Transcriptome Profiles of Highly Pathogenic Pure Avian H7N9 Virus-Infected Lungs of BALB/c Mice
 Wenxiao Gong1,2,3 Kun Huang1,2,3 Yufei Zhang1,2,3 Xinglin He1,2,3 Chengfei Li1,2,3 Haiying Mao1,2,3 Yanming Wei2* Zhong Zou1,2,3*
Wenxiao Gong1,2,3 Kun Huang1,2,3 Yufei Zhang1,2,3 Xinglin He1,2,3 Chengfei Li1,2,3 Haiying Mao1,2,3 Yanming Wei2* Zhong Zou1,2,3*  Meilin Jin1,2,3*
Meilin Jin1,2,3*- 1State Key Laboratory of Agricultural Microbiology, Huazhong Agricultural University, Wuhan, China
- 2College of Veterinary Medicine, Huazhong Agricultural University, Wuhan, China
- 3Key Laboratory of Development of Veterinary Diagnostic Products, Ministry of Agriculture, Wuhan, China
Avian influenza A (H7N9) viruses emerged in China in 2013 and caused a zoonotic disease associated with a high case-fatality ratio of more than 30%. Transcriptional profiles obtained using animal models reveal host responses to the disease, thereby providing insights into disease pathogenesis. Therefore, we aimed to characterize the host responses of the H7N9 virus infected-mouse lungs in this study. First, we isolated an avian-originated H7N9 strain, which was shown to be highly pathogenic to both chickens and mice. Genomic analysis results suggested that a 12-nucleotide-insertion was present at the hemagglutinin cleavage site, and both the hemagglutinin and neuraminidase genes belonged to the Yangtze River Delta lineage. RNA sequencing results revealed 566 differentially expressed genes in the H7N9-infected lungs. Moreover, transcriptome analysis revealed that over-activated antiviral signals and intense interferon-stimulated gene products possibly contributed to the high virulence of the virus in mice. Importantly, lung concentrations of inflammatory cytokines, including interleukin-1β and interleukin-6, interferon-β, and tumor necrosis factor-α, were upregulated in response to H7N9 virus infection. Overall, the present study provided a comprehensive understanding of H7N9 virus pathogenicity and correlated host immune responses.
Introduction
Influenza A virus (IAV) is an enveloped, segmented, negative-strand RNA virus belonging to the Orthomyxoviridae family (1). IAVs have a wide range of hosts, including humans, wild birds, poultry, and pigs; wild waterfowl are generally considered IAV reservoirs (2). The segmented pattern of the IAV genome and the proofreading activity-lacking nature of the viral polymerase allowed the accumulation of nucleotide changes via antigenic shift and drift, resulting in the added diversity and emergence of novel influenza viruses. This further created the possibility of cross-species transmission (3). Of note, avian influenza viruses (AIVs), such as H5N1, H6N1, H7N9, H9N2, and H10N8, can occasionally break the species barrier and spread from domestic poultry to humans, posing a great threat to public health (4–7).
The H7N9 virus is a reassortant AIV consisting of the H9N2 viruses that circulate in the poultry in China and the Eurasian wild bird H7 and N9 viruses (8). In March 2013, the H7N9 virus was first isolated from patients in eastern China (9). Since then, 1,564 patients with H7N9 disease have been reported, and 612 of them were deceased, demonstrating an overall case-fatality ratio of ~38% (10). Sufficient epidemiological data indicate that human infections are directly linked to live-poultry markets (11). Due to several host adaptation mutations and the exchange of internal genes with the H9N2 viruses circulating in the poultry, highly pathogenic AIV (HPAIV) H7N9 viruses emerged during the fifth wave of the H7N9 virus outbreak, characteristically showing a four-amino-acid-insertion at the hemagglutinin (HA) cleavage site (12). During this period, high and low pathogenic H7N9 AIVs co-existed in the poultry.
Studies on highly virulent and pandemic viruses, such as H5N1, have demonstrated specific hallmarks of viral infection, including viral replication rate enhancement and hyper-cytokinemia or “cytokine storm” induction (13, 14). An increasing number of microarray studies performed using animal models have revealed that early and sustained innate immune responses are characteristic of lethal IAV infection (15, 16). Evidence for such responses has been confirmed by counting the lung immune cells via flow cytometry, specifically showing a remarkably increased number of macrophages and neutrophils destined to play critical roles in viral clearance. However, as far as we know, only a cynomolgus macaque model has been used to conduct a study on the gene expression profile changes that occur in H7N9 virus infection (15). Therefore, transcriptome profiles of other animal models are still needed to elucidate H7N9 pathogenesis.
In the present study, we successfully isolated HPAIV H7N9 from the poultry samples collected from Guangxi province. Interestingly, a 12-nucleotide-insertion was observed at the HA cleavage site, and both the HA and neuraminidase (NA) genes were found to belong to the Yangtze River Delta lineage by genomic analysis. This virus not only showed high pathogenicity toward chickens but could also cause lethal infection without prior adaptation in mice. Additionally, it was associated with high pulmonary levels of pro-inflammatory cytokines. Therefore, we aimed to identify host transcriptome responses associated with virological outcomes in IAV H7N9-infected mice. To the best of our knowledge, this study is the first microarray analysis of H7N9-infected mouse lungs, thereby providing a basis for a better understanding of H7N9 pathogenicity in the mouse model.
Materials and Methods
Virus
During the fifth wave of the H7N9 outbreak, AIV A/chicken/Guangxi/YL01/2017 (abbreviated as GX/YL01) (accession number EPI_ISL_436706) was isolated from the lungs of dead chickens obtained from Guangxi provinces. The virus was amplified using 10-day-old specific-pathogen-free (SPF) embryonic chicken eggs and then titrated by determining the log10[egg infective dose 50% (EID50) per milliliter] values. Aliquots of viral stock were stored at −80°C until use. All experiments using the H7N9 virus were performed in a Biosafety Level 3 laboratory (17). This study was performed in accordance with the recommendations by the Biosafety Level 3 laboratory at Huazhong Agricultural University. All procedures were approved by the Institutional Biosafety Committee of Huazhong Agricultural University.
Genomic Sequencing and Phylogenetic Analysis
Viral RNA was obtained from samples of virus-infected allantoic fluids via the lysis of the viruses using TRIzol LS Reagent (Life Technologies, Inc., CA, USA), as described in a previous study (18). RNA was reverse-transcribed into complementary DNA (cDNA) using 12-base universal primers for IAVs (U12 A/G: AGCG/AAAAGCAGG) and M-MuLV reverse transcriptase (Promega, WI, USA). All segments were amplified using Ex Taq™ DNA polymerase (Takara, Japan) and segment-specific primers (19). The polymerase chain reaction (PCR) products were purified using a Cycle-Pure Kit and a Gel Extraction Kit (Omega Bio-Tek, USA), and the fragments were cloned into a pGEM-T easy vector and sequenced by the dideoxy method using an ABI 3730 DNA sequencer (Applied Biosystems, CA, USA) (19). Data were edited and aligned by BioEdit (version 7.0.5.2).
Phylogenetic analysis was performed based on the open reading frame of the HA and NA genes. Multiple alignments were constructed using the ClustalW multiple alignment tool of BioEdit (version 7.0.5.2). Phylogenetic trees were generated by neighbor-joining bootstrap analysis (1,000 replicates) using the Construct/Test Neighbor-Joining Tree tool of MEGA (version 7.0.14) (20).
Viral Pathogenicity in Chicken and Mouse Models
In the chicken pathogenicity test, groups of 5 5-week-old SPF white Leghorn chickens were intranasally infected with 10-fold serial dilutions (50 μl) of the H7N9/GX virus (1 ml) at 103-106 EID50. The mortality rate was observed for 14 days. Lethal dose 50% (LD50) titers were calculated using the method of Reed and Muench, and the LD50 value of the virus was expressed as log10EID50. At 106 EID50 infected groups, brain, lungs, spleen, kidneys, and cecum of chicken were collected when they died and stored below −80°C. Then we measured the virus titers in the tissue according to the previous study (12).
In the mouse pathogenicity test, 6-week-old female BALB/c mice (weight, 16–18 g) were intranasally inoculated with 10-fold serial dilutions (50 μl) of the virus at 104-108 EID50. Mouse body weight, symptoms, and survival rate were monitored daily for 14 days after virus inoculation. Groups of 6 mice were intranasally inoculated with serial 10-fold dilutions of the virus. Mouse lethal dose 50% (MLD50) titers were calculated using the method of Reed and Muench and expressed as the log10EID50 required to give one unit of MLD50 (12).
To compare morbidity (measured by weight loss), mortality, and viral distribution in different tissues, additional mice were inoculated with the virus at 106 EID50. Mouse weight loss and mortality rate were monitored daily for 14 days. Mice (n = 3 from each group) were killed 1, 3, and 5 days post-infection (dpi), and their blood and lungs were collected. The collected organs were separated into two sets; one set was frozen at −80°C for RNA extraction and other analyses, whereas the other set was fixed in 10% neutral formalin for histopathological study. Viral titers in the tissue homogenates were determined via the EID50 titer assay in the SPF chicken eggs (21).
Determination of the Interferon and Cytokine Levels in the Homogenized Lung Specimens
The pulmonary messenger RNA (mRNA) expression levels of interferon (IFN-β), IFN-γ, interleukin (IL)-1β, IL-6, and tumor necrosis factor (TNF)-α were determined by real-time reverse transcription (RT)-PCR using oligo(dT) transcribed-cDNA obtained from lung tissue homogenates. Glyceraldehyde 3-phosphate dehydrogenase expression was quantified by real-time RT-PCR and used for RNA normalization, and the ΔΔCt method (where Ct is threshold cycle) was used to estimate the differential gene expression between samples (22). Primers for real-time PCR are listed in Table 1.

Table 1. Primers used in this study.
Protein levels of IFN-γ, IL-1β, IL-6, and TNF-α in the clarified lung homogenates were determined by enzyme-linked immunosorbent assay (ELISA) using Magnetic Luminex® Assay Multiplex Kits (R&D Systems, USA), as described previously (23). Additionally, IFN-β levels in the lung homogenates were determined using a VeriKine Mouse IFN-β ELISA Kit (BioLegend, CA, USA).
Histopathological Staining
To characterize the pathological features in a mouse infected with the virus at 106 EID50, lung tissues were obtained 3 dpi. To examine the pathological changes in the infected mouse lungs 3 dpi, the right side of the lung was fixed in 10% formalin. All four lung lobes were embedded in paraffin and sectioned (5-mm thickness) for hematoxylin and eosin (H&E) staining (23). All lung fields of the four lobes in each sample were examined at 20× magnification.
RNA Isolation, Library Preparation, and Sequencing
Total RNA was extracted from the samples stored in RNAstore reagent using TRIzol (Invitrogen, USA), according to the manufacturer's protocol. The purity and concentration of the extracted RNA were determined by measuring the optical density at 260 nm (OD260) and 280 nm (OD280) using the NanoDrop 2000 System (Thermo Scientific, MA, USA). RNA integrity was assessed using the RNA Nano 6000 Assay Kit on the Agilent Bioanalyzer 2100 System (Agilent Technologies, CA, USA). RNA (3 μg per sample) was used as the input material, and sequencing libraries were generated using the NEBNext® Ultra™ Directional RNA Library Prep Kit for Illumina® (NEB, MA, USA), following the manufacturer's instructions. High-quality cDNA libraries were then sequenced on an Illumina Hiseq 4000 Sequencer (Illumina, CA, USA), and the base-calling was performed using the software CASAVA (version 1.8.2) (Illumina); as a result, 150-bp paired-end reads were generated (24).
Transcriptome Data Analysis
Raw data (raw reads) of FASTQ format were first processed using in-house Perl scripts. Clean data (clean reads) were obtained by removing low-quality reads and reads that contained the adapter or ploy-N from the raw data. At the same time, the Q30 value and the GC content of the clean data were calculated. Clean reads were then mapped to the mouse reference genome (GRCm38.p6) downloaded from the National Center for Biotechnology Information (https://www.ncbi.nlm.nih.gov/assembly/GCF_000001635.26) using the software HISAT2 (version 2.0.1). To detect the differentially expressed genes (DEGs), the number of clean reads assigned to a gene was determined using HTSeq (version 0.6.1) and then normalized to the fragments per kilobase of exon per million fragments mapped value. The differential expression levels among the groups were analyzed using the DEseq2 R package (version 1.20.0). DEGs were identified by setting the corrected P < 0.05 and |log2 fold change (FC)| >1 as the threshold parameters (25).
Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) analyses of DEGs were performed using the clusterProfiler package, with P < 0.05 set as the threshold. GO groups essential for viral immunity with adjusted P < 0.05 were visualized using bubble plots. Details of the genes that belonged to particular GO terms and their FC values were presented as circos plots using the “GOplot” library (26).
Quantitative Real-Time Reverse Transcription-Polymerase Chain Reaction Analysis
RNA was extracted using TRIzol and purified from the final aqueous phase using the RNeasy Mini Kit (Qiagen, Germany). RNA was treated with DNase (Promega), and first-strand cDNA was synthesized using All-in-One cDNA Synthesis SuperMix (Bimake, TX, USA). Relative mRNA expression was determined via SYBR Green-based quantitative RT (qRT)-PCR using SYBR Green SuperMix (Bimake) on an ABI ViiA 7 Real-Time PCR System (Applied Biosystems) (22). Obtained mRNA levels were normalized to the glyceraldehyde 3-phosphate dehydrogenase mRNA level. Gene-specific primers for qRT-PCR are listed in Table 1.
Statistical Analysis
The results were expressed as the mean ± standard deviation, and all the data were representative of at least three independent experiments. Data analysis was performed using Student's t-test. Differences between the mean values were considered significant at P < 0.05.
Results
Nucleotide Sequencing and Phylogenetic Analysis
The GX/YL01 H7N9 virus was isolated from a dead chicken obtained from the chicken farms in Guangxi province and passaged in SPF embryos. First, the entire genome of the isolated virus was sequenced. Full-genome sequence data for this virus (A/chick/Guangxi/YL01/2017) were submitted to GISAID (https://www.gisaid.org/) and are available under accession number EPI_ISL_436706. According to the BLAST analysis performed for each sequence (https://blast.ncbi.nlm.nih.gov/Blast.cgi), the novel isolated virus belonged to the AIV H7N9 subtype.
Previously, the genetic analysis indicated that the dynamic reassortment of the internal and surface genes of the Yangtze River Delta and Pearl River Delta lineages between LP-H7N9/H9N2/H6Ny and HP-H7N9 resulted in at least 36 genotypes (12, 27, 28). Using BLAST search in GenBank, we found that six internal genes of the GX/YL01 virus were highly homologous with poultry H9N2 virus (98.46–99.40%) (Supplementary Data 1). To understand the evolutionary relationships in detail, a phylogenetic analysis of the coding regions of the HA and NA genes was conducted. As shown in Figure 1, both HA and NA of the GX/YL01 H7N9 isolate belonged to the Yangtze River Delta lineage.
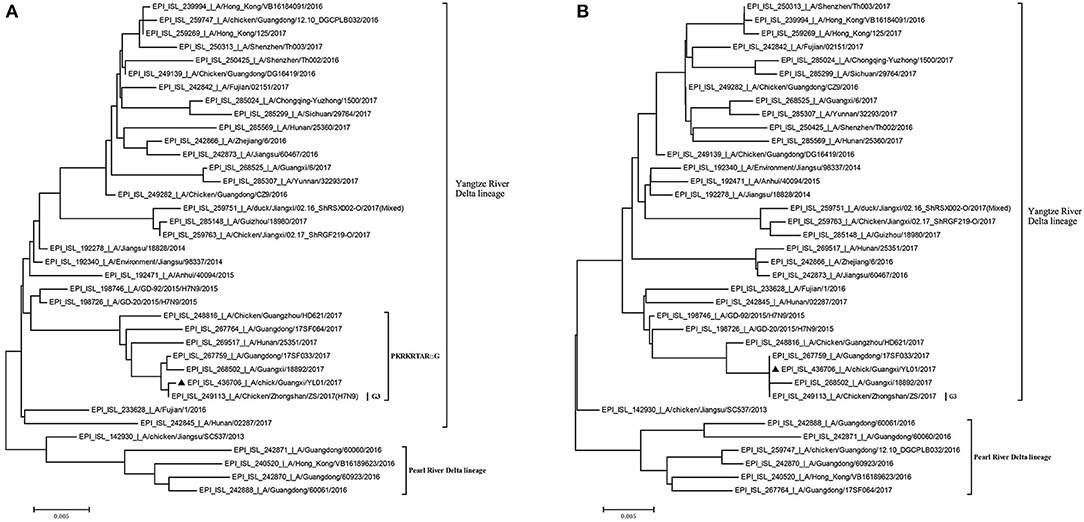
Figure 1. Phylogenies of HA and NA genes of H7N9 viruses from different genotype. All of the H7N9 viruses that represent different genotype viruses prevalent in wave5, including our one new isolate, were used to perform the phylogenetic analysis. Tree was constructed by using the neighbor-joining method with the maximum composite likelihood model in MEGA version 7.0.14 (http://www.megasoftware.net) with 1,000 bp replicates, based on the nucleotide sequence 1–1,695 (HA) and 1–1,398 (NA). Virus lineages are shown at right. Scale bar indicates nucleotide substitutions per site. (A) shows the HA gene tree, and (B) shows the NA gene tree.
Similar to the previously isolated HPAIV H7N9, GX/YL01 possessed an HA gene that belonged to the G3 genotype (the represented strain: A/Chicken/Zhongshan/ZS/2017-like) and a four-amino-acid-insertion at the cleavage sites (Pkrkrtarg) (Table 2). This insertion is characteristic of highly pathogenic avian species (12, 29). Unlike the A/Anhui/40094/2015 (GISAID ID: EPI_ISL_192471) HA protein, which showed an amino acid mutation (Q226L) at the HA receptor-binding site, increasing its binding affinity to 2,6-linked sialic acids (characteristic of human cell-surface receptors), the GX/YL01 HA gene showed a Q226 amino acid mutation (30).
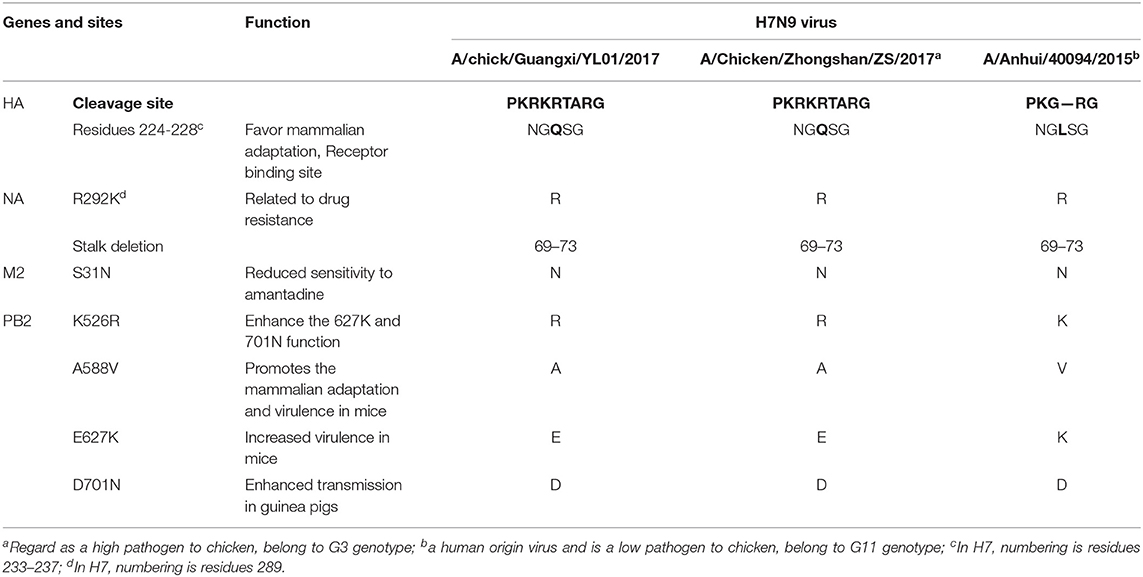
Table 2. Molecular characteristics of the H7N9 viruses isolated from poultry.
Similarly, the isolated GX/YL01 H7N9 virus possessed the amantadine resistance mutation (S31N) in M2 but not the NA inhibitor resistance mutation (R292K). Additionally, a five-amino-acid-deletion at positions 69–73, often observed in other H7N9 viruses, was observed in the NA stalk of GX/YL01. It has been documented that all the emerging human isolates possessed at least one of the four mammalian adaptation mutations, i.e., K526R, A588V, E627K, or D701N, in PB2. Nevertheless, the GX/YL01 H7N9 virus of avian origin possessed only one mutation (K526R) (Table 2).
H7N9 Viral Replication and Virulence in Chickens
The EID50 value of GX/YL01 was 8.17, which was calculated using the method of Reed and Muench and expressed as log10. To determine GX/YL01 viral replication and pathogenicity in chickens, groups of 5-week-old SPF white leg-horn chickens were intranasally inoculated with 10-fold serial dilutions (50 μl) of the GX/YL01 virus at 103-106 EID50. As shown in Figure 2A, the LD50 value in SPF chicken was 4.5, and 100% of the chickens infected with the virus at 106 EID50 succumbed by 4 dpi.
Figure 2. Challenge of chickens and mice with isolated H7N9 viruses. (A) Mortality of inoculated chickens. Groups of five 5-week-old SPF white leg-horn chickens were inoculated intranasally with 50 μl 10-fold serial dilutions containing 103-106EID50 of H7N9/GX. (B) Virus titers in the lungs, brains, spleen, kidney, and cloaca of the first four dead chickens in 106EID50 of H7N9/GX infected group. (C) Mortality of inoculated mouse. Groups of eight 6-week-old SPF BALB/c mice were inoculated intranasally with 50 μl 10-fold serial dilutions containing 103-106EID50 of H7N9/GX. (D) Weight changes in the mice. (E) Virus titers in the lungs, brains, spleen, and kidney of the first four dead mice in 106EID50 of H7N9/GX-infected group.
We collected the brain, lungs, spleen, kidneys, and cecum of each chicken inoculated with the virus at 106 EID50 and determined the viral titers in the eggs. Mean viral titers ranging from 3.7 to 6.0 log10EID50/g were detected in all the organs mentioned earlier (Figure 2B). Therefore, we concluded that GX/YL01 could systemically infect chickens after intranasal viral inoculation, thereby indicating that the GX/YL01 virus was highly pathogenic toward chickens.
Analysis of Viral Pathogenicity in Mice
The multiple basic amino acid motifs in the HA cleavage site of AIV H7N9 is a prerequisite for its lethality in mammals (12, 31, 32). To evaluate whether the four-amino-acid-insertion at the HA cleavage site increased GX/YL01 virulence in mammals, we tested viral replication and virulence in mice. Weight loss of infected mouse was monitored daily for 14 dpi, and MLD50 values were determined (Figure 2C). Consistent with previous studies, HPAIV GX/YL01 inoculation caused significant weight loss, and the MLD50 value was calculated to be 4.25 log10EID50/ml (Figure 2D). We collected the brain, lungs, spleen, and kidneys of each mouse inoculated with the virus at 106 EID50 and determined the viral titers in the eggs. As expected, high levels of infectious GX/YL01 were successfully rescued from the lung samples but not the spleen, kidney, or brain samples (Figure 2E).
To characterize the pathological features in mice infected with the virus at 106 EID50, lung tissues were obtained 3 dpi and subjected to histopathological analysis via H&E staining. Histopathological changes in mouse lungs were similar to those exhibited in human lungs; the GX/YL01 virus induced severe lung injury, represented by pulmonary alveolar hemorrhage, bronchial mucosa injury with accompanying inflammatory cell infiltration around the bronchus, and obvious interstitial hyperplasia with accompanying lymphocyte and dust cell infiltration (Figures 3C,D). On the contrary, no obvious histopathology was observed in the PBS-incubated mouse lungs (Figures 3A,B). Altogether, these results demonstrated that the GX/YL01 virus was virulent in mice, showing high lung viral titers and severe lung injury.
Figure 3. Histopathological changes in hematoxylin and eosin-stained lung tissues. Representative histopathological changes in hematoxylin and eosin-stained lung tissues from a mouse injected with PBS (A,B) or 106EID50 of H7N9/GX virus (C,D) 3 days postinfection. Pulmonary alveolar hemorrhage, bronchial mucosa injury, and accompanied by inflammatory cells infiltration around the bronchus (shown as a black arrow), obviously interstitial hyperplasia is captured, and accompany with lymphocytes and dust cells infiltration (shown as asterisk) (C,D). No obvious histopathology was observed in the PBS-incubated mouse lung (A,B).
Identification and Enrichment Analysis of Differentially Expressed Genes
To elucidate global host responses specifically associated with virus-induced lung injury sites in GX/YL01-infected mice, we used microarrays to assess the transcriptional profiles of the lung lesions. Mice (n = 3 from each group) were killed 3 dpi, and lungs were collected to determine the transcriptional cytokine response. Six cDNA libraries prepared using GX/YL01 virus-infected (GX_72) and non-infected (CK_72) mouse lungs were sequenced and filtered, yielding ~87–108 million clean reads per sample (Table 3). The transcripts were filtered by setting P ≤ 0.05 and |log2 FC| ≥ 1 as the thresholds. In total, 54,298 transcripts were identified via RNA sequencing analysis, and 566 of them were identified as DEGs between the virus-infected and non-infected mice (Supplementary Data 2). Among these, 400 genes were upregulated, showing log2 FC values ranging from 1 to 7.8, whereas the remaining 166 genes were downregulated, showing log2 FC values ranging from −4.3 to −1, as shown in the volcano map (Figures 4A,B). Meanwhile, the expression levels of 17 representative DEGs were further verified by qRT-PCR, confirming the accuracy and reliability of the transcriptome data (Table 4).
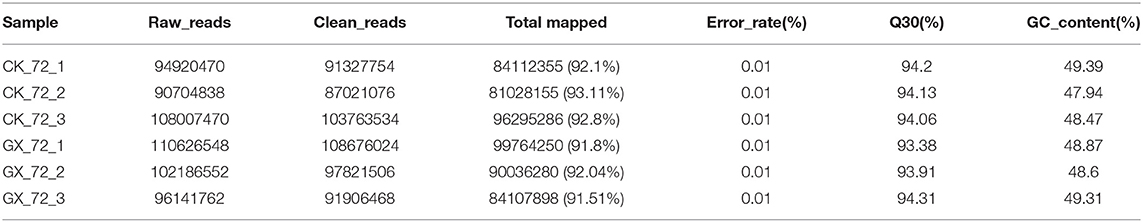
Table 3. Statistics of the RNA-seq datasets.
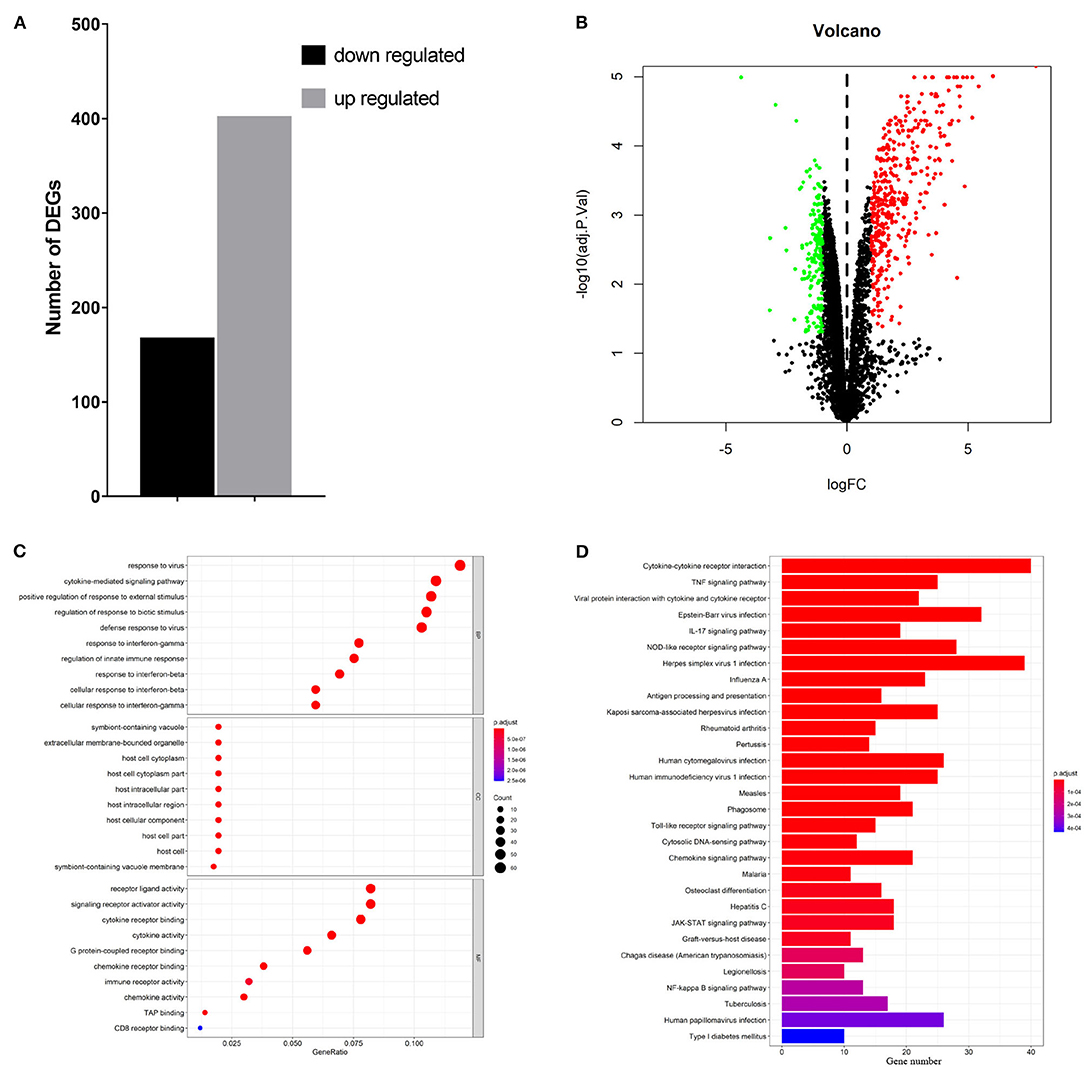
Figure 4. Transcriptome data profile generated by Illumina sequencing and differential expression analysis. (A) Numbers of differentially expressed genes. (B) Volcano map showing the significant differentially expressed genes between PBS or 106EID50 of H7N9/GX virus-infected mouse lung tissue. Red and green dots represent upregulated and downregulated genes, respectively, whereas the black dots represent microRNAs with no significant difference (P < 0.05 and |log2FC| ≥ 1 as the threshold cutoff). (C) Gene Ontology functional annotation and classification of differentially expressed genes. Functional classification of DEGs was performed using GO analysis (http://www.geneontology.org/) and the categories “biological process (BP),” “molecular function (MF),” and “cellular component (CC)” were analyzed. X-axis is the enrichment ratio (gene ratio); calculated as the ratio of the number of genes annotated to an entry in the selected gene set to the total number of genes annotated to the entry in the species. Size of the bubble represents the number of genes annotated to GO terms. Color represents the enrichment Q value. (D) Kyoto Encyclopedia of Genes and Genomes (https://www.kegg.jp/) functional annotations of the DEGs (top 30). A bar chart shows enrichment of the DEGs in the signaling pathways, the horizontal axis shows the number of genes, and the vertical axis shows each pathway. Color represents the enrichment value.
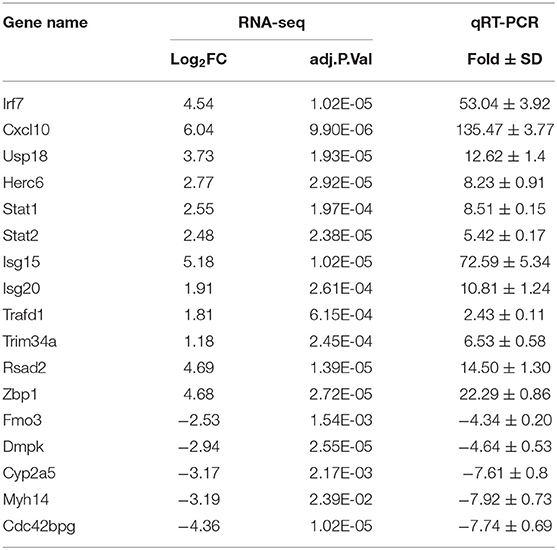
Table 4. Validation of DEGs in RNA-seq by qRT-PCR.
To better understand the host's response to the GX/YL01 virus infection, GO analysis was conducted (Figure 4C). The genes that were differentially expressed during this experiment are involved in biological processes, such as “response to virus,” “cytokine-mediated signaling pathway,” “positive regulation of cytokine production,” “regulation of response to biotic stimulus,” “defense response to virus,” and “response to interferon-beta” (Supplementary Data 3). Then, to identify the biological pathways in which the DEGs participated, KEGG pathway/enrichment analysis was performed. As shown in Figure 4D, DEGs were primarily involved in either host immune response or disease pathways, including “cytokine–cytokine receptor interaction,” “TNF signaling pathway,” “viral protein interaction with cytokine and cytokine receptor,” “Epstein–Barr virus infection,” “herpes simplex virus 1 infection,” and “influenza A” (Supplementary Data 4).
Interferon-Stimulating Genes (ISGs) and Cytokines Associated With Multi-Gene Ontology Terms Were Significantly Upregulated
Based on the earlier mentioned KEGG analysis results, we found that the DEGs were mainly related to immunity and antivirus pathways; thus, GO plot analysis was performed to present further details of the genes, such as the most popular genes related to the top GO terms and their gene expression levels. As shown in Figure 5A, genes associated with at least two GO terms were listed. These genes included IFN-stimulating genes (ISGs), z-DNA-binding protein 1, C-X-C motif chemokine 10, IFN regulatory factor 7, myeloid differentiation primary response gene 88, and IFN-induced transmembrane protein 3. On the other hand, cytokines, including IL-6, IL-1β, IFN-β, and TNF-α, were upregulated in response to H7N9 infection. Finally, the mRNA and protein levels of the selected cytokines in the GX/YL01 virus-infected lungs were tested by qPCR and ELISA, respectively. Significantly increased levels were exhibited in response to viral infection (Figures 5B,C).
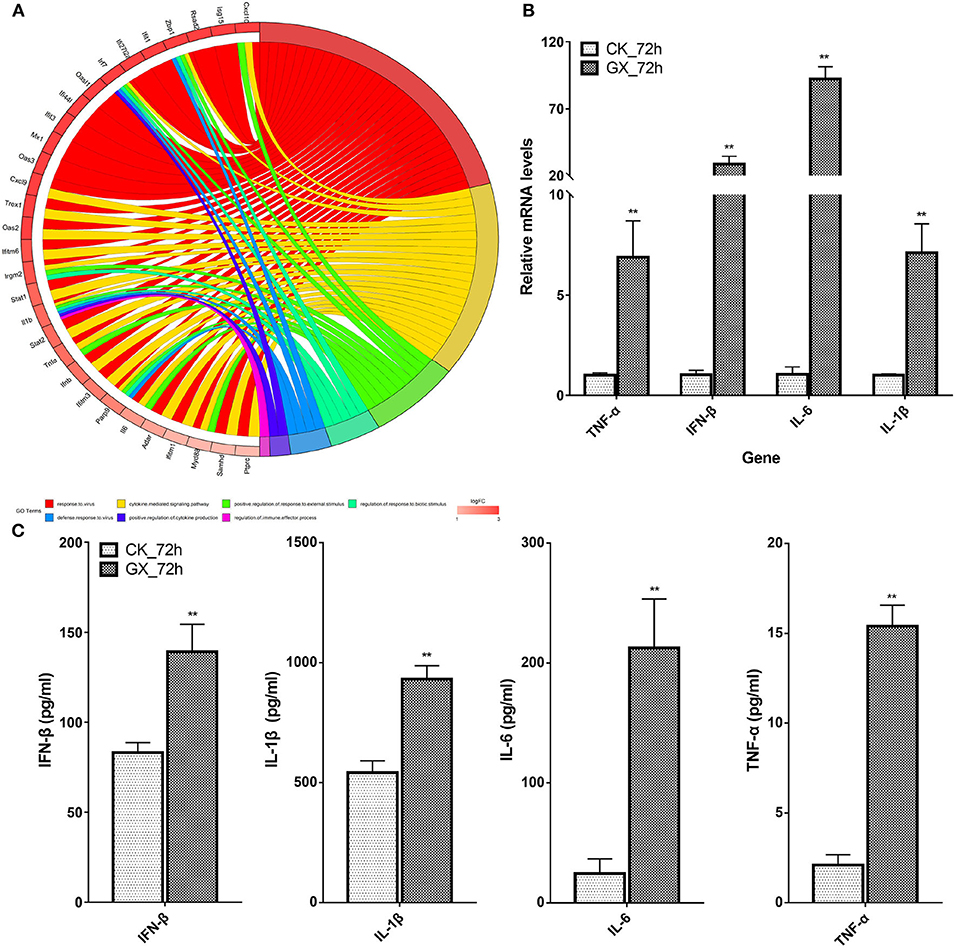
Figure 5. Most frequent genes in the Top7 Gene Ontology terms. (A) Circos plots of virus immunity-related Gene Ontology terms. Line color represents different Gene Ontology terms listed in legend; box color grid indicates the gene expression levels. (B) Relative messenger RNA levels of interferon-β, interleukin (IL)-1β, interleukin-6, and tumor necrosis factor-α genes in mouse lung were determined by real-time reverse transcription-polymerase chain reaction. Mouse glyceraldehyde 3-phosphate dehydrogenase messenger RNA was used for RNA concentration normalization. Error bar indicates ± SD. (means ± SD from three independent experiments) (*P < 0.05; **P < 0.01; all by two-tailed Student's t test). (C) Protein levels of cytokines interferon-β, IL-1β, IL-6, and tumor necrosis factor-α presented in mouse lung homogenates were determined by enzyme-linked immunosorbent assay. Lungs from infected mice (3–5 mice from each group) were homogenized in 1 ml of phosphate-buffered saline. Clarified homogenates were used for cytokine detection.
Discussion
Since its first emergence in China in 2013, the novel AIV of the H7N9 subtype has been of great concern worldwide. The human H7N9 virus is a reassortant virus with the surface HA and NA genes derived from duck and migratory bird viruses, respectively, and six internal genes originating from two different groups of H9N2 viruses circulating in the poultry in China. In this study, we isolated a highly pathogenic pure avian H7N9 virus (GX/YL01), which could replicate effectively in the mouse lung, causing severe pneumonia. Additionally, host innate immune response dysregulation and cytokine induction in mice were found to be similar to that in humans. Importantly, we found that over-activated antiviral signals and intense ISG products possibly contributed to the high virulence of the virus in mice.
Initially, H7N9 viruses were non-pathogenic in chickens; however, they evolved into highly pathogenic viruses, causing severe outbreaks in chickens during the fifth wave of the H7N9 virus outbreak. Indeed, the virus isolated in this study was highly pathogenic in SPF chickens (Figures 2A,B). The increased viral virulence resulted from several adaptive mutations. First, a four-amino-acid-insertion at the HA cleavage site, regarded as characteristic of highly pathogenic avian viruses, was observed in the GX/YL01 HA. Additionally, other adaptive mutations in the gene fragments contributed to the high pathogenicity of avian influenza strains. For example, the HA receptor-binding site mutation (Q226L), NA stalk deletion, and PB2 mutations (K526R, A588V, E627K, and D701N) promoted mammalian adaptation and increased viral virulence in mice (33). Unlike the L226 mutation at the receptor-binding site in the low pathogenic avian H7N9 virus, the Q226 mutation in the GX/YL01 HA resulted in a high binding affinity toward 2,3-linked sialic acids; this is characteristic of avian cell surface receptors. Taken together, the insertion at the HA cleavage site and the high binding affinity of avian cell receptors might explain the high pathogenicity of the virus in poultry.
Patients infected with H7N9 viruses often display a severe and lethal disease characterized by rapidly progressive pneumonia that results in acute respiratory distress syndrome and respiratory failure (34). To check whether the virus-infected mice developed pneumonia, we detected the histopathological changes in the H&E-stained lung tissues of the mice intranasally infected with the GX/YL01 virus at 106 EID50. As expected, diffuse alveolar damage, pulmonary alveolar hemorrhage, bronchial mucosa injury, inflammatory cell infiltration around the bronchus, and obvious interstitial hyperplasia could be observed in the virus-infected mouse lung tissues (Figure 3). However, clinically and histopathologically, H7N9 infection was not as severe in mice as it is in humans. Although viral characteristics could most likely be responsible for the differences in disease severity between mice and humans, underlying medical complications in humans might be another reason.
Several studies have demonstrated that high viral replication, lymphopenia, “cytokine storm” induction, macrophage replication, and viral dissemination beyond the respiratory tract are associated with increased severity of the H5 and H7 disease in humans and animal models (29, 35). Investigation of these features in pure HPAIV H7N9 infection warrants further study. To detail the host response to the infection, a transcriptome analysis of the virus-infected or control mouse lung was performed. Enrichment analysis of DEGs showed that some antiviral biological pathways, including “response to virus,” “cytokine-mediated signaling pathway,” “positive regulation of cytokine production,” “regulation of response to biotic stimulus,” “defense response to virus,” and “response to IFN-β” were enriched. GO plot analysis results showed that some genes, which we call “hot genes,” participated in multiple pathways. These genes could be divided into two categories, ISGs and cytokines, according to their functions. It has been shown that cytokine dysregulation is a contributory factor to H5N1- and H7N9-related disease pathogenesis. Our results also showed that pure HPAIV H7N9 infection induced high pulmonary levels of cytokines, including IL-6, IL-1β, IFN-β, and TNF-α. These results are consistent with the published report (15).
In conclusion, the present study reported the mRNA and cytokine expression profiles of pure HPAIV H7N9-infected mouse models for the first time. The results suggested that host innate immune response dysregulation and cytokine induction might play key roles in enhancing viral virulence in mice. This information will be critical for advancing our understanding of the pathogenicity of avian-origin H7N9 viruses in mammals.
Data Availability Statement
The datasets generated for this study can be found in NCBI SRA, NCBI Accession No. PRJNA662664.
Ethics Statement
The animal study was reviewed and approved by All animal experiments were approved by the Research Ethics Committee, Huazhong Agricultural University, Hubei, China (HZAUMO-2016-022) and were performed in accordance with the Guidelines for the Care and Use of Laboratory Animals of the Research Ethics Committee, Huazhong Agricultural University, Hubei, China.
Author Contributions
WG, HK, and ZZ: conceptualization and methodology. YZ, CL, and KH: software. WG and ZZ: formal analysis and writing—original draft preparation. WG, ZZ, and HM: investigation. ZZ, MJ, and YW: writing—review and editing. XYZ and MJ: supervision. WG, ZZ, MJ, and YW: project administration. ZZ and MJ: funding acquisition. All authors: have read and agreed to the published version of the manuscript.
Funding
This research was supported by the National Key Research and Development Program of China (2016YFD0501600) and the National Natural Science Foundation of China (31702255 and 31820103015).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fvets.2020.603584/full#supplementary-material
References
1. David PMH, Knipe M. Orthomyxoviruses. In: Palese MSP, editor. Fields Virology 6thedition. Philadelphia, PA: Lippincott Williams & Wilkins (2013). p. 1151–81.
2. Nelson MI, Vincent AL. Reverse zoonosis of influenza to swine: new perspectives on the human-animal interface. Trends Microbiol. (2015) 23:142–53. doi: 10.1016/j.tim.2014.12.002
3. Su S, Bi Y, Wong G, Gray GC, Gao GF, Li S. epidemiology evolution, and recent outbreaks of avian influenza virus in China. J Virol. (2015) 89:8671–6. doi: 10.1128/JVI.01034-15
4. Chen H, Yuan H, Gao R, Zhang J, Wang D, Xiong Y, et al. Clinical and epidemiological characteristics of a fatal case of avian influenza A H10N8 virus infection: a descriptive study. Lancet. (2014) 383:714–21. doi: 10.1016/S0140-6736(14)60111-2
5. Wei SH, Yang JR, Wu HS, Chang MC, Lin JS, Lin CY, et al. Human infection with avian influenza A H6N1 virus: an epidemiological analysis. Lancet Respir Med. (2013) 1:771–8. doi: 10.1016/S2213-2600(13)70221-2
6. Pan Y, Cui S, Sun Y, Zhang X, Ma C, Shi W, et al. Human infection with H9N2 avian influenza in northern China. Clin Microbiol Infect. (2018) 24:321–3. doi: 10.1016/j.cmi.2017.10.026
7. Poovorawan Y, Pyungporn S, Prachayangprecha S, Makkoch J. Global alert to avian influenza virus infection: from H5N1 to H7N9. Pathogens Global Health. (2013) 107:217–23. doi: 10.1179/2047773213Y.0000000103
8. Babakir-Mina M, Dimonte S, Lo Presti A, Cella E, Perno CF, Ciotti M, et al. Phylogenetic and evolutionary analysis of influenza A H7N9 virus. New Microbiol. (2014) 37:369–76.
9. Su S, Gu M, Liu D, Cui J, Gao GF, Zhou J, et al. Epidemiology evolution, and pathogenesis of H7N9 influenza viruses in five epidemic waves since 2013 in China. Trends Microbiol. (2017) 25:713–28. doi: 10.1016/j.tim.2017.06.008
10. Ding X, Luo J, Quan L, Wu A, Jiang T. Evolutionary genotypes of influenza A (H7N9) viruses over five epidemic waves in China. Infect Gene Evol. (2017) 55:269–76. doi: 10.1016/j.meegid.2017.09.027
11. Cui L, Liu D, Shi W, Pan J, Qi X, Li X, et al. Dynamic reassortments and genetic heterogeneity of the human-infecting influenza A (H7N9) virus. Nat Commun. (2014) 5:3142. doi: 10.1038/ncomms4142
12. Qi W, Jia W, Liu D, Li J, Bi Y, Xie S, et al. Emergence and adaptation of a novel highly pathogenic H7N9 influenza virus in birds and humans from a 2013 human-infecting low-pathogenic ancestor. J Virol. (2018) 92:1–12. doi: 10.1128/JVI.00921-17
13. Cline TD, Karlsson EA, Seufzer BJ, Schultz-Cherry S. The hemagglutinin protein of highly pathogenic H5N1 influenza viruses overcomes an early block in the replication cycle to promote productive replication in macrophages. J Virol. (2013) 87:1411–9. doi: 10.1128/JVI.02682-12
14. Writing Committee of the Second World Health Organization Consultation on Clinical Aspects of Human Infection with Avian Influenza, Abdel-Ghafar AN, Chotpitayasunondh T, Gao Z, Hayden FG, Nguyen DH, et al. Update on avian influenza A (H5N1) virus infection in humans. N Engl J Med. (2008) 358:261–73. doi: 10.1056/NEJMra0707279
15. de Wit E, Rasmussen AL, Feldmann F, Bushmaker T, Martellaro C, Haddock E, et al. Influenza virus A/Anhui/1/2013 (H7N9) replicates efficiently in the upper and lower respiratory tracts of cynomolgus macaques. mBio. (2014) 5:e01331-14. doi: 10.1128/mBio.01331-14
16. Zhang Q, Shi J, Deng G, Guo J, Zeng X, He X, et al. H7N9 influenza viruses are transmissible in ferrets by respiratory droplet. Science. (2013) 341:410–4. doi: 10.1126/science.1240532
17. Zhou H, Jin M, Chen H, Huag Q, Yu Z. Genome-sequence analysis of the pathogenic H5N1 avian influenza A virus isolated in China in 2004. Virus Genes. (2006) 32:85–95. doi: 10.1007/s11262-005-5849-9
18. Xu XJ, Xu GY, Zhou HB, Yu ZJ, Zhang AD, Song YF, et al. Evolutionary characterization of influenza virus A/duck/Hubei/W1/2004 (H9N2) isolated from central China. Virus Genes. (2008) 36:79–83. doi: 10.1007/s11262-007-0123-y
19. Hoffmann E, Stech J, Guan Y, Webster RG, Perez DR. Universal primer set for the full-length amplification of all influenza A viruses. Arch Virol. (2001) 146:2275–89. doi: 10.1007/s007050170002
20. Kumar S, Stecher G, Tamura K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. (2016) 33:1870–4. doi: 10.1093/molbev/msw054
21. Ma S, Zhang B, Shi J, Yin X, Wang G, Cui P, et al. Amino acid mutations A286V and T437M in the nucleoprotein attenuate H7N9 viruses in mice. J Virol. (2020) 94:e01530-19. doi: 10.1128/JVI.01530-19
22. Yu S, Mao H, Jin M, Lin X. Transcriptomic analysis of the chicken MDA5 response genes. Genes. (2020) 11:308. doi: 10.3390/genes11030308
23. Cai W, Chen S, Li Y, Zhang A, Zhou H, Chen H, Jin M. 14-Deoxy-11,12-didehydroandrographolide attenuates excessive inflammatory responses and protects mice lethally challenged with highly pathogenic A(H5N1) influenza viruses. Antiviral Res. (2016) 133:95–105. doi: 10.1016/j.antiviral.2016.07.020
24. Guo Y, Huang N, Tian M, Fan M, Liu Q, Liu Z, et al. Integrated analysis of microRNA-mRNA expression in mouse lungs infected with H7N9 influenza virus: a direct comparison of host-adapting PB2 mutants. Front Microbiol. (2020) 11:1762. doi: 10.3389/fmicb.2020.01762
25. He Y, Wang A, Chen S, Wu Z, Zhang J, Wang M, et al. Differential immune-related gene expression in the spleens of duck Tembusu virus-infected goslings. Veterinary Microbiol. (2017) 212:39–47. doi: 10.1016/j.vetmic.2017.08.002
26. Jopek K, Tyczewska M, Celichowski P, Malendowicz LK, Rucinski M. Transcriptome profile in unilateral adrenalectomy-induced compensatory adrenal growth in the rat. Int J Mol Sci. (2018) 19:1111. doi: 10.3390/ijms19041111
27. Quan C, Shi W, Yang Y, Yang Y, Liu X, Xu W, et al. New threats from H7N9 influenza virus: spread and evolution of high- and low-pathogenicity variants with high genomic diversity in wave five. J Virol. (2018) 92:e00301-18. doi: 10.1128/JVI.00301-18
28. Wang N, Sun M, Wang W, Ouyang G, Chen Z, Zhang Y, et al. Avian influenza (H7N9) viruses co-circulating among chickens, Southern China. Emerg Infect Dis. (2017) 23:2100–2. doi: 10.3201/eid2312.170782
29. Zhang F, Bi Y, Wang J, Wong G, Shi W, Hu F, et al. Human infections with recently-emerging highly pathogenic H7N9 avian influenza virus in China. J Infect. (2017) 75:71–5. doi: 10.1016/j.jinf.2017.04.001
30. Ramos, Krammer F, Hai R, Aguilera D, Bernal-Rubio D, Steel J, et al. H7N9 influenza viruses interact preferentially with alpha2,3-linked sialic acids and bind weakly to alpha2,6-linked sialic acids. J Gener Virol. (2013) 94:2417–23. doi: 10.1099/vir.0.056184-0
31. Hatta M, Gao P, Halfmann P, Kawaoka Y. Molecular basis for high virulence of Hong Kong H5N1 influenza A viruses. Science. (2001) 293:1840–2. doi: 10.1126/science.1062882
32. Suguitan AL, Jr., Matsuoka Y, Lau YF, Santos CP, Vogel L, et al. The multibasic cleavage site of the hemagglutinin of highly pathogenic A/Vietnam/1203/2004 (H5N1) avian influenza virus acts as a virulence factor in a host-specific manner in mammals. J Virol. (2012) 86:2706–14. doi: 10.1128/JVI.05546-11
33. Shi J, Deng G, Kong H, Gu C, Ma S, Yin X, et al. H7N9 virulent mutants detected in chickens in China pose an increased threat to humans. Cell Res. (2017) 27:1409–21. doi: 10.1038/cr.2017.129
34. Guan W, Yang Z, Wu NC, Lee HHY, Li Y, Jiang W, et al. Clinical correlations of transcriptional profile in patients infected with avian influenza H7N9 virus. J Infect Dis. (2018) 218:1238–48. doi: 10.1093/infdis/jiy317
Keywords: highly pathogenic H7N9 avian influenza virus, yangtze river delta lineage, over-activated antiviral signals, transcriptome, pathogenicity
Citation: Gong W, Huang K, Zhang Y, He X, Li C, Mao H, Wei Y, Zou Z and Jin M (2020) Transcriptome Profiles of Highly Pathogenic Pure Avian H7N9 Virus-Infected Lungs of BALB/c Mice. Front. Vet. Sci. 7:603584. doi: 10.3389/fvets.2020.603584
Received: 07 September 2020; Accepted: 20 November 2020;
Published: 21 December 2020.
Edited by:
Jesus Hernandez, Consejo Nacional de Ciencia y Tecnología (CONACYT), MexicoReviewed by:
Anamika Mishra, ICAR-National Institute of High Security Animal Diseases (ICAR-NIHSAD), IndiaAhmed Ali, Beni-Suef University, Egypt
Copyright © 2020 Gong, Huang, Zhang, He, Li, Mao, Wei, Zou and Jin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yanming Wei, 370551650@qq.com; Zhong Zou, zz19841024@126.com; Meilin Jin, jml8328@126.com