH2 and Bmi-1, induce the senescence of human dermal fibroblasts and reduce hyaluronic acid (HA) synthesis. The use of GSK126, an inhibitor of the EZH2-methyl transferase activity, restores dermal fibroblasts growth after UV-A irradiation by modulating the expression of enzymes involved in HA synthesis and inhibiting the transcription of various photo-aging-related genes (e.g., Smad2, Smad4, MMP-1) [102,103]. Moreover, UV-B radiations induce human dermal fibroblast senescence through miR-101 induction and EZH2 reduction [104]. Globally, UV irradiation results in increased levels of the active H3K4me3 mark and decreased amounts of the repressive H3K9me2 mark as well as a concomitant increase of MMP-1 and MMP-3 secretion by human dermal fibroblasts [105].
Finally, the histone methyltransferase SETD8 (KMT5A) catalyzes the monomethylation of histone H4 at lysine 20 (H4K20me1), which represents a mark of gene activation. SETD8 is a Myc-target gene that implicated in epidermal homeostasis and adult human keratinocyte self-renewal. The loss of Setd8 in mice results in loss of p63 and increased levels of p53 proteins [106]. In human epithelial cells, SETD8 is down-regulated both in oncogene-induced and replicative senescence. Moreover, the inhibition of SETD8 is sufficient to trigger cell senescence [107].
4.3. Histone Acetylation
Histone acetyltransferases (HATs) and histone deacetylases (HDACs) modify histone tails by displaying opposite activities. HATs catalyze the acetylation of ε-amino groups of lysine residues promoting chromatin relax and subsequent gene transactivation. HDACs remove the acetyl groups and allow chromatin structure compaction and gene silencing. HATs are broadly categorized into three families: p300/CBP, Gcn5-related N-acetyltransferases (GNAT), and MYST [90].
p300 (also named EP300, E1A-binding protein p300) and CBP are closely related co-activators that acetylate multiple lysines on histone H3 (K14, K18, and K23) and on histone H4 (K5, K8, and K12). p300 and CBP enzymes play several roles in skin homeostasis: They control cell growth and early differentiation in keratinocytes through the stabilization of p63 and the p53-mediated regulation of p21Waf1/CIP1 expression [108,109], they modulate p16INK4a expression [110,111], maintain the integrity of skin barrier [112], and regulate the expression of pro-inflammatory and ECM genes in fibroblasts [90,113].
HDAC enzymes are classified in four classes: Class I, including HDACs 1–3 and 8; Class II, including HDACs 4–7, 9, and 10; Class III or Sirtuins (Sirt1-7), and Class IV, containing HDAC11 [90]. Whereas Class I HDACs play important roles in maintaining skin homeostasis, Class II is mainly involved in the wound healing process [28]. Among Class I, HDAC1 and HDAC2 are required for ΔNp63-mediated repression of the INK4a/ARF locus and, therefore, play an important role in epidermal SC self-renewal maintenance [114].
Class III HDACs or Sirtuins (Sirts 1–7) are NAD+ dependent enzymes relevant for several skin cell functions and processes, including control of energy metabolism and oxidative stress, cell survival, UV damage response, DNA repair, tissue regeneration and inflammation [115]. Specifically, SIRTs 1, 6, and 7 play a key role in epigenetic regulation and transcription facilitation. SIRT1 deacetylates histones H1 (H1K26), H3 (H3K9), and H4 (H4K16) and regulates the acetylation status of several transcription factors (e.g., nuclear factor-κB (NF-κB)), DNA repair proteins (e.g., poly-ADP-ribose polymerase 1 (PARP1) and NER factors), and the Werner syndrome ATP-dependent helicase (WRN). SIRT1 works in concert with p63 to regulate keratinocyte proliferation and the maintenance of epidermal progenitor cells by inhibiting cell senescence [21]. Moreover, SIRT1 inhibits collagen degradation suppressing the IL-1β-mediated induction of MMPs [116]. SIRT6 deacetylates the histone H3 (H3K9 and H3K56) and PARP1 to promote DNA repair. Specifically, SIRT6 may regulate DNA repair pathways, NFkB activity, and α-1-type 1 collagen (COL1A1) production in human dermal fibroblasts [117,118,119].
The modulation of HDACs and HATs enzyme activity is strongly implicated in skin cell senescence. The epigenetic regulator ING1 is part of both HAT and HDAC complexes and mediates chromatin remodeling and gene expression modification through the binding to H3K4m3. ING1 gene encodes two major splicing isoforms: p33ING1b that induces apoptosis and p47ING1a that induces senescence. Although both INGl-isoforms have been found associated with HDAC complexes, INGlb exhibits a binding preference for the p300/CBP HAT complexes [120]. ING1a expression and ING1a-associated HDAC1 activity increase during fibroblast replicative senescence whereas ING1b expression decreases. Moreover, in primary human fibroblasts and HaCat cells, the overexpression of ING1a induces multiple markers of senescence, including SAHF containing HP1γ, flat morphology with large nuclei, inhibition of PCNA expression, and cell cycle arrest in the G1 phase, which strongly correlate with the activation of p16INK4a/Rb pathway [57,121]. Loss of function of both HDAC1 and HDAC2 determines p21 and p16INK4a upregulation and extinguishment of SC proliferation [114].
Age-dependent changes of histone acetylation are coupled to the decline of metabolic activities and may influence global gene expression, which, in turn, affects health and longevity. SIRT1 and 6 seems to act as metabolic sensors regulating both transcription and genome stability depending on changes in the NAD+/NADH ratio [115]. Histone acetylation patterns are susceptible to quantitative alterations of key metabolites such NAD+ as well as mitochondrial metabolic activity, thus, allowing chromatin to function as a sensor of cellular metabolism [122]. Although mice moderately overexpressing SIRT1 do not live longer under standard diet conditions, they exhibit healthy aging features, such as improved glucose homeostasis, reduced tumor incidence, and reduced levels of the aging molecular marker, p16INK4a or DNA damage [123]. Moreover, SIRT6-deficient mice display numerous progeroid and aging-like phenotypes [124].
Oxidative stress increases with age in human skin and leads to a PARP-mediated decline of NAD+ levels. A significant concomitant decrease of NAD+ levels and SIRT1 activity has been found in human non-sun exposed skin [125]. SIRT1 levels are consistently reduced during replicative senescence and epidermal aging. Moreover, SIRT1 silencing induces senescence similarly to p63 in keratinocytes. Specifically, ΔNp63 inhibits the expression of miR-138, miR-181a, and miR-181b that directly target SIRT1 [12]. SIRT1 also modulates H2O2-induced premature senescence in human primary keratinocytes. Although SIRT1 is not expressed in senescent keratinocytes, following forced expression its results are localized only in the cytosol suggesting a nuclear import dysfunction [126].
In addition, in human dermal fibroblasts, SIRT1 expression is significantly reduced with age [42,127]. Moreover, the expression of both SIRT1 and SIRT6 decreases in human dermal fibroblasts during replicative senescence in parallel with the appearance of aging biomarkers [128]. Notably, SIRT1 up-regulation or down-regulation results in delayed or accelerated fibroblast senescence, respectively [129]. The SIRT1-promoted cell proliferation and antagonized cellular senescence in human diploid fibroblasts appears partially mediated by the activation of ERK/ S6K1 signaling pathway [130]. Furthermore, SIRT1 mediates the inhibition of MMP transcription and, therefore, the preservation of collagens in the ECM of the dermis. Treatment of human dermal fibroblasts with the SIRT1-activator Resveratrol inhibits MMP-1 expression, whereas SIRT1 down-regulation results in increased levels of MMP-1 and -3 [116].
As previously mentioned, also SIRT6 is involved in cell senescence and aging as it modulates the accessibility of DNA repair proteins to chromatin, represses nuclear factor (NF)-κB promoter through the histone deacetylation and, in turn, inhibits NF-κB target genes expression. Thus, SIRT6 down-regulation in human dermal fibroblasts results in increased DNA damage and NF-κB activity [118,131]. It has been shown that the activity of the DNA repair pathway BER declines with the aging of keratinocytes [132] and fibroblasts [131]. Dysfunctional BER contributes to the accumulation of mutations and, therefore, impacts on the age-related tissue homeostasis impairment. Notably, SIRT6 overexpression rescues the decline of BER in aged fibroblasts [131]. Similar to SIRT1, SIRT6 is implicated in the synthesis or degradation of skin collagen. SIRT6 down-regulation in human dermal fibroblasts reduces COLA1 transcription, induces NF-κB activation, and increases MMP-1 secretion, suggesting that the reduced expression of SIRT6 in aged skin is directly implicated to the reduced levels of collagen fibrils [117].
Histone H3 hyperacetylation appears to be a relevant epigenetic mechanism in skin photo-aging. In comparison with non sun-exposed skin, solar irradiated skin samples exhibit a global increase acetylation of histone H3 in concomitance with increased p300 activity and reduced HDAC1 and SIRT1 expression. Gene promoters implicated in collagen degradation (MMP-1, -3 and -9, and Ahr receptor), apoptosis (PDCD5), and inflammation (ITIH5) are hyperacetylated in the sun-exposed area of the skin [133]. In keeping with these data, UV irradiation induces histone H3K9/14 acetylation within the promoter regions of ATF3, COX2, IL-8, MKP1, and MnSOD genes in human keratinocytes [134]. Moreover, UV irradiation induces the acetylation of histone H3 by decreasing HDAC and increasing HAT activity in human dermal fibroblasts. This H3 hyperacetylation may be inhibited by p300 down-regulation, which plays a critical role in the transcriptional regulation of MMP-1 upon UV exposure [135].
SIRTs play key roles in the cellular response to UV irradiation by reducing the oxidative stress, decreasing DNA damage signaling, and inhibiting MMP expression. Upon UV irradiation, SIRT1 deacetylates the NER factor XPA, thus, optimizing the activity of the NER repair pathway and, hopefully, protecting from photo-aging [136]. Photo-exposed skin displays increased levels of SIRT1 [137]. Moreover, SIRT1 confers protection against UVB-induced keratinocytes death via modulation of p53 and JNK [128,138]. Notably, it has been reported that UV radiation down-regulates SIRT1 expression through ROS-mediated JNK pathway activation in keratinocytes [138]. UV-B radiation down-regulates SIRT1 in a time- and dose-dependent manner in human immortalized HaCat keratinocytes.
Furthermore, in human fibroblasts, UV-B radiation results in decreased SIRT1 protein levels [139] whereas SIRT1 overexpression protects dermal fibroblasts against UV-B-induced senescence. This protection is mediated by the suppression of p53 acetylation and, at the same time, by the deacetylation of the transcription factor FOXO3a that increases the cellular resistance to oxidative stress [140]. Resveratrol treatment inhibits MMP-9 expression, thus, preventing collagen degradation in human fibroblasts or mouse skin after UV radiation exposure [141].
4.4. Higher-Order Chromatin Remodeling and Three-Dimensional Genome or Ganization
The higher-order organization of chromatin and the spatial distribution of genes within the nuclear space, as well as the nuclear compartmentalization of chromatin-remodeling complexes and transcription machinery, play a key role in regulating gene expression and driving cell differentiation [24,145]. During epidermal regeneration and keratinocyte differentiation, the transcription factor p63 coordinates several chromatin-remodeling pathways by regulating the activity and expression of specific target genes. Among the p63-target genes are included proteins regulating covalent histone modifications (HDAC1/2), ATP-dependent chromatin remodeling factors (Brg1, Lsh/HELLS), higher-order chromatin remodeling proteins (SATB1,) and nuclear envelop assembly molecules (Lamins) [24,114,146,147,148,149].
Whereas the epigenetic mechanisms driven by DNMT1 or PRC complexes are conserved among different tissues and their de-regulation during aging results in self-renewal impairment of several SC types (i.e., hematopoietic, neural, epidermal SCs) [28,150], p63 is an epidermal master regulator and its age-related down-regulation specifically affect keratinocyte SC functions by its epigenetic targets [11,12].
ATP-dependent chromatin remodelers—Chromatin remodeling complexes play a direct role in gene expression regulation by coordinating DNA accessibility and dynamic of nucleosomes organization. Their activity depends on the hydrolysis of ATP as a source of energy [151].
The multisubunit SWI/SNF family complexes contain SWI2/SNF2-like ATPases (Brm/Smarca2/Snf2α or Brg1/Smarca4/Snf2β) that hydrolyze ATP to alter the histone-DNA interactions within nucleosome, leading to an open chromatin state and transcriptional activation. The SWI/SNF complexes also contain several regulatory components, called BAFs, endowed with different DNA target affinities and functions [151]. Brg1 is a p63 target gene, which is required for gene activation during epidermal barrier formation [152] and whose activity is prevented in epidermal SCs. Indeed, its subunit actin-like 6a (ACTL6a/BAF53a) inhibits the binding of the remodeler complex Brg1 to the gene promoter of Krupper-like factor 4 (KLF4) transcription factor, whose function is to drive skin barrier formation [153,154]. Notably, the expression of KLF4 decreases during chronological epidermal aging and keratinocyte replicative senescence. Specifically, KLF4 silencing is sufficient to induce a senescent phenotype in primary keratinocytes [155]. Furthermore, Brg1 drives SAHF formation during senescence via its chromatin remodeling activity and/or by up-regulating p16INK4a expression. Indeed, Brg1 may interact with pRB to drive SAHF formation [156]. SWI/SNF complexes are critical for the phosphorylation of H2AX and the formation of γ-H2AX foci following DNA damage. In particular, they facilitate DNA double-strand breaks (DSB) repair through the binding to γ-H2AX [157].
Lsh (HELLS) is a chromatin remodeler that belongs to the SNF2/helicase family, contains an ATP binding and a C-terminal helicase domains and is a meCpG stabilizer [158]. Lsh acts by recruiting DNMTs or through interaction with PRC1 complex and HDACs [159]. Furthermore, Lsh is a direct target of both p63 [147] and DNMT1 [160] that may cooperate to repress p16INK4a expression and maintain a proliferative state in epidermal progenitors. Thus, p63 requires HDAC1/2 and Lsh/HELLS to repress the expression of anti-proliferative genes [114,147]. Notably, Lsh represses p16INK4a expression by recruiting HDAC1 at its promoter and, through this event, it delays cell senescence in human fibroblasts [161]. Lsh transcription is activated by E2F1. Thus, E2F repression may be relevant for Lsh transcription decrease in senescent cells [162]. The loss of Lsh induces senescence in human keratinocytes and leads to accelerated skin aging in p63 heterozygous mice [12,147].
Genome organizers and nuclear architecture remodelers—CTCF is a zinc finger transcription factor that functions as genome organizer through the recruitment of cohesin, a protein essential for proper chromosome segregation, location, and gene expression. The genome organizer CTCF forms thousands of inter-chromosomal loops of different sizes and brings into close proximity to specific genes located on different chromosomes [163]. CTFC binds the INK4/ARF locus and inhibits its epigenetic silencing through chromatin remodeling. Thus, CTCF maintains high p16INK4a levels and limits keratinocyte growth [164,165].
The specialized AT-rich binding protein 1 (SATB1) is a genome organizer that recruits chromatin remodelers at AT-rich sequences to regulate chromatin structure and gene expression [166]. SATB1 may activate or repress multiple genes depending on its posttranslational modifications and protein interactions. Indeed, SATB1 is a p63 target that allows the expression of EDC genes by remodeling chromatin architecture in the nucleus. However, SATB1 is also a direct target of miR-191 that induces keratinocyte senescence. Notably, SATB1 expression decreases during keratinocytes replicative senescence, and its silencing induces a G1 block in cell cycle and an increment of senescence-associated markers [167]. Furthermore, SATB1 interacts also with pRB/E2F1 complex to repress the promoter of the p16INK4a encoding gene [146,167,168]. 16INK4a and the pRB/E2F pathway are critical for SATB1-induced cell cycle arrest [168].
The nuclear lamina is a protein structure that lines the inner membrane of the nuclear envelope (NE) and contributes to the size, shape, and mechanical stability of the nucleus. Besides its structural role, the nuclear lamina acts as an anchor for the peripheral elements of chromatin, thus, regulating the spatial organization of chromosome territories and the epigenetic state of the chromatin. Proteins of the nuclear lamina may be implicated in DNA replication, and DNA repair and transcription [169,170]. Notably, p63 regulates the nuclear shape and the expression of NE-associated genes, determining changes in heterochromatin organization and intranuclear position of keratin loci in keratinocytes [149]. The major structural elements of the nuclear lamina are the lamins [169]. Both A- and B-type lamins are required for chromatin organization and proper gene expression in skin cells [170,171]. During cellular senescence, the nuclear lamina undergoes dramatic remodeling [172,173]. Specifically, aberrant prelamin A isoform called progerin is present in cultured aged fibroblasts as well as in the dermis or few differentiated keratinocytes of elderly individuals, suggesting that senescence may be associated to the accumulation of an altered form of Lamin A [174]. Notably, overexpression of progerin in primary dermal fibroblasts results in reduced cell proliferation and premature senescence of a large number of cells [175]. Moreover, Lamin B1 levels decline during senescence, both in vitro cultures of human dermal fibroblasts and keratinocytes [172,176,177] and in vivo during the chronological aging of human skin [173,176]. Interestingly, loss of Lamin B1 is an early sensitive marker for the in vitro and in vivo quantification of UV-B induced keratinocyte senescence. It is worth noting, Lamin B1 levels are restored upon skin regeneration [177].
5. Epigenetic Regulation of Skin Premature Aging Diseases
5.1. Premature Aging Diseases
Progeroid syndromes (PSs) represent a group of clinical and genetically heterogeneous pathological entities characterized by an accelerated aging process affecting various (but not all) tissues and organs. This aspect earned them the name of segmental progeroid syndromes, and they feature many of the clinical and molecular hallmarks associated with the naturally occurring aging process.
Because of these similarities, investigation on the PSs contributes to the understanding of the causal mechanisms that underlie aging. PSs do not show gender or ethnic preferences. Most of them appear in the first years of life and lead to life shortening. Major clinical features include short stature, alopecia, lipodystrophy, skin alterations, osteoporosis, and cardiovascular abnormalities [178]. Even though PSs display a large variety of clinical features and different propensities towards precious aging, the accumulation of DNA damage, increased genome instability, and epigenetic changes are clearly visible in all patient skins. Due to its easy accessibility, skin is becoming an ideal tissue in which to investigate the molecular processes responsible for aging or carcinogenesis. Even more, skin biopsies and primary skin cell cultures from patients affected by PSs represent a precious and irreplaceable source of biological material whose analysis is shedding light on the processes regulating senescence surveillance and progression [179,180].
According to the functional alteration responsible for the pathological symptoms, PSs can be classified as associated to DNA repair defects, to alterations of the nuclear envelope or telomere metabolism. This implies that the pathological mechanisms leading to premature aging are different, even though genomic instability is a common feature to all PSs.
5.1.1. Progeroid Syndromes Resulting from DNA Repair Alterations
It is a consolidated notion that DNA repair defects lead to genome instability and to age-related pathological conditions, such as cancer, neurodegeneration, and decline of organ functionality. In humans, five genes encode the RNAQ helicases, a family of highly conserved proteins capable of hydrolyzing ATP to unwind double-stranded DNA during DNA replication, recombination, repair, and transcription [181]. Mutations in three of the RECQL genes, namely RECQL2/WRN, RECQL3/BLM and RECQL4, are responsible for the progeroid Werner (WS), Bloom (BS) and Rothmund–Thomson (RTS) syndromes, respectively. Concerning skin manifestations, all of them are associated with accelerated-aging features and cancer predisposition. In detail, WS patients reveal an increased tendency to melanoma formation, skin tightness, and ulcerations on ankles and elbows; BS is characterized by cancer proneness, UV-hypersensitivity leading to skin lesions, hypo and hyperpigmented area whereas RTS patients display poikiloderma, sparse grey hair, and increased susceptibility to cutaneous epithelial neoplasms, such as squamous and basal cell carcinomas [180].
Cocakyne (CS) syndrome, xeroderma pigmentosum (XP), and trichothiodystrophy (TTD) are caused by mutations in genes implicated in nucleotide excision repair (NER), the only pathway in human cells that removes the bulky lesions distorting the DNA double helix. Major targets of the NER pathway are the DNA photolesions (CPDs and 6-4PPs), caused by UV irradiation. Eleven different NER genes with different enzymatic activities are responsible for the XP, TTD, and CS clinical features, or the very rare cases showing a combination of XP and CS (XPCS) or XP and TTD (XPTTD) clinical features. XP is characterized by unusual skin photosensitivity, marked freckle-like pigmentation in the sun-exposed area, high skin cancer proneness and, in some cases, neurodegeneration, whereas CS and TTD are multisystemic disorders showing growth and mental delay, progressive functional deterioration, cutaneous photosensitivity but no skin cancer susceptibility despite the persistence of UV-induced DNA damage. In addition, TTD patients are characterized by brittle hair, and dry and scaly skin (ichthyosis) [182]. Accumulation of unrepaired DNA lesions in the epidermal stem or progenitor cells may explain the high skin cancer risk in XP patients. Indeed, the retroviral-mediated expression of wild-type XPC gene in primary keratinocytes from XP patients with mutations in XPC is, per se sufficient to restore the DNA repair capacity as well as to regenerate a fully differentiated epidermis [183]. Nevertheless, the same NER defects and mutation accumulation are observed in epidermal stem cells from TTD or CS patients, who do not develop skin cancer. The observation that many NER proteins are not only DNA repair factors but also key players in transcription provides a rationale to explain the distinct clinical features of NER disorders [182,184].
Mutations in 21 different genes (FANC genes) encoding proteins implicated in repairing interstrand cross-linked DNA give rise to Fanconi anaemia (FA) characterized by progressive bone marrow failure, short stature, skeletal and skin abnormalities, and high risk of hematopoietic and epithelial malignancies. Cells from FA patients are hypersensitive to DNA-cross-linking agents, which cause chromosomal breakage and genomic instability. Until recently, it was thought that all FA features were due to the accumulation of unrepaired DNA lesions in hematopoietic stem cells (HSCs). The discovery that FA proteins are involved in additional non-canonical biochemical functions, such as signaling pathways engaged in the cellular response to oxidative stress, viral infection or inflammation, has opened the possibility that additional failures contribute to FA pathogenesis [185,186].
The progeroid ataxia telangiectasia (AT) and Seckel syndrome (SS) result from mutations in genes encoding two related phospoinositol-3 kinases (PIKKs), the AT Mutated protein (ATM) and the AT and Rad3-related protein (ATR), respectively. Whereas the serine/threonine kinase activity of ATM is activated by the appearance of DSBs in the human genome, ATR responds to a wide range of lesions interfering with DNA replication [187,188]. Mutations leading to the inactivation of the ATM kinase result in early childhood ataxia (caused by progressive cerebellar neurodegeneration), telangiectasia (usually in the eyes), immune dysfunction, sterility, and cancer proneness. At the level of the skin, AT patients display pigmentary abnormalities and hair greying. Differently, loss of ATR function in SS patients is characterized by severe intrauterine growth retardation, neurodevelopmental abnormalities, dwarfism, and microcephaly. The cellular activation of either ATM or ATR leads to the phosphorylation of specific substrates acting as effectors of the DNA damage response (DDR) signaling pathway, which involves a broad spectrum of cellular processes relevant for genomic stability. Alterations of ATM effectors are observed in patients affected by other progeroid disorders: The AT-like disorder (ATLD) caused by mutations in the hMRE11 gene and the Nijmegen breakage (NBN) syndrome due to mutations in Nbs1 gene. Whereas ATLD appears as a partial phenocopy of the AT clinical features, NBS is characterized by growth retardation, microcephaly and cancer proneness. Notably, Mre11 and Nbs1 proteins are subunits of the same Mre11/Rad50/Nbs1protein complex [189,190].
Even though defects in the DNA repair pathways that account for the progressive aging features of PSs derive from different factors, they all flow into the common feature of chromosomal alterations and genomic instability. These events likely impair the expression of essential genes and result in metabolic dysfunctions, proteostasis failure, and impaired tissue homeostasis. Nevertheless, the molecular mechanisms linking DNA repair defects to aging is still elusive.
5.1.2. Progeroid Syndromes Associated to Alterations of the Nuclear Architecture
There is a group of segmental PSs that are generally referred to as laminopathies because they result from alterations in components of the nuclear envelope. The Hutchinson-Gilford progeria (HGPS), the atypical progeria (APS), the atypical Werner (a-WS) syndromes, and the mandibuloacral dysplasia with type A (MADA) are all caused by mutations in the LMNA gene. By alternative splicing the LMNA gene encodes lamins A and C proteins, which are intermediate filament components of the nuclear lamina [191]. Several post-translation modifications of the prelamin A polypeptide are required for the generation of a mature lamin A. This includes the farnesylation of the cysteine residue in the C-terminal domain CaaX of the protein, cleavage of the aaX peptide by the zinc metallopeptidase STE24 (ZMPSTE24), carboxyl-methylation of the farnesylated cysteine, and excision of the most 15 terminal residues by ZMPSTE24 [192]. In about 80% of HGPS patients, a mutation in LMNA gene causing the activation of a cryptic donor splice site results in a truncated isoform of prelamin A, known as progerin, which is farnesylated but not proteolytically cleaved by the metalloproteinase. The accumulation of progerin at the nuclear envelope is the primary cause of nuclear alterations in HGPS patients [193]. It is worth noting, progerin has also been found in some skin fibroblasts of young adults and in larger subsets of dermal fibroblasts, and in few terminally differentiated keratinocytes of older people [174]. Moreover, alterations of the lamin A maturation process also give rise to premature aging features as attested by the restrictive dermopathy (RD) and the mandibuloacral dysplasia with type B lipodystrophy (MADB), which arise from mutations in the ZMPSTE24 gene. Finally, mutations in the BANF1 gene give rise to the Néstor–Guillermo progeria syndrome (NGPS). The barrier to autointegration factor (BAF) encoded by the BANF1 gene interacts with lamin A during the process of mitotic and post-mitotic nuclear assembly and was shown to influence the chromatin organization and gene expression regulation [194].
Besides the abnormal processing of lamin A, all progeroid laminopathies are characterized by altered nuclear shape (known as “nuclear bleb”) and persistence of DNA damage, which are associated with abnormal patterns of epigenetic modifications, altered recruitment of DNA repair proteins to the site of damage as well as unrepaired double-strand breaks and oxidative DNA lesions by intracellular ROS. The diversity of laminopathies is rather striking, even though they share common features (i.e., premature aging, axonal neuropathy, lipoatrophy, and myopathy). Skin alterations are observed in some of the LMNA-related disorders. In particular, hyperkeratosis of the epidermis, thinning of the dermis, and severe reduction of the elastic fibers are the cause of rigid and tight skin in RD patients. Moreover, the first signs of HGPS are skin alterations, including patches of hyper or hypopigmentation and sclerodermatitis, which are progressively associated with alopecia and skin wrinkling, reminiscent of the normal aging process [195].
Even though the nuclear envelop defects of laminopathies are not found in the PSs associated to DNA repair alterations, all these disorders share many clinical/cellular features, indicating that the molecular events causing the progeroid pathophysiological manifestations are still largely unknown.
5.1.3. PSs Associated to Alterations of Telomere Metabolism
The lack of the RNA-dependent DNA polymerase telomerase in most mammalian somatic cells results in the progressive shortening of telomeric sequences at the end of chromosomes and leads to a permanent cell cycle arrest, known as replicative senescence. Thus, attrition of telomeric sequence length represents an aging marker [196]. The progeroid disorders dyskeratosis congenita (DKC) and Hoyeraal–Hreidarsson syndrome (HHS) are monogenic disorders caused by mutations in genes encoding components of the telomerase complex. In particular, DKC is caused by defects in TERC, TERT or WRAP53 genes that are engaged in telomere replication or by mutations in CTC1 gene involved in telomere maintenance. HHS is caused by defects in RTEL1 or ACD genes implicated in telomere-length regulation. Patients with DKC present skin defects, bone marrow failure, cancer predisposition, premature hair greying, and osteoporosis. Nowadays, HHS is considered as a severe form of DKC, which manifest early in life and is associated with additional symptoms usually not reported in DKC [197].
Notably, the lack of the WRN helicase in WS cells, as well as the accumulation of progerin in HGPS primary dermal fibroblasts, result in telomere attrition. This event can be counteracted by the ectopic expression of the catalytic subunit of the telomerase (hTERT), which results in HGPS immortalization, even if with a lower efficiency compared to the dermal fibroblasts from healthy donors [198]. Similar findings and an increased cellular lifespan were achieved by the hTERT retroviral transduction of human fibroblasts over-expressing progerin [175].
Therefore, investigations on PSs suggest a close relationship between DNA repair defects, nuclear envelope defects and telomere attrition. Even though triggered by different functional failure, physiological and premature aging share several molecular hallmarks among which are genomic instability, telomere shortening, loss of proteostasis, mitochondrial dysfunction, epigenetic alterations, deregulated nutrient sensing, stem cell exhaustion, cellular senescence and altered intercellular communication.
5.2. Cellular Senescence in Premature Aging Diseases
The decline in DNA repair ability, the increase of oxidative stress, the shortening of the telomeres, the production of progerin or the loss of lamin B1 may drive cells towards senescence [199]. Similarly, skin cells from PS patients frequently show premature cellular senescence characterized by the expression of senescence-associated markers, such as loss of lamin B1, increased levels of p16INK4a, the presence of heterochromatin and γ-H2AX foci, senescence-associated secretory phenotype, impaired redox balance, and mitochondrial dysfunction [11,178,200,201].
Primary keratinocytes obtained from CS, TTD, and XPC patients exhibit senescent features in concomitance with high p16 and low p63 levels, similar to aged keratinocytes. These cells undergo a sudden clonal conversion that limits their life-span in culture [11,201]. Notably, the retroviral-mediated expression of wild-type CSA gene shows that CSA protein plays a relevant role in protecting keratinocytes from senescence by facilitating DNA damage processing, maintaining physiological redox status and keratinocyte clonogenic ability and reducing the senescence-associated secretory phenotype [85]. CS fibroblasts are characterized by increased levels of ROS, intense glycolytic metabolism and mitochondria abnormalities [202,203,204]. Moreover, these cells display an increased and persisting high levels of γ-H2AX foci, which co-localize with 53BP1 following infrared radiation (IR) exposure [200]. The loss of CSB in fibroblasts leads to activation of the p53/p21 pathway and expression of inflammatory genes [205]. Furthermore, primary dermal fibroblasts from TTD patients are characterized by the over-expression of MMP-1 leading to the partial loss of dermal interstitial collagens and wound healing defects, in agreement with the progeroid aging features of this disorder [206].
Fibroblasts from an HGPS mouse model present an up-regulation of p53-target genes. Notably, inhibition of p53 protein by the human papillomavirus E6 oncoprotein or the overexpression of hTERT can revert the proliferative defects associated with progerin expression [207,208]. The causative role of progerin in telomere shortening is still not fully elucidated. It has been observed that more than half of the cell telomeres localize to the nuclear lamina where they interact with the lamina-associated polypeptide-α (LAP2α). In HGPS cells the LAP2α interaction with telomeres is impaired, thus, suggesting a direct role of progerin in telomere structure and maintenance [209]. Indeed, the overexpression of wild-type lamin A leads to accelerated telomere loss and reduced lifespan [210]. Furthermore, progerin overexpression in human mesenchymal stem cells results in sporadic differentiation and premature exhaustion of the stem cell pool, thus, pointing to premature aging of the skin dermal layer and vasculature that relays on the Notch signaling pathways [211]. Similarly, the progeroid Zmpste24-knockout mouse model, which fails to process the lamin A protein, reveals an increased number of epidermal stem cells with a decreased proliferative activity in the hair follicle [212]. Overall, these finding suggest the idea that increased progerin levels in the skin of progeroid patients, as well as naturally aged individuals, may affect stem cell maintenance and directly impair skin regeneration.
5.3. Epigenetic Changes in Premature Aging Diseases
Several lines of evidence support the idea that the molecular mechanisms leading to progeroid features are also implicated in the normal aging processes. Epigenetic changes affecting the status of the chromatin were shown to mark the age-related decline of progeroid cells (Figure 2) [29]. Indeed, chromatin disorganization is one of the major alterations associated to the impairment of prelamin A processing, as shown by cells from patients with HGPS and NGPS [213]. A large fraction (about 70%) of primary fibroblasts from HGPS patients show aberrant nuclear morphology, reduced levels of the heterochromatin protein HP1α, which acts as adaptor between the nuclear lamina and the chromatin, reduced levels of multiple members of the lamina-associated polypeptides (LAP2) as well as reduced levels of the heterochromatin-specific marker H3K9me3 [214]. A similar pattern of reductions is observed in about 61% of fibroblasts from normally aged individuals, with an age ranging between 81 and 96 years [208], strongly supporting the concept that progeroid disorders mimic the natural aging events. A global loss of H3K9me3 and changes in heterochromatin architecture have also been observed in human embryonic stem cells lacking the WRN protein functionality and induced to differentiate into mesoderm lineages. A similar decrease of heterochromatin marks and WRN protein are detected in the mesenchymal stem cells isolated from normally aged individuals [215].
Besides H3K9me3, progerin expression in HGPS cells affects the epigenetic control of facultative and constitutive heterochromatin, as shown by the reduced levels of H3K27me3 in the inactive X chromosome of HGPS females as well as by the up-regulation of H4K20me3, a marker for constitutive heterochromatin [216]. Both events have been found associated with natural cellular senescence [217,218].
Recent findings demonstrate an altered DNA replication timing in HGPS and RTS cells. The temporal order of genome duplication in cells is strictly associated with the chromatin organization and epigenetic marks, which are modulated during development. HGPS and RTS cells reveal a progeroid-specific signature of DNA replication timing, which is associated with the altered ratio of TP63 isoform expression and resembling that of cellular senescence defects [219].
Altered epigenetic marks have also been observed in NER-defective cells [220]. Indeed, additional functions for the NER proteins XPC, XPA, RPA, XPG, and XPF have been demonstrated outside the DNA repair pathway. Even in the absence of a genotoxic attack, the recruitment of these NER factors at the promoter of active genes is necessary to achieve DNA methylation as well as histone post-translation modifications including methylation (H3K4 and H3K9) and acetylation (H3K9/14) [220,221]. Moreover, XPG and XPF are essential for the DNA demethylation and the formation of a CTCF-dependent chromatin looping between the promoter and the terminator of the active RARβ2 gene [222].
Among the NER factors, the CS causative protein CSB is an SWI/SNF-like DNA-dependent ATPase that exhibits ATP-dependent chromatin remodeling activity in vitro. CSB disrupts the regulation of many genes involved in human aging, such as those genes affected by inhibitors of histone deacetylase and DNA methylation, as well as by defects in PARP-1 function and RNA polymerase II elongation [177]. Mouse and human CSB deficient cells display PARP-1 hyperactivation that leads to SIRT1 attenuation and, in turn, drives mitochondrial dysfunction and pro-inflammatory profile. PARP inhibition or NAD+ replenishment rescue the CS associated phenotypes in human cells and mice [176]. CSB modulates p53 activity and competes with p300 for p53 binding. The absence of CSB in patient fibroblasts induces the binding of p53 to p300 causing the stabilization of p53 and the activation of its target genes [179].
Moreover, an active heterochromatinization process following UV irradiation has been observed in NER-defective cells from progeroid patients presenting a combination of XP and CS clinical features. The heterochromatin alterations in these cells are due to the action of SIRT1 and results in wide transcription repression [178]. Since the generation of mouse models deficient for or over-expressing specific genes of interests, it has become well accepted that the SIRT family protects from age-related features and extends healthy life. Mice deficient for SIRT6 exhibit progeroid symptoms. Moreover, SIRT6 prevents telomere dysfunction by deacetylating H3K9 at the telomeric loci of human cells. Because SIRT6 is a chromatin-associated protein that promotes resistance to DNA damage and suppresses genomic instability in mouse cells, these findings link once more DNA repair pathways to the aging processes [116]. Nevertheless, SIRT6 also interacts with the stress-responsive transcription factor NF-κB and regulates the expression of some dependent NF-κB target genes, opening the possibility that the role of SIRT6 in aging may be ascribed to its ability to regulate the expression of aging-related genes including those implicated in cell survival, senescence, inflammation and immunity [223].
Primary keratinocytes obtained from TTD and XPC patients display Bmi-1 downregulation in concomitance with early expression of p16INK4a and SC senescence. Notably, Bmi-1 expression restores keratinocyte clonogenic potential and delays cell senescence by modulating p16INK4a transcription [11].
Finally, evidence for heterochromatin instability has also been reported in cells from patients affected by FA [224]. Even though epigenetic changes have not yet been identified in all progeroid syndromes, it is plausible to hypothesize that chromatin alteration associated with epigenetic and transcriptional deregulations typify progeroid features.
6. Conclusions and Perspectives
Epigenetic code and chromatin status are strictly interconnected and exhibit their effects on cell proliferation and differentiation by regulating the gene expression profile of every single cell. During normal skin homeostasis and tissue renewal, epigenetic mechanisms govern the decision between epidermal SC self-renewal and differentiation toward the fully differentiated keratinocytes.
By regulating the expression of several chromatin remodelers and histone modifying enzymes, the transcription factor p63 is a key player in this process. Furthermore, epigenetic modifications maintain cellular stemness by tightly regulating the expression of the tumor suppressor p16INK4a, which orchestrates the cell cycle exit and senescence response. Indeed, epigenetic mechanisms also specify cell senescence and, therefore, delineate the naturally occurring aging process of both epidermal cells as well as dermal fibroblasts. Some of these aging-related epigenetic marks have been found in the skin cells of young individuals, who are affected by premature aging disorders. This finding emphasizes the relevance of PSs for the identification of longevity pathways and potential therapeutic approaches for aging. Most of the progeroid disorders are characterized by genome instability and high cancer susceptibility. Therefore, investigations on PSs will hopefully lead to the identification of novel therapeutic approaches relevant to aging and carcinogenesis. The easy accessibility of the skin makes this tissue the favorite model system to investigate the molecular alterations and dysfunctions of aging-associated disorders.
Since epigenetic modifications are reversible, several studies point to the potential of epigenetic therapies as a tool to reprogram cell fate. Epigenetic therapies are successfully applied for the treatment of some hematological malignancies. However, a deeper knowledge and further studies on skin epigenetic mechanisms are required to achieve the realization of epigenetic therapeutic approaches directed to alleviate the signs of aging in the human population.
Author Contributions
D.O. and E.D. conceived the ideas, wrote and edited the manuscript.
Funding
This research was funded by grants from the Italian Ministry of Health (Ricerca corrente) (E.D.) and the Associazione Italiana per la Ricerca sul Cancro Grant IG 17710 (D.O.).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Murphree, R.W. Impairments in Skin Integrity. Nurs. Clin. N. Am. 2017, 52, 405–417. [Google Scholar] [CrossRef] [PubMed]
- Hsu, Y.; Li, L.; Fuchs, E. Emerging interactions between skin stem cells and their niches. Nat. Med. 2014, 20, 847–856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrandon, Y.; Grasset, N.; Zaffalon, A.; Gorostidi, F.; Claudinot, S.; Droz-Georget, S.L.; Nanba, D.; Rochat, A. Capturing epidermal stemness for regenerative medicine. Semin. Cell Dev. Biol. 2012, 23, 937–944. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, T.; Rothoeft, T.; Teig, N.; Bauer, J.W.; Pellegrini, G.; De Rosa, L.; Scaglione, D.; Reichelt, J.; Klausegger, A.; Kneisz, D.; et al. Regeneration of the entire human epidermis using transgenic stem cells. Nature 2017, 551, 327–332. [Google Scholar] [CrossRef] [PubMed]
- De Rosa, L.; De Luca, M. Cell biology: Dormant and restless skin stem cells. Nature 2012, 489, 215–217. [Google Scholar] [CrossRef] [PubMed]
- Arseni, L.; Lombardi, A.; Orioli, D. From Structure to Phenotype: Impact of Collagen Alterations on Human Health. Int. J. Mol. Sci. 2018, 19. [Google Scholar] [CrossRef] [PubMed]
- Sriram, G.; Bigliardi, P.L.; Bigliardi-Qi, M. Fibroblast heterogeneity and its implications for engineering organotypic skin models in vitro. Eur. J. Cell Biol. 2015, 94, 483–512. [Google Scholar] [CrossRef] [PubMed]
- Driskell, R.R.; Lichtenberger, B.M.; Hoste, E.; Kretzschmar, K.; Simons, B.D.; Charalambous, M.; Ferron, S.R.; Herault, Y.; Pavlovic, G.; Ferguson-Smith, A.C.; et al. Distinct fibroblast lineages determine dermal architecture in skin development and repair. Nature 2013, 504, 277–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eckert, R.L.; Adhikary, G.; Rorke, E.A.; Chew, Y.C.; Balasubramanian, S. Polycomb Group Proteins Are Key Regulators of Keratinocyte Function. J. Investig. Dermatol. 2011, 131, 295–301. [Google Scholar] [CrossRef] [Green Version]
- Maurelli, R.; Zambruno, G.; Guerra, L.; Abbruzzese, C.; Dimri, G.; Gellini, M.; Bondanza, S.; Dellambra, E. Inactivation of p16 INK4a ( inhibitor of cyclin-dependent kinase 4A ) immortalizes primary human keratinocytes by maintaining cells in the stem cell compartment. FASEB J. 2006, 20, 1516–1518. [Google Scholar] [CrossRef]
- Cordisco, S.; Maurelli, R.; Bondanza, S.; Stefanini, M.; Zambruno, G.; Guerra, L.; Dellambra, E. Bmi-1 reduction plays a key role in physiological and premature aging of primary human keratinocytes. J. Investig. Dermatol. 2010, 130, 1048–1062. [Google Scholar] [CrossRef] [PubMed]
- Rivetti di Val Cervo, P.; Lena, A.M.; Nicoloso, M.; Rossi, S.; Mancini, M.; Zhou, H.; Saintigny, G.; Dellambra, E.; Odorisio, T.; Mahé, C.; et al. p63-microRNA feedback in keratinocyte senescence. Proc. Natl. Acad. Sci. USA 2012, 109, 1133–1138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huschtscha, L.I. p16INK4a and the control of cellular proliferative life span. Carcinogenesis 1999, 20, 921–926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharpless, N.; DePinho, R. Telomeres, stem cells, senescence, and cancer. J. Clin. Investig. 2004, 113. [Google Scholar] [CrossRef] [PubMed]
- Iglesias-Bartolome, R.; Callejas-Valera, J.L.; Gutkind, J.S. Control of the epithelial stem cell epigenome: The shaping of epithelial stem cell identity. Curr. Opin. Cell Biol. 2013, 25, 162–169. [Google Scholar] [CrossRef] [PubMed]
- D’Arcangelo, D.; Tinaburri, L.; Dellambra, E. The Role of p16INK4a Pathway in Human Epidermal Stem Cell Self-Renewal, Aging and Cancer. Int. J. Mol. Sci. 2017, 18, 1591. [Google Scholar] [CrossRef] [PubMed]
- Mills, A.A.; Zheng, B.; Wang, X.J.; Vogel, H.; Roop, D.R.; Bradley, A. p63 is a p53 homologue required for limb and epidermal morphogenesis. Nature 1999, 398, 708–713. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.; Schweitzer, R.; Sun, D.; Kaghad, M.; Walker, N.; Bronson, R.T.; Tabin, C.; Sharpe, A.; Caput, D.; Crum, C.; et al. p63 is essential for regenerative proliferation in limb, craniofacial and epithelial development. Nature 1999, 398, 714–718. [Google Scholar] [CrossRef]
- Missero, C.; Antonini, D. Crosstalk among p53 family members in cutaneous carcinoma. Exp. Dermatol. 2014, 23, 143–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pellegrini, G.; Dellambra, E.; Golisano, O.; Martinelli, E.; Fantozzi, I.; Bondanza, S.; Ponzin, D.; McKeon, F.; De Luca, M. p63 identifies keratinocyte stem cells. Proc. Natl. Acad. Sci. USA 2001, 98, 3156–3161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Candi, E.; Amelio, I.; Agostini, M.; Melino, G. MicroRNAs and p63 in epithelial stemness. Cell Death Differ. 2015, 22, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Ellisen, L.W.; Ramsayer, K.D.; Johannessen, C.M.; Yang, A.; Beppu, H.; Minda, K.; Oliner, J.D.; McKeon, F.; Haber, D.A. REDD1, a developmentally regulated transcriptional target of p63 and p53, links p63 to regulation of reactive oxygen species. Mol. Cell 2002, 10, 995–1005. [Google Scholar] [CrossRef]
- Truong, A.B.; Kretz, M.; Ridky, T.W.; Kimmel, R.; Khavari, P.A. p63 regulates proliferation and differentiation of developmentally mature keratinocytes. Genes Dev. 2006, 20, 3185–3197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Botchkarev, V.A.; Gdula, M.R.; Mardaryev, A.N.; Sharov, A.A.; Fessing, M.Y. Epigenetic Regulation of Gene Expression in Keratinocytes. J. Investig. Dermatol. 2012, 132, 2505–2521. [Google Scholar] [CrossRef] [PubMed]
- Frye, M.; Benitah, S.A. Chromatin regulators in mammalian epidermis. Semin. Cell Dev. Biol. 2012, 23, 897–905. [Google Scholar] [CrossRef] [PubMed]
- Perdigoto, C.N.; Valdes, V.J.; Bardot, E.S.; Ezhkova, E. Epigenetic Regulation of Epidermal Differentiation. Cold Spring Harb. Perspect. Med. 2014, 4, a015263. [Google Scholar] [CrossRef]
- Adam, R.C.; Fuchs, E. The Yin and Yang of Chromatin Dynamics In Stem Cell Fate Selection. Trends Genet. 2016. [CrossRef] [PubMed]
- Avgustinova, A.; Benitah, S.A. Epigenetic control of adult stem cell function. Nat. Rev. Mol. Cell Biol. 2016, 17, 643–658. [Google Scholar] [CrossRef]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef]
- Muñoz-Espín, D.; Serrano, M. Cellular senescence: From physiology to pathology. Nat. Rev. Mol. Cell Biol. 2014, 15, 482–496. [Google Scholar] [CrossRef]
- van Deursen, J.M. The role of senescent cells in ageing. Nature 2014, 509, 439–446. [Google Scholar] [CrossRef] [Green Version]
- Krishnamurthy, J.; Torrice, C.; Ramsey, M.R.; Kovalev, G.I.; Al-Regaiey, K.; Su, L.; Sharpless, N.E. Ink4a/Arf expression is a biomarker of aging. J. Clin. Investig. 2004, 114, 1299–1307. [Google Scholar] [CrossRef]
- Baker, D.J.; Wijshake, T.; Tchkonia, T.; LeBrasseur, N.K.; Childs, B.G.; van de Sluis, B.; Kirkland, J.L.; van Deursen, J.M. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 2011, 479, 232–236. [Google Scholar] [CrossRef] [Green Version]
- Keyes, W.M.; Wu, Y.; Vogel, H.; Guo, X.; Lowe, S.W.; Mills, A.A. p63 deficiency activates a program of cellular senescence and leads to accelerated aging. Genes Dev. 2005, 19, 1986–1999. [Google Scholar] [CrossRef] [Green Version]
- Fisher, G.J.; Kang, S.; Varani, J.; Bata-Csorgo, Z.; Wan, Y.; Datta, S.; Voorhees, J.J. Mechanisms of photoaging and chronological skin aging. Arch. Dermatol. 2002, 138, 1462–1470. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Park, H.J. Molecular Mechanisms of Skin Aging and Rejuvenation. In Molecular Mechanisms of the Aging Process and Rejuvenation; InTech: Manila, Philippines, 2016. [Google Scholar] [Green Version]
- Kang, S.; Fisher, G.J.; Voorhees, J.J. Photoaging: Pathogenesis, prevention, and treatment. Clin. Geriatr. Med. 2001, 17, 643–659. [Google Scholar] [CrossRef]
- Fuchs, E. Epithelial Skin Biology. In Current Topics in Developmental Biology; Elsevier: Amsterdam, The Netherlands, 2016; pp. 357–374. [Google Scholar]
- Sharpless, N.E.; DePinho, R.A. How stem cells age and why this makes us grow old. Nat. Rev. Mol. Cell Biol. 2007, 8, 703–713. [Google Scholar] [CrossRef] [PubMed]
- d’Adda di Fagagna, F.; Reaper, P.M.; Clay-Farrace, L.; Fiegler, H.; Carr, P.; Von Zglinicki, T.; Saretzki, G.; Carter, N.P.; Jackson, S.P. A DNA damage checkpoint response in telomere-initiated senescence. Nature 2003, 426, 194–198. [Google Scholar] [CrossRef]
- Ressler, S.; Bartkova, J.; Niederegger, H.; Bartek, J.; Scharffetter-Kochanek, K.; Jansen-Dürr, P.; Wlaschek, M. p16INK4A is a robust in vivo biomarker of cellular aging in human skin. Aging Cell 2006, 5, 379–389. [Google Scholar] [CrossRef]
- Tigges, J.; Krutmann, J.; Fritsche, E.; Haendeler, J.; Schaal, H.; Fischer, J.W.; Kalfalah, F.; Reinke, H.; Reifenberger, G.; Stühler, K.; et al. The hallmarks of fibroblast ageing. Mech. Ageing Dev. 2014, 138, 26–44. [Google Scholar] [CrossRef]
- Krutmann, J.; Bouloc, A.; Sore, G.; Bernard, B.A.; Passeron, T. The skin aging exposome. J. Dermatol. Sci. 2017, 85, 152–161. [Google Scholar] [CrossRef] [PubMed]
- Narita, M.; Nũnez, S.; Heard, E.; Narita, M.; Lin, A.W.; Hearn, S.A.; Spector, D.L.; Hannon, G.J.; Lowe, S.W. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell 2003, 113, 703–716. [Google Scholar] [CrossRef]
- Herbig, U.; Sedivy, J.M. Regulation of growth arrest in senescence: Telomere damage is not the end of the story. Mech. Ageing Dev. 2006, 127, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Rodier, F.; Campisi, J. Four faces of cellular senescence. J. Cell Biol. 2011, 192, 547–556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quan, T.; Qin, Z.; Voorhees, J.J.; Fisher, G.J. Cysteine-rich protein 61 (CCN1) mediates replicative senescence-associated aberrant collagen homeostasis in human skin fibroblasts. J. Cell. Biochem. 2012, 113, 3011–3018. [Google Scholar] [CrossRef] [PubMed]
- Yaar, M.; Gilchrest, B.A. Photoageing: Mechanism, prevention and therapy. Br. J. Dermatol. 2007, 157, 874–877. [Google Scholar] [CrossRef] [PubMed]
- Imokawa, G.; Ishida, K. Biological mechanisms underlying the ultraviolet radiation-induced formation of skin wrinkling and sagging I: Reduced skin elasticity, highly associated with enhanced dermal elastase activity, triggers wrinkling and sagging. Int. J. Mol. Sci. 2015, 16, 7753–7775. [Google Scholar] [CrossRef]
- Bernerd, F.; Vioux, C.; Asselineau, D. Evaluation of the Protective Effect of Sunscreens on In Vitro Reconstructed Human Skin Exposed to UVB or UVA Irradiation. Photochem. Photobiol. 2000, 71, 314. [Google Scholar] [CrossRef]
- Bernerd, F.; Asselineau, D. An organotypic model of skin to study photodamage and photoprotection in vitro. J. Am. Acad. Dermatol. 2008, 58, 8–12. [Google Scholar] [CrossRef]
- Duval, C.; Cohen, C.; Chagnoleau, C.; Flouret, V.; Bourreau, E.; Bernerd, F. Key Regulatory Role of Dermal Fibroblasts in Pigmentation as Demonstrated Using a Reconstructed Skin Model: Impact of Photo-Aging. PLoS ONE 2014, 9, e114182. [Google Scholar] [CrossRef]
- Fagot, D.; Asselineau, D.; Bernerd, F. Matrix metalloproteinase-1 production observed after solar-simulated radiation exposure is assumed by dermal fibroblasts but involves a paracrine activation through epidermal keratinocytes. Photochem. Photobiol. 2004, 79, 499–505. [Google Scholar] [CrossRef] [PubMed]
- Bernerd, F.; Marionnet, C.; Duval, C. Solar ultraviolet radiation induces biological alterations in human skin in vitro: Relevance of a well-balanced UVA/UVB protection. Indian J. Dermatol. Venereol. Leprol. 2012, 78, 152. [Google Scholar] [CrossRef] [PubMed]
- D’Errico, M.; Lemma, T.; Calcagnile, A.; Proietti De Santis, L.; Dogliotti, E. Cell type and DNA damage specific response of human skin cells to environmental agents. Mutat. Res. 2007, 614, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Sidler, C.; Kovalchuk, O.; Kovalchuk, I. Epigenetic Regulation of Cellular Senescence and Aging. Front. Genet. 2017, 8, 138. [Google Scholar] [CrossRef] [PubMed]
- Rajarajacholan, U.K.; Riabowol, K. Aging with ING: A comparative study of different forms of stress induced premature senescence. Oncotarget 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Rossetto, D.; Truman, A.W.; Kron, S.J.; Cote, J. Epigenetic Modifications in Double-Strand Break DNA Damage Signaling and Repair. Clin. Cancer Res. 2010, 16, 4543–4552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Contrepois, K.; Coudereau, C.; Benayoun, B.A.; Schuler, N.; Roux, P.-F.; Bischof, O.; Courbeyrette, R.; Carvalho, C.; Thuret, J.-Y.; Ma, Z.; et al. Histone variant H2A.J accumulates in senescent cells and promotes inflammatory gene expression. Nat. Commun. 2017, 8, 14995. [Google Scholar] [CrossRef] [Green Version]
- Rishi, V.; Bhattacharya, P.; Chatterjee, R.; Rozenberg, J.; Zhao, J.; Glass, K.; Fitzgerald, P.; Vinson, C. CpG methylation of half-CRE sequences creates C/EBP binding sites that activate some tissue-specific genes. Proc. Natl. Acad. Sci. USA 2010, 107, 20311–20316. [Google Scholar] [CrossRef] [Green Version]
- Rinaldi, L.; Datta, D.; Serrat, J.; Morey, L.; Solanas, G.; Avgustinova, A.; Blanco, E.; Pons, J.I.; Matallanas, D.; Von Kriegsheim, A.; et al. Dnmt3a and Dnmt3b Associate with Enhancers to Regulate Human Epidermal Stem Cell Homeostasis. Cell Stem Cell 2016, 19, 491–501. [Google Scholar] [CrossRef] [Green Version]
- Zampieri, M.; Ciccarone, F.; Calabrese, R.; Franceschi, C.; Bürkle, A.; Caiafa, P. Reconfiguration of DNA methylation in aging. Mech. Ageing Dev. 2015, 151, 60–70. [Google Scholar] [CrossRef]
- Feng, S.; Jacobsen, S.E.; Reik, W. Epigenetic Reprogramming in Plant and Animal Development. Science 2010, 330, 622–627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sen, G.L.; Reuter, J.A.; Webster, D.E.; Zhu, L.; Khavari, P.A. DNMT1 maintains progenitor function in self-renewing somatic tissue. Nature 2010, 463, 563–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bock, C.; Beerman, I.; Lien, W.H.; Smith, Z.D.; Gu, H.; Boyle, P.; Gnirke, A.; Fuchs, E.; Rossi, D.J.; Meissner, A. DNA Methylation Dynamics during In Vivo Differentiation of Blood and Skin Stem Cells. Mol. Cell 2012, 47, 633–647. [Google Scholar] [CrossRef] [PubMed]
- Mulder, K.W.; Wang, X.; Escriu, C.; Ito, Y.; Schwarz, R.F.; Gillis, J.; Sirokmány, G.; Donati, G.; Uribe-Lewis, S.; Pavlidis, P.; et al. Diverse epigenetic strategies interact to control epidermal differentiation. Nat. Cell Biol. 2012, 14, 753–763. [Google Scholar] [CrossRef] [PubMed]
- Teschendorff, A.E.; West, J.; Beck, S. Age-associated epigenetic drift: Implications, and a case of epigenetic thrift? Hum. Mol. Genet. 2013, 22, R7–R15. [Google Scholar] [CrossRef]
- Fraga, M.F.; Ballestar, E.; Paz, M.F.; Ropero, S.; Setien, F.; Ballestar, M.L.; Heine-Suñer, D.; Cigudosa, J.C.; Urioste, M.; Benitez, J.; et al. Epigenetic differences arise during the lifetime of monozygotic twins. Proc. Natl. Acad. Sci. USA 2005, 102, 10604–10609. [Google Scholar] [CrossRef] [PubMed]
- Poulsen, P.; Esteller, M.; Vaag, A.; Fraga, M.F. The epigenetic basis of twin discordance in age-related diseases. Pediatr. Res. 2007, 61, 38R–42R. [Google Scholar] [CrossRef] [PubMed]
- Grönniger, E.; Weber, B.; Heil, O.; Peters, N.; Stäb, F.; Wenck, H.; Korn, B.; Winnefeld, M.; Lyko, F. Aging and chronic sun exposure cause distinct epigenetic changes in human skin. PLoS Genet. 2010, 6, 6. [Google Scholar] [CrossRef]
- Raddatz, G.; Hagemann, S.; Aran, D.; Söhle, J.; Kulkarni, P.P.; Kaderali, L.; Hellman, A.; Winnefeld, M.; Lyko, F. Aging is associated with highly defined epigenetic changes in the human epidermis. Epigenetics Chromatin 2013, 6, 1–12. [Google Scholar] [CrossRef]
- Bormann, F.; Rodríguez-Paredes, M.; Hagemann, S.; Manchanda, H.; Kristof, B.; Gutekunst, J.; Raddatz, G.; Haas, R.; Terstegen, L.; Wenck, H.; et al. Reduced DNA methylation patterning and transcriptional connectivity define human skin aging. Aging Cell 2016, 15, 563–571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koch, C.M.; Suschek, C.V.; Lin, Q.; Bork, S.; Goergens, M.; Joussen, S.; Pallua, N.; Ho, A.D.; Zenke, M.; Wagner, W. Specific Age-Associated DNA Methylation Changes in Human Dermal Fibroblasts. PLoS ONE 2011, 6, e16679. [Google Scholar] [CrossRef] [PubMed]
- Landan, G.; Cohen, N.M.; Mukamel, Z.; Bar, A.; Molchadsky, A.; Brosh, R.; Horn-Saban, S.; Zalcenstein, D.A.; Goldfinger, N.; Zundelevich, A.; et al. Epigenetic polymorphism and the stochastic formation of differentially methylated regions in normal and cancerous tissues. Nat. Genet. 2012, 44, 1207–1214. [Google Scholar] [CrossRef] [PubMed]
- Lowe, R.; Overhoff, M.G.; Ramagopalan, S.V.; Garbe, J.C.; Koh, J.; Stampfer, M.R.; Beach, D.H.; Rakyan, V.K.; Bishop, C.L. The senescent methylome and its relationship with cancer, ageing and germline genetic variation in humans. Genome Biol. 2015, 16, 194. [Google Scholar] [CrossRef] [PubMed]
- Pérez, R.F.; Tejedor, J.R.; Bayón, G.F.; Fernández, A.F.; Fraga, M.F. Distinct chromatin signatures of DNA hypomethylation in aging and cancer. Aging Cell 2018, 17. [Google Scholar] [CrossRef] [PubMed]
- Vandiver, A.R.; Irizarry, R.A.; Hansen, K.D.; Garza, L.A.; Runarsson, A.; Li, X.; Chien, A.L.; Wang, T.S.; Leung, S.G.; Kang, S.; et al. Age and sun exposure-related widespread genomic blocks of hypomethylation in nonmalignant skin. Genome Biol. 2015, 16, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Pastor, W.A.; Shen, Y.; Tahiliani, M.; Liu, D.R.; Rao, A. The Behaviour of 5-Hydroxymethylcytosine in Bisulfite Sequencing. PLoS ONE 2010, 5, e8888. [Google Scholar] [CrossRef] [PubMed]
- Yuan, T.; Jiao, Y.; de Jong, S.; Ophoff, R.A.; Beck, S.; Teschendorff, A.E. An integrative multi-scale analysis of the dynamic DNA methylation landscape in aging. PLoS Genet. 2015, 11, e1004996. [Google Scholar] [CrossRef]
- Li, J.; Jiang, T.-X.; Hughes, M.W.; Wu, P.; Widelitz, R.B.; Fan, G.; Chuong, C. Progressive Alopecia Reveals Decreasing Stem Cell Activation Probability during Aging of Mice with Epidermal Deletion of DNA Methyltransferase 1. J. Investig. Dermatol. 2012, 132, 2681–2690. [Google Scholar] [CrossRef] [Green Version]
- Xie, H.F.; Liu, Y.Z.; Du, R.; Wang, B.; Chen, M.T.; Zhang, Y.Y.; Deng, H.L.; Li, J. Mir-377 induces senescence in human skin fibroblasts by targeting DNA methyltransferase 1. Cell Death Dis. 2017, 8, 1–11. [Google Scholar] [CrossRef]
- Zheng, Q.H.; Ma, L.W.; Zhu, W.G.; Zhang, Z.Y.; Tong, T.J. p21Waf1/Cip1 plays a critical role in modulating senescence through changes of DNA methylation. J. Cell. Biochem. 2006, 98, 1230–1248. [Google Scholar] [CrossRef]
- Jung, H.J.; Byun, H.O.; Jee, B.A.; Min, S.; Jeoun, U.W.; Lee, Y.K.; Seo, Y.; Woo, H.G.; Yoon, G. The ubiquitin-like with PHD and ring finger domains 1 (UHRF1)/DNA methyltransferase 1 (DNMT1) axis is a primary regulator of cell senescence. J. Biol. Chem. 2017, 292, 3729–3739. [Google Scholar] [CrossRef] [PubMed]
- Moulin, L.; Cenizo, V.; Antu, A.N.; André, V.; Pain, S.; Sommer, P.; Debret, R. Methylation of LOXL1 Promoter by DNMT3A in Aged Human Skin Fibroblasts. Rejuvenation Res. 2017, 20, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Yi, Y.; Xie, H.; Xiao, X.; Wang, B.; Du, R.; Liu, Y.; Li, Z.; Wang, J.; Sun, L.; Deng, Z.; et al. Ultraviolet a irradiation induces senescence in human dermal fibroblasts by down-regulating DNMT1 via ZEB1. Aging (Albany NY) 2018, 10, 212–228. [Google Scholar] [CrossRef] [PubMed]
- Lahtz, C.; Kim, S.-I.; Bates, S.E.; Li, A.X.; Wu, X.; Pfeifer, G.P. UVB irradiation does not directly induce detectable changes of DNA methylation in human keratinocytes. F1000Research 2013, 2, 45. [Google Scholar] [CrossRef]
- Wang, D.; Huang, J.H.; Zeng, Q.H.; Gu, C.; Ding, S.; Lu, J.Y.; Chen, J.; Yang, S.B. Increased 5-hydroxymethylcytosine and ten-eleven translocation protein expression in ultraviolet B-irradiated HaCaT cells. Chin. Med. J. 2017, 130, 594–599. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Bardot, E.; Ezhkova, E. Epigenetic regulation of skin: Focus on the Polycomb complex. Cell. Mol. Life Sci. 2012, 69, 2161–2172. [Google Scholar] [CrossRef]
- Gil, J.; O’Loghlen, A. PRC1 complex diversity: Where is it taking us? Trends Cell Biol. 2014, 24, 632–641. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, K.; O’Neil, K.; Capell, B.C. Histone modifiers: Dynamic regulators of the cutaneous transcriptome. J. Dermatol. Sci. 2017, 89, 226–232. [Google Scholar] [CrossRef] [PubMed]
- Blackledge, N.P.; Farcas, A.M.; Kondo, T.; King, H.W.; McGouran, J.F.; Hanssen, L.L.P.; Ito, S.; Cooper, S.; Kondo, K.; Koseki, Y.; et al. Variant PRC1 complex-dependent H2A ubiquitylation drives PRC2 recruitment and polycomb domain formation. Cell 2014, 157, 1445–1459. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, G.; Szabo, V.; Pirity, M.K. Absence of Rybp Compromises Neural Differentiation of Embryonic Stem Cells. Stem Cells Int. 2016, 2016, 4034620. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Tong, H.; Huang, Y.; Yan, Y.; Teng, H.; Xia, Y.; Jiang, Q.; Qin, J. Essential Role for Polycomb Group Protein Pcgf6 in Embryonic Stem Cell Maintenance and a Noncanonical Polycomb Repressive Complex 1 (PRC1) Integrity. J. Biol. Chem. 2017, 292, 2773–2784. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Liu, M.; Ji, H.; Zhu, Y.; Wang, C.; Huang, Y.; Ma, X.; Xing, G.; Xia, Y.; Jiang, Q.; et al. The polycomb group protein Yaf2 regulates the pluripotency of embryonic stem cells in a phosphorylation-dependent manner. J. Biol. Chem. 2018, 293, 12793–12804. [Google Scholar] [CrossRef] [PubMed]
- Sen, G.L.; Webster, D.E.; Barragan, D.I.; Chang, H.Y.; Khavari, P.A. Control of differentiation in a self-renewing mammalian tissue by the histone demethylase JMJD3. Genes Dev. 2008, 22, 1865–1870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dimri, M.; Carroll, J.D.; Cho, J.H.; Dimri, G.P. MicroRNA-141 regulates BMI1 expression and induces senescence in human diploid fibroblasts. Cell Cycle 2013, 12, 3537–3546. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, J.J.; Kieboom, K.; Marino, S.; DePinho, R.A.; van Lohuizen, M. The oncogene and Polycomb-group gene bmi-1 regulates cell proliferation and senescence through the ink4a locus. Nature 1999, 397, 164–168. [Google Scholar] [CrossRef] [PubMed]
- Tschen, S.-I.; Dhawan, S.; Gurlo, T.; Bhushan, A. Age-dependent decline in beta-cell proliferation restricts the capacity of beta-cell regeneration in mice. Diabetes 2009, 58, 1312–1320. [Google Scholar] [CrossRef]
- Schuettengruber, B.; Martinez, A.-M.; Iovino, N.; Cavalli, G. Trithorax group proteins: Switching genes on and keeping them active. Nat. Rev. Mol. Cell Biol. 2011, 12, 799–814. [Google Scholar] [CrossRef]
- Hopkin, A.S.; Gordon, W.; Klein, R.H.; Espitia, F.; Daily, K.; Zeller, M.; Baldi, P.; Andersen, B. GRHL3/GET1 and trithorax group members collaborate to activate the epidermal progenitor differentiation program. PLoS Genet. 2012, 8. [Google Scholar] [CrossRef]
- Kotake, Y.; Zeng, Y.; Xiong, Y. DDB1-CUL4 and MLL1 mediate oncogene-induced p16INK4a activation. Cancer Res. 2009, 69, 1809–1814. [Google Scholar] [CrossRef]
- Wu, Z.; Zhang, L. Polycomb group proteins: Novel molecules associated with ultraviolet A-induced photoaging of human skin. Exp. Ther. Med. 2017, 14, 2554–2562. [Google Scholar] [CrossRef] [Green Version]
- Qin, H.; Zhang, G.; Zhang, L. GSK126 (EZH2 inhibitor) interferes with ultraviolet A radiation-induced photoaging of human skin fibroblast cells. Exp. Ther. Med. 2018, 15, 3439–3448. [Google Scholar] [CrossRef] [PubMed]
- Greussing, R.; Hackl, M.; Charoentong, P.; Pauck, A.; Monteforte, R.; Cavinato, M.; Hofer, E.; Scheideler, M.; Neuhaus, M.; Micutkova, L.; et al. Identification of microRNA-mRNA functional interactions in UVB-induced senescence of human diploid fibroblasts. BMC Genom. 2013, 14, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Gesumaria, L.; Matsui, M.S.; Kluz, T.; Costa, M. Solar-simulated ultraviolet radiation induces histone 3 methylation changes in the gene promoters of matrix metalloproteinases 1 and 3 in primary human dermal fibroblasts. Exp. Dermatol. 2015, 24, 384–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Driskell, I.; Oda, H.; Blanco, S.; Nascimento, E.; Humphreys, P.; Frye, M. The histone methyltransferase Setd8 acts in concert with c-Myc and is required to maintain skin. EMBO J. 2012, 31, 616–629. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Takebayashi, S.-I.; Sakamoto, A.; Igata, T.; Nakatsu, Y.; Saitoh, N.; Hino, S.; Nakao, M. The SETD8/PR-Set7 Methyltransferase Functions as a Barrier to Prevent Senescence-Associated Metabolic Remodeling. Cell Rep. 2017, 18, 2148–2161. [Google Scholar] [CrossRef] [PubMed]
- Restelli, M.; Molinari, E.; Marinari, B.; Conte, D.; Gnesutta, N.; Costanzo, A.; Merlo, G.R.; Guerrini, L. FGF8, c-Abl and p300 participate in a pathway that controls stability and function of the ΔNp63 α protein. Hum. Mol. Genet. 2015, 24, 4185–4197. [Google Scholar] [CrossRef] [PubMed]
- Wong, P.-P.; Pickard, A.; McCance, D.J. p300 Alters Keratinocyte Cell Growth and Differentiation through Regulation of p21Waf1/CIP1. PLoS ONE 2010, 5, e8369. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Poi, M.J.; Tsai, M. Regulatory mechanisms of tumor suppressor P16(INK4A) and their relevance to cancer. Biochemistry 2011, 50, 5566–5582. [Google Scholar] [CrossRef]
- Wang, W.; Pan, K.; Chen, Y.; Huang, C.; Zhang, X. The acetylation of transcription factor HBP1 by p300/CBP enhances p16INK4A expression. Nucleic Acids Res. 2012, 40, 981–995. [Google Scholar] [CrossRef]
- Bin, B.-H.; Lee, S.-H.; Bhin, J.; Irié, T.; Kim, S.; Seo, J.; Mishima, K.; Lee, T.R.; Hwang, D.; Fukada, T.; et al. The epithelial zinc transporter ZIP10 epigenetically regulates human epidermal homeostasis by modulating histone acetyltransferase activity. Br. J. Dermatol. 2018. [Google Scholar] [CrossRef]
- Cheng, H.-H.; Wang, K.-H.; Chu, L.; Chang, T.-C.; Kuo, C.-C.; Wu, K.K. Quiescent and proliferative fibroblasts exhibit differential p300 HAT activation through control of 5-methoxytryptophan production. PLoS ONE 2014, 9, e88507. [Google Scholar] [CrossRef] [PubMed]
- LeBoeuf, M.; Terrell, A.; Trivedi, S.; Sinha, S.; Epstein, J.A.; Olson, E.N.; Morrisey, E.E.; Millar, S.E. Hdac1 and Hdac2 act redundantly to control p63 and p53 functions in epidermal progenitor cells. Dev. Cell 2010, 19, 807–818. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Peterson, L.M.; Wilking-Busch, M.J.; Ndiaye, M.A.; Philippe, C.G.A.C.G.A.; Setaluri, V.; Ahmad, N. Sirtuins in Skin and Skin Cancers. Skin Pharmacol. Physiol. 2017, 30, 216–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohguchi, K.; Itoh, T.; Akao, Y.; Inoue, H.; Nozawa, Y.; Ito, M. SIRT1 modulates expression of matrix metalloproteinases in human dermal fibroblasts. Br. J. Dermatol. 2010, 163, 689–694. [Google Scholar] [CrossRef] [PubMed]
- Baohua, Y.; Li, L. Effects of SIRT6 silencing on collagen metabolism in human dermal fibroblasts. Cell Biol. Int. 2012, 36, 105–108. [Google Scholar] [CrossRef] [PubMed]
- Goyarts, E.C.; Dong, K.; Pelle, E.; Pernodet, N. Effect of SIRT6 knockdown on NF-κB induction and on residual DNA damage in cultured human skin fibroblasts. J. Cosmet. Sci. 2017, 68, 25–33. [Google Scholar] [PubMed]
- Joo, D.; An, S.; Choi, B.G.; Kim, K.; Choi, Y.M.; Ahn, K.J.; An, I.-S.; Cha, H.J. MicroRNA-378b regulates α-1-type 1 collagen expression via sirtuin 6 interference. Mol. Med. Rep. 2017, 16, 8520–8524. [Google Scholar] [CrossRef]
- Vieyra, D.; Loewith, R.; Scott, M.; Bonnefin, P.; Boisvert, F.-M.; Cheema, P.; Pastyryeva, S.; Meijer, M.; Johnston, R.N.; Bazett-Jones, D.P.; et al. Human ING1 proteins differentially regulate histone acetylation. J. Biol. Chem. 2002, 277, 29832–29839. [Google Scholar] [CrossRef]
- Soliman, M.A.; Berardi, P.; Pastyryeva, S.; Bonnefin, P.; Feng, X.; Colina, A.; Young, D.; Riabowol, K. ING1a expression increases during replicative senescence and induces a senescent phenotype. Aging Cell 2008, 7, 783–794. [Google Scholar] [CrossRef] [Green Version]
- Sen, P.; Shah, P.P.; Nativio, R.; Berger, S.L. Epigenetic mechanisms regulating longevity and aging. Cell 2016, 166, 822–839. [Google Scholar] [CrossRef]
- Herranz, D.; Muñoz-Martin, M.; Cañamero, M.; Mulero, F.; Martinez-Pastor, B.; Fernandez-Capetillo, O.; Serrano, M. Sirt1 improves healthy ageing and protects from metabolic syndrome-associated cancer. Nat. Commun. 2010, 1, 3. [Google Scholar] [CrossRef] [PubMed]
- Mostoslavsky, R.; Chua, K.F.; Lombard, D.B.; Pang, W.W.; Fischer, M.R.; Gellon, L.; Liu, P.; Mostoslavsky, G.; Franco, S.; Murphy, M.M.; et al. Genomic instability and aging-like phenotype in the absence of mammalian SIRT6. Cell 2006, 124, 315–329. [Google Scholar] [CrossRef] [PubMed]
- Massudi, H.; Grant, R.; Braidy, N.; Guest, J.; Farnsworth, B.; Guillemin, G.J. Age-associated changes in oxidative stress and NAD+ metabolism in human tissue. PLoS ONE 2012, 7, 1–9. [Google Scholar] [CrossRef]
- Ido, Y.; Duranton, A.; Lan, F.; Weikel, K.A.; Breton, L.; Ruderman, N.B. Resveratrol prevents oxidative stress-induced senescence and proliferative dysfunction by activating the AMPK-FOXO3 cascade in cultured primary human keratinocytes. PLoS ONE 2015, 10, 1–18. [Google Scholar] [CrossRef]
- Carlomosti, F.; D’Agostino, M.; Beji, S.; Torcinaro, A.; Rizzi, R.; Zaccagnini, G.; Maimone, B.; Di Stefano, V.; De Santa, F.; Cordisco, S.; et al. Oxidative Stress-Induced miR-200c Disrupts the Regulatory Loop Among SIRT1, FOXO1, and eNOS. Antioxid. Redox Signal. 2017, 27, 328–344. [Google Scholar] [CrossRef]
- Kim, K.S.; Park, H.-K.; Lee, J.-W.; Kim, Y.I.; Shin, M.K. Investigate correlation between mechanical property and aging biomarker in passaged human dermal fibroblasts. Microsc. Res. Tech. 2015, 78, 277–282. [Google Scholar] [CrossRef] [PubMed]
- de Cabo, R.; Liu, L.; Ali, A.; Price, N.; Zhang, J.; Wang, M.; Lakatta, E.; Irusta, P.M. Serum from calorie-restricted animals delays senescence and extends the lifespan of normal human fibroblasts in vitro. Aging (Albany NY) 2015, 7, 152–166. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Gan, Q.; Han, L.; Li, J.; Zhang, H.; Sun, Y.; Zhang, Z.; Tong, T. SIRT1 overexpression antagonizes cellular senescence with activated ERK/S6k1 signaling in human diploid fibroblasts. PLoS ONE 2008, 3. [Google Scholar] [CrossRef]
- Xu, Z.; Zhang, L.; Zhang, W.; Meng, D.; Zhang, H.; Jiang, Y.; Xu, X.; Meter, M.V.; Seluanov, A.; Gorbunova, V.; et al. Van SIRT6 rescues the age related decline in base excision repair in a PARP1-dependent manner SIRT6 rescues the age related decline in base excision repair in a PARP1-dependent manner. Cell Cycle 2015, 4101, 1551–4005. [Google Scholar] [CrossRef]
- Tinaburri, L.; D’Errico, M.; Sileno, S.; Maurelli, R.; Degan, P.; Magenta, A.; Dellambra, E. miR-200a Modulates the Expression of the DNA Repair Protein OGG1 Playing a Role in Aging of Primary Human Keratinocytes. Oxid. Med. Cell. Longev. 2018, 2018, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Ding, S.; Chen, J.; Zeng, Q.; Lu, J.; Tan, L.; Guo, A.; Kang, J.; Yang, S.; Xiang, Y.; Zuo, C.; et al. Chronic sun exposure is associated with distinct histone acetylation changes in human skin. Br. J. Dermatol. 2018, 179, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Pollack, B.P.; Sapkota, B.; Boss, J.M. Ultraviolet Radiation-induced Transcription is Associated with Gene-specific Histone Acetylation. Photochem. Photobiol. 2009, 85, 652–662. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.K.; Shin, J.M.; Eun, H.C.; Chung, J.H. The role of p300 histone acetyltransferase in UV-induced histone modifications and MMP-1 gene transcription. PLoS ONE 2009, 4, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Fan, W.; Luo, J. SIRT1 Regulates UV-Induced DNA Repair through Deacetylating XPA. Mol. Cell 2010, 39, 247–258. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Liu, W.; Niu, T.; Dai, C.; Li, X.; Cui, C.; Zhao, X.; E, Y.; Lu, H. The injury and cumulative effects on human skin by UV exposure from artificial fluorescence emission. Photochem. Photobiol. 2014, 90, 1433–1438. [Google Scholar] [CrossRef] [PubMed]
- Cao, C.; Lu, S.; Kivlin, R.; Wallin, B.; Card, E.; Bagdasarian, A.; Tamakloe, T.; Wang, W.; Song, X.; Chu, W.; et al. SIRT1 confers protection against UVB- and H2O2-induced cell death via modulation of p53 and JNK in cultured skin keratinocytes. J. Cell. Mol. Med. 2009, 13, 3632–3643. [Google Scholar] [CrossRef] [PubMed]
- Wahedi, H.M.; Lee, T.H.; Moon, E.-Y.; Kim, S.Y. Juglone up-regulates sirt1 in skin cells under normal and UVB irradiated conditions. J. Dermatol. Sci. 2016, 81, 210–212. [Google Scholar] [CrossRef] [PubMed]
- Chung, K.W.; Choi, Y.J.; Park, M.H.; Jang, E.J.; Kim, D.H.; Park, B.H.; Yu, B.P.; Chung, H.Y. Molecular insights into SIRT1 protection against UVB-induced skin fibroblast senescence by suppression of oxidative stress and p53 acetylation. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2015, 70, 959–968. [Google Scholar] [CrossRef]
- Lee, J.-S.; Park, K.-Y.; Min, H.-G.; Lee, S.J.; Kim, J.-J.; Choi, J.-S.; Kim, W.-S.; Cha, H.-J. Negative regulation of stress-induced matrix metalloproteinase-9 by Sirt1 in skin tissue. Exp. Dermatol. 2010, 19, 1060–1066. [Google Scholar] [CrossRef]
- Ming, M.; Soltani, K.; Shea, C.R.; Li, X.; He, Y.Y. Dual role of SIRT1 in UVB-induced skin tumorigenesis. Oncogene 2014, 34, 1–7. [Google Scholar] [CrossRef]
- Benavente, C.A.; Schnell, S.A.; Jacobson, E.L. Effects of niacin restriction on sirtuin and PARP responses to photodamage in human skin. PLoS ONE 2012, 7, e42276. [Google Scholar] [CrossRef] [PubMed]
- Lang, A.; Grether-Beck, S.; Singh, M.; Kuck, F.; Jakob, S.; Kefalas, A.; Altinoluk-Hambüchen, S.; Graffmann, N.; Schneider, M.; Lindecke, A.; et al. MicroRNA-15b regulates mitochondrial ROS production and the senescence-associated secretory phenotype through sirtuin 4/SIRT4. Aging (Albany NY) 2016, 8, 484–505. [Google Scholar] [CrossRef] [PubMed]
- Botchkarev, V.A. The Molecular Revolution in Cutaneous Biology: Chromosomal Territories, Higher-Order Chromatin Remodeling, and the Control of Gene Expression in Keratinocytes. J. Investig. Dermatol. 2017, 137, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Fessing, M.Y.; Mardaryev, A.N.; Gdula, M.R.; Sharov, A.A.; Sharova, T.Y.; Rapisarda, V.; Gordon, K.B.; Smorodchenko, A.D.; Poterlowicz, K.; Ferone, G.; et al. p63 regulates Satb1 to control tissue-specific chromatin remodeling during development of the epidermis. J. Cell Biol. 2011, 194, 825–839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keyes, W.M.; Pecoraro, M.; Aranda, V.; Vernersson-Lindahl, E.; Li, W.; Vogel, H.; Guo, X.; Garcia, E.L.; Michurina, T.V.; Enikolopov, G.; et al. A ΔNp63α is an oncogene that targets chromatin remodeler Lsh to drive skin stem cell proliferation and tumorigenesis. Cell Stem Cell 2011, 8, 164–176. [Google Scholar] [CrossRef] [PubMed]
- Mardaryev, A.N.; Gdula, M.R.; Yarker, J.L.; Emelianov, V.U.; Emelianov, V.N.; Poterlowicz, K.; Sharov, A.A.; Sharova, T.Y.; Scarpa, J.A.; Joffe, B.; et al. p63 and Brg1 control developmentally regulated higher-order chromatin remodelling at the epidermal differentiation complex locus in epidermal progenitor cells. Development 2014, 141, 101–111. [Google Scholar] [CrossRef]
- Rapisarda, V.; Malashchuk, I.; Asamaowei, I.E.; Poterlowicz, K.; Fessing, M.Y.; Sharov, A.A.; Karakesisoglou, I.; Botchkarev, V.A.; Mardaryev, A. p63 Transcription Factor Regulates Nuclear Shape and Expression of Nuclear Envelope-Associated Genes in Epidermal Keratinocytes. J. Investig. Dermatol. 2017, 137, 2157–2167. [Google Scholar] [CrossRef] [Green Version]
- Rinaldi, L.; Benitah, S.A. Epigenetic regulation of adult stem cell function. FEBS J. 2015, 282, 1589–1604. [Google Scholar] [CrossRef]
- Clapier, C.R.; Cairns, B.R. The biology of chromatin remodeling complexes. Annu. Rev. Biochem. 2009, 78, 273–304. [Google Scholar] [CrossRef]
- Indra, A.K.; Dupé, V.; Bornert, J.-M.; Messaddeq, N.; Yaniv, M.; Mark, M.; Chambon, P.; Metzger, D. Temporally controlled targeted somatic mutagenesis in embryonic surface ectoderm and fetal epidermal keratinocytes unveils two distinct developmental functions of BRG1 in limb morphogenesis and skin barrier formation. Development 2005, 132, 4533–4544. [Google Scholar] [CrossRef] [Green Version]
- Jaubert, J.; Cheng, J.; Segre, J.A. Ectopic expression of kruppel like factor 4 (Klf4) accelerates formation of the epidermal permeability barrier. Development 2003, 130, 2767–2777. [Google Scholar] [CrossRef] [PubMed]
- Bao, X.; Tang, J.; Lopez-Pajares, V.; Tao, S.; Qu, K.; Crabtree, G.R.; Khavari, P.A. ACTL6a Enforces the Epidermal Progenitor State by Suppressing SWI/SNF-Dependent Induction of KLF4. Cell Stem Cell 2013, 12, 193–203. [Google Scholar] [CrossRef]
- Panatta, E.; Lena, A.M.; Mancini, M.; Affinati, M.; Smirnov, A.; Annicchiarico-Petruzzelli, M.; Piro, M.C.; Campione, E.; Bianchi, L.; Mazzanti, C.; et al. Kruppel-like factor 4 regulates keratinocyte senescence. Biochem. Biophys. Res. Commun. 2018, 499, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Tu, Z.; Zhuang, X.; Yao, Y.-G.; Zhang, R. BRG1 is required for formation of senescence-associated heterochromatin foci induced by oncogenic RAS or BRCA1 loss. Mol. Cell. Biol. 2013, 33, 1819–1829. [Google Scholar] [CrossRef] [PubMed]
- Park, J.-H.; Park, E.-J.; Lee, H.-S.; Kim, S.J.; Hur, S.-K.; Imbalzano, A.N.; Kwon, J. Mammalian SWI/SNF complexes facilitate DNA double-strand break repair by promoting gamma-H2AX induction. EMBO J. 2006, 25, 3986–3997. [Google Scholar] [CrossRef]
- Muegge, K. Lsh: A guardian of heterochromatin at repeat elements. Biochem. Cell Biol. 2005, 83, 548–554. [Google Scholar] [CrossRef] [PubMed]
- Xi, S.; Zhu, H.; Xu, H.; Schmidtmann, A.; Geiman, T.M.; Muegge, K. Lsh controls Hox gene silencing during development. Proc. Natl. Acad. Sci. USA 2007, 104, 14366–14371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Myant, K.; Stancheva, I. LSH cooperates with DNA methyltransferases to repress transcription. Mol. Cell. Biol. 2008, 28, 215–226. [Google Scholar] [CrossRef]
- Zhou, R.; Han, L.; Li, G.; Tong, T. Senescence delay and repression of p16INK4a by Lsh via recruitment of histone deacetylases in human diploid fibroblasts. Nucleic Acids Res. 2009, 37, 5183–5196. [Google Scholar] [CrossRef]
- Niu, J.; Chen, T.; Han, L.; Wang, P.; Li, N.; Tong, T. Transcriptional activation of the senescence regulator Lsh by E2F1. Mech. Ageing Dev. 2011, 132, 180–186. [Google Scholar] [CrossRef]
- Ohlsson, R.; Bartkuhn, M.; Renkawitz, R. CTCF shapes chromatin by multiple mechanisms: The impact of 20 years of CTCF research on understanding the workings of chromatin. Chromosoma 2010, 119, 351–560. [Google Scholar] [CrossRef] [PubMed]
- Witcher, M.; Emerson, B.M. Epigenetic silencing of the p16(INK4a) tumor suppressor is associated with loss of CTCF binding and a chromatin boundary. Mol. Cell 2009, 34, 271–284. [Google Scholar] [CrossRef] [PubMed]
- Rosa-Garrido, M.; Ceballos, L.; Alonso-Lecue, P.; Abraira, C.; Delgado, M.D.; Gandarillas, A. A cell cycle role for the epigenetic factor CTCF-L./BORIS. PLoS ONE 2012, 7, e39371. [Google Scholar] [CrossRef] [PubMed]
- Han, H.-J.; Russo, J.; Kohwi, Y.; Kohwi-Shigematsu, T. SATB1 reprogrammes gene expression to promote breast tumour growth and metastasis. Nature 2008, 452, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Lena, A.M.; Mancini, M.; Rivetti di Val Cervo, P.; Saintigny, G.; Mahé, C.; Melino, G.; Candi, E. MicroRNA-191 triggers keratinocytes senescence by SATB1 and CDK6 downregulation. Biochem. Biophys. Res. Commun. 2012, 423, 509–514. [Google Scholar] [CrossRef] [PubMed]
- Agrelo, R.; Kishimoto, H.; Novatchkova, M.; Peraza, V.; Paolino, M.; Souabni, A.; Wutz, A. SATB1 collaborates with loss of p16 in cellular transformation. Oncogene 2013, 32, 5492–5500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dechat, T.; Pfleghaar, K.; Sengupta, K.; Shimi, T.; Shumaker, D.K.; Solimando, L.; Goldman, R.D. Nuclear lamins: Major factors in the structural organization and function of the nucleus and chromatin. Genes Dev. 2008, 22, 832–853. [Google Scholar] [CrossRef]
- Shimi, T.; Pfleghaar, K.; Kojima, S.; Pack, C.-G.; Solovei, I.; Goldman, A.E.; Adam, S.A.; Shumaker, D.K.; Kinjo, M.; Cremer, T.; et al. The A- and B-type nuclear lamin networks: Microdomains involved in chromatin organization and transcription. Genes Dev. 2008, 22, 3409–3421. [Google Scholar] [CrossRef]
- Jung, H.-J.; Tatar, A.; Tu, Y.; Nobumori, C.; Yang, S.H.; Goulbourne, C.N.; Herrmann, H.; Fong, L.G.; Young, S.G. An absence of nuclear lamins in keratinocytes leads to ichthyosis, defective epidermal barrier function, and intrusion of nuclear membranes and endoplasmic reticulum into the nuclear chromatin. Mol. Cell. Biol. 2014, 34, 4534–4544. [Google Scholar] [CrossRef]
- Shimi, T.; Butin-Israeli, V.; Adam, S.A.; Hamanaka, R.B.; Goldman, A.E.; Lucas, C.A.; Shumaker, D.K.; Kosak, S.T.; Chandel, N.S.; Goldman, R.D. The role of nuclear lamin B1 in cell proliferation and senescence. Genes Dev. 2011, 25, 2579–2593. [Google Scholar] [CrossRef] [Green Version]
- Dreesen, O.; Chojnowski, A.; Ong, P.F.; Zhao, T.Y.; Common, J.E.; Lunny, D.; Lane, E.B.; Lee, S.J.; Vardy, L.A.; Stewart, C.L.; Colman, A. Lamin B1 fluctuations have differential effects on cellular proliferation and senescence. J. Cell Biol. 2013, 200, 605–617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McClintock, D.; Ratner, D.; Lokuge, M.; Owens, D.M.; Gordon, L.B.; Collins, F.S.; Djabali, K. The mutant form of Lamin A that causes Hutchinson-Gilford progeria is a biomarker of cellular aging in human skin. PLoS ONE 2007, 2. [Google Scholar] [CrossRef] [PubMed]
- Kudlow, B.A.; Stanfel, M.N.; Burtner, C.R.; Johnston, E.D.; Kennedy, B.K. Suppression of proliferative defects associated with processing-defective lamin A mutants by hTERT or inactivation of p53. Mol. Biol. Cell 2008, 19, 5238–5248. [Google Scholar] [CrossRef] [PubMed]
- Dreesen, O.; Ong, P.F.; Chojnowski, A.; Colman, A. The contrasting roles of lamin B1 in cellular aging and human disease. Nucleus 2013, 4, 283–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, A.S.; Dreesen, O. Biomarkers of cellular senescence and skin aging. Front. Genet. 2018, 9, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Carrero, D.; Soria-Valles, C.; López-Otín, C. Hallmarks of progeroid syndromes: Lessons from mice and reprogrammed cells. Dis. Model. Mech. 2016, 9, 719–735. [Google Scholar] [CrossRef]
- Navarro, C.L.; Cau, P.; Lévy, N. Molecular bases of progeroid syndromes. Hum. Mol. Genet. 2006, 15, 151–161. [Google Scholar] [CrossRef]
- Capell, B.C.; Tlougan, B.E.; Orlow, S.J. From the rarest to the most common: Insights from progeroid syndromes into skin cancer and aging. J. Investig. Dermatol. 2009, 129, 2340–2350. [Google Scholar] [CrossRef]
- Croteau, D.L.; Popuri, V.; Opresko, P.L.; Bohr, V.A. Human RecQ Helicases in DNA Repair, Recombination, and Replication. Annu. Rev. Biochem. 2014, 83, 519–552. [Google Scholar] [CrossRef] [Green Version]
- Stefanini, M.; Botta, E.; Lanzafame, M.; Orioli, D. Trichothiodystrophy: From basic mechanisms to clinical implications. DNA Repair 2010, 9, 2–10. [Google Scholar] [CrossRef]
- Warrick, E.; Garcia, M.; Chagnoleau, C.; Chevallier, O.; Bergoglio, V.; Sartori, D.; Mavilio, F.; Angulo, J.F.; Avril, M.-F.; Sarasin, A.; et al. Preclinical Corrective Gene Transfer in Xeroderma Pigmentosum Human Skin Stem Cells. Mol. Ther. 2012, 20, 798–807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanzafame, M.; Vaz, B.; Nardo, T.; Botta, E.; Orioli, D.; Stefanini, M. From laboratory tests to functional characterisation of Cockayne syndrome. Mech. Ageing Dev. 2013, 134, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Ceccaldi, R.; Sarangi, P.; D’Andrea, A.D. The Fanconi anaemia pathway: New players and new functions. Nat. Rev. Mol. Cell Biol. 2016, 17, 337–349. [Google Scholar] [CrossRef] [PubMed]
- Bagby, G. Recent advances in understanding hematopoiesis in Fanconi Anemia. F1000Research 2018, 7, 105. [Google Scholar] [CrossRef] [Green Version]
- Alderton, G.K.; Joenje, H.; Varon, R.; Børglum, A.D.; Jeggo, P.A.; O’Driscoll, M. Seckel syndrome exhibits cellular features demonstrating defects in the ATR-signalling pathway. Hum. Mol. Genet. 2004, 13, 3127–3138. [Google Scholar] [CrossRef] [Green Version]
- Marechal, A.; Zou, L. DNA Damage Sensing by the ATM and ATR Kinases. Cold Spring Harb. Perspect. Biol. 2013, 5, a012716. [Google Scholar] [CrossRef]
- Taylor, A.M.R.A.M.R.; Groom, A.; Byrd, P.J. Ataxia-telangiectasia-like disorder (ATLD)-Its clinical presentation and molecular basis. DNA Repair 2004, 3, 1219–1225. [Google Scholar] [CrossRef]
- McKinnon, P.J. ATM and the Molecular Pathogenesis of Ataxia Telangiectasia. Annu. Rev. Pathol. Mech. Dis. 2012, 7, 303–321. [Google Scholar] [CrossRef]
- Lin, F.; Worman, H.J. Structural organization of the human gene encoding nuclear lamin A and nuclear lamin C. J. Biol. Chem. 1993, 268, 16321–16326. [Google Scholar] [CrossRef]
- Ramírez, C.L.; Cadiñanos, J.; Varela, I.; Freije, J.M.P.J.M.P.; López-Otín, C. Human progeroid syndromes, aging and cancer: New genetic and epigenetic insights into old questions. Cell. Mol. Life Sci. 2007, 64, 155–170. [Google Scholar] [CrossRef]
- Goldman, R.D.; Shumaker, D.K.; Erdos, M.R.; Eriksson, M.; Goldman, A.E.; Gordon, L.B.; Gruenbaum, Y.; Khuon, S.; Mendez, M.; Varga, R.; et al. Accumulation of mutant lamin A causes progressive changes in nuclear architecture in Hutchinson-Gilford progeria syndrome. Proc. Natl. Acad. Sci. USA 2004, 101, 8963–8968. [Google Scholar] [CrossRef] [PubMed]
- Jamin, A.; Wiebe, M.S. Barrier to Autointegration Factor (BANF1): Interwoven roles in nuclear structure, genome integrity, innate immunity, stress responses and progeria. Curr. Opin. Cell Biol. 2015, 34, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Rankin, J.; Ellard, S. The laminopathies: A clinical review. Clin. Genet. 2006, 70, 261–274. [Google Scholar] [CrossRef]
- Shawi, M.; Autexier, C. Telomerase, senescence and ageing. Mech. Ageing Dev. 2008, 129, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Glousker, G.; Touzot, F.; Revy, P.; Tzfati, Y.; Savage, S.A. Unraveling the pathogenesis of Hoyeraal-Hreidarsson syndrome, a complex telomere biology disorder. Br. J. Haematol. 2015, 170, 457–471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallis, C.V.; Sheerin, A.N.; Green, M.H.L.M.H.L.; Jones, C.J.; Kipling, D.; Faragher, R.G.A. Fibroblast clones from patients with Hutchinson-Gilford progeria can senesce despite the presence of telomerase. Exp. Gerontol. 2004, 39, 461–467. [Google Scholar] [CrossRef] [PubMed]
- Campisi, J.; d’Adda di Fagagna, F. Cellular senescence: When bad things happen to good cells. Nat. Rev. Mol. Cell Biol. 2007, 8, 729–740. [Google Scholar] [CrossRef] [PubMed]
- Pascucci, B.; Fragale, A.; Marabitti, V.; Leuzzi, G.; Salvatore Calcagnile, A.; Parlanti, E.; Franchitto, A.; Dogliotti, E.; D’Errico, M. CSA and CSB play a role in the response to DNA breaks. Oncotarget 2018, 9, 11581–11591. [Google Scholar] [CrossRef]
- Cordisco, S.; Tinaburri, L.; Teson, M.; Orioli, D.; Cardin, R.; Degan, P.; Stefanini, M.; Zambruno, G.; Guerra, L.; Dellambra, E. Cockayne Syndrome Type A Protein Protects Primary Human Keratinocytes from Senescence. J. Investig. Dermatol. 2018, 1–13. [Google Scholar] [CrossRef]
- Pascucci, B.; Lemma, T.; Iorio, E.; Giovannini, S.; Vaz, B.; Iavarone, I.; Calcagnile, A.; Narciso, L.; Degan, P.; Podo, F.; et al. An altered redox balance mediates the hypersensitivity of Cockayne syndrome primary fibroblasts to oxidative stress. Aging Cell 2012, 11, 520–529. [Google Scholar] [CrossRef] [Green Version]
- Scheibye-Knudsen, M.; Croteau, D.L.; Bohr, V.A. Mitochondrial deficiency in Cockayne syndrome. Mech. Ageing Dev. 2013, 134, 275–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheibye-Knudsen, M.; Mitchell, S.J.; Fang, E.F.; Iyama, T.; Ward, T.; Wang, J.; Dunn, C.A.; Singh, N.; Veith, S.; Hasan-Olive, M.M.; et al. A high-fat diet and NAD(+) activate Sirt1 to rescue premature aging in cockayne syndrome. Cell Metab. 2014, 20, 840–855. [Google Scholar] [CrossRef] [PubMed]
- Newman, J.C.; Bailey, A.D.; Weiner, A.M. Cockayne syndrome group B protein (CSB) plays a general role in chromatin maintenance and remodeling. Proc. Natl. Acad. Sci. USA 2006, 103, 9613–9618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arseni, L.; Lanzafame, M.; Compe, E.; Fortugno, P.; Afonso-Barroso, A.; Peverali, F.A.; Lehmann, A.R.; Zambruno, G.; Egly, J.-M.; Stefanini, M.; et al. TFIIH-dependent MMP-1 overexpression in trichothiodystrophy leads to extracellular matrix alterations in patient skin. Proc. Natl. Acad. Sci. USA 2015, 112, 1499–1504. [Google Scholar] [CrossRef] [PubMed]
- Varela, I.; Cadiñanos, J.; Pendás, A.M.; Gutiérrez-Fernández, A.; Folgueras, A.R.; Sánchez, L.M.; Zhou, Z.; Rodríguez, F.J.; Stewart, C.L.; Vega, J.A.; et al. Accelerated ageing in mice deficient in Zmpste24 protease is linked to p53 signalling activation. Nature 2005, 437, 564–568. [Google Scholar] [CrossRef] [PubMed]
- Scaffidi, P.; Misteli, T. Lamin A-dependent nuclear defects in human aging. Science 2006, 312, 1059–1063. [Google Scholar] [CrossRef] [PubMed]
- Chojnowski, A.; Ong, P.F.; Wong, E.S.M.E.S.M.; Lim, J.S.Y.J.S.Y.; Mutalif, R.A.; Navasankari, R.; Dutta, B.; Yang, H.; Liow, Y.Y.; Sze, S.K.; et al. Progerin reduces LAP2α-telomere association in Hutchinson-Gilford progeria. Elife 2015, 4. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.; Risques, R.A.; Martin, G.M.; Rabinovitch, P.S.; Oshima, J. Accelerated telomere shortening and replicative senescence in human fibroblasts overexpressing mutant and wild-type lamin A. Exp. Cell Res. 2008, 314, 82–91. [Google Scholar] [CrossRef] [Green Version]
- Scaffidi, P.; Misteli, T. Lamin A-dependent misregulation of adult stem cells associated with accelerated ageing. Nat. Cell Biol. 2008, 10, 452–459. [Google Scholar] [CrossRef] [Green Version]
- Espada, J.; Varela, I.; Flores, I.; Ugalde, A.P.; Cadiñanos, J.; Pendás, A.M.; Stewart, C.L.; Tryggvason, K.; Blasco, M.A.; Freije, J.M.P.J.M.P.; et al. Nuclear envelope defects cause stem cell dysfunction in premature-aging mice. J. Cell Biol. 2008, 181, 27–35. [Google Scholar] [CrossRef] [Green Version]
- Loi, M.; Cenni, V.; Duchi, S.; Squarzoni, S.; Otin, C.L.; Foisner, R.; Lattanzi, G.; Capanni, C. Barrier-to-Autointegration Factor (BAF) involvement in prelamin A-related chromatin organization changes. Oncotarget 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Scaffidi, P.; Misteli, T. Reversal of the cellular phenotype in the premature aging disease Hutchinson-Gilford progeria syndrome. Nat. Med. 2005, 11, 440–445. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Li, J.; Suzuki, K.; Qu, J.; Wang, P.; Zhou, J.; Liu, X.; Ren, R.; Xu, X.; Ocampo, A.; et al. A Werner syndrome stem cell model unveils heterochromatin alterations as a driver of human aging. Science 2015, 348, 1160–1163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shumaker, D.K.; Dechat, T.; Kohlmaier, A.; Adam, S.A.; Bozovsky, M.R.; Erdos, M.R.; Eriksson, M.; Goldman, A.E.; Khuon, S.; Collins, F.S.; et al. Mutant nuclear lamin A leads to progressive alterations of epigenetic control in premature aging. Proc. Natl. Acad. Sci. USA 2006, 103, 8703–8708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarg, B.; Koutzamani, E.; Helliger, W.; Rundquist, I.; Lindner, H.H. Postsynthetic trimethylation of histone H4 at lysine 20 in mammalian tissues is associated with aging. J. Biol. Chem. 2002, 277, 39195–39201. [Google Scholar] [CrossRef] [PubMed]
- Nelson, D.M.; Jaber-Hijazi, F.; Cole, J.J.; Robertson, N.A.; Pawlikowski, J.S.; Norris, K.T.; Criscione, S.W.; Pchelintsev, N.A.; Piscitello, D.; Stong, N.; et al. Mapping H4K20me3 onto the chromatin landscape of senescent cells indicates a function in control of cell senescence and tumor suppression through preservation of genetic and epigenetic stability. Genome Biol. 2016, 17, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Rivera-Mulia, J.C.; Desprat, R.; Trevilla-Garcia, C.; Cornacchia, D.; Schwerer, H.; Sasaki, T.; Sima, J.; Fells, T.; Studer, L.; Lemaitre, J.-M.; et al. DNA replication timing alterations identify common markers between distinct progeroid diseases. Proc. Natl. Acad. Sci. USA 2017, 2017, 11613. [Google Scholar] [CrossRef] [PubMed]
- Le May, N.; Mota-Fernandes, D.; Vélez-Cruz, R.; Iltis, I.; Biard, D.; Egly, J.M. NER Factors Are Recruited to Active Promoters and Facilitate Chromatin Modification for Transcription in the Absence of Exogenous Genotoxic Attack. Mol. Cell 2010, 38, 54–66. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, K.M.; Schmitt, N.; Hoffmann-Rohrer, U.; Schäfer, A.; Grummt, I.; Mayer, C. TAF12 Recruits Gadd45a and the Nucleotide Excision Repair Complex to the Promoter of rRNA Genes Leading to Active DNA Demethylation. Mol. Cell 2009, 33, 344–353. [Google Scholar] [CrossRef] [PubMed]
- Le May, N.; Fradin, D.; Iltis, I.; Bougnères, P.; Egly, J.-M. XPG and XPF Endonucleases Trigger Chromatin Looping and DNA Demethylation for Accurate Expression of Activated Genes. Mol. Cell 2012, 47, 622–632. [Google Scholar] [CrossRef] [PubMed]
- Kawahara, T.L.A.T.L.A.; Rapicavoli, N.A.; Wu, A.R.; Qu, K.; Quake, S.R.; Chang, H.Y. Dynamic chromatin localization of sirt6 shapes stress- and aging-related transcriptional networks. PLoS Genet. 2011, 7, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.J.; Edelman, J.R. Unstable Heterochromatin in Fanconi ’s Anemia Cells. Cytologia (Tokyo) 2001, 66, 403–407. [Google Scholar] [CrossRef]
Figure 1.
Morphological features of young, chronologically- and photo-aged skin. Schematic organization of the young, chronologically aged or photo-aged skin tissue. Human skin is constituted by three tissue layers: epidermis, dermis, and hypodermis. The epidermis is a stratified epithelium composed of keratinocytes organized into four major layers (basal, spinous, granular, and cornified layers) at progressive differentiation stages including melanocytes. The dermis is populated by fibroblasts embedded by the components of the extracellular matrix made of collagen, elastic fibers, glycoproteins, and proteoglycans. Fibroblasts are the main producers of the extracellular matrix components. The hypodermis is mainly populated by adipocytes. Morphological changes occurring with age are indicated in chronologically- and photo-aged skin.
Figure 1.
Morphological features of young, chronologically- and photo-aged skin. Schematic organization of the young, chronologically aged or photo-aged skin tissue. Human skin is constituted by three tissue layers: epidermis, dermis, and hypodermis. The epidermis is a stratified epithelium composed of keratinocytes organized into four major layers (basal, spinous, granular, and cornified layers) at progressive differentiation stages including melanocytes. The dermis is populated by fibroblasts embedded by the components of the extracellular matrix made of collagen, elastic fibers, glycoproteins, and proteoglycans. Fibroblasts are the main producers of the extracellular matrix components. The hypodermis is mainly populated by adipocytes. Morphological changes occurring with age are indicated in chronologically- and photo-aged skin.

Figure 2.
De-regulation of epigenetic modifiers affecting senescent epidermal keratinocytes and senescent dermal fibroblasts in chronologically-, photo- and premature-aged skin. Senescent cells are characterized by alterations of several cellular processes (DNA damage response, inflammation, and cell-cycle exit). Senescent fibroblasts display a modified metabolism of collagen and elastic fibers (elastic fiber maturation, collagen synthesis, and collagen degradation). Epigenetic modifiers inhibiting the cellular processes are indicated in light red when down-regulated and in dark red when up-regulated (or not down-regulated). Epigenetic modifiers activating the cellular processes are indicated in light green when down-regulated and in dark green when up-regulated (or not down-regulated).
Figure 2.
De-regulation of epigenetic modifiers affecting senescent epidermal keratinocytes and senescent dermal fibroblasts in chronologically-, photo- and premature-aged skin. Senescent cells are characterized by alterations of several cellular processes (DNA damage response, inflammation, and cell-cycle exit). Senescent fibroblasts display a modified metabolism of collagen and elastic fibers (elastic fiber maturation, collagen synthesis, and collagen degradation). Epigenetic modifiers inhibiting the cellular processes are indicated in light red when down-regulated and in dark red when up-regulated (or not down-regulated). Epigenetic modifiers activating the cellular processes are indicated in light green when down-regulated and in dark green when up-regulated (or not down-regulated).

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Orioli, D.; Dellambra, E. Epigenetic Regulation of Skin Cells in Natural Aging and Premature Aging Diseases. Cells 2018, 7, 268. https://doi.org/10.3390/cells7120268
AMA Style
Orioli D, Dellambra E. Epigenetic Regulation of Skin Cells in Natural Aging and Premature Aging Diseases. Cells. 2018; 7(12):268. https://doi.org/10.3390/cells7120268
Chicago/Turabian StyleOrioli, Donata, and Elena Dellambra. 2018. "Epigenetic Regulation of Skin Cells in Natural Aging and Premature Aging Diseases" Cells 7, no. 12: 268. https://doi.org/10.3390/cells7120268
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.